DNA methylation signature of cognitive reserve moderates CSF tau pathology in prodromal Alzheimer’s disease
David Lukacsovich, Juan I. Young, Lissette Gomez, Brian W. Kunkle, Zhixin Mao, Wei Zhang, X. Steven Chen, Deirdre M. O’Shea, Tatjana Rundek, Eden R. Martin, Lily Wang

TL;DR
Blood DNA methylation patterns can act as a biomarker for cognitive resilience in Alzheimer's disease, helping to predict memory decline.
Contribution
The study identifies a DNA methylation signature that moderates the impact of tau pathology on memory in early Alzheimer's disease.
Findings
Six DNA methylation sites were found to interact with CSF pTau181 to influence memory scores.
A methylation-based memory reserve score predicts slower memory decline in MCI patients.
The identified DNA methylation loci are linked to synaptic, vascular, immune, and metabolic pathways.
Abstract
Cognitive reserve (CR) refers to differences in the adaptability of cognitive processes that modify the impact of Alzheimer’s disease (AD) pathology on cognitive performance. Currently there are no established blood-based biomarkers of CR in prodromal AD. In this study, we operationalize CR as memory reserve, defined as moderation (attenuation) of the CSF pTau181-memory association. DNA methylation (DNAm) integrates genetic and environmental influences and may capture biological processes that mitigate the impact of AD pathology on memory. We aimed to identify blood DNAm loci that moderate the association between cerebrospinal fluid (CSF) phosphorylated tau (pTau181) and memory in mild cognitive impairment (MCI). We also sought to determine if a DNAm-based signature of memory reserve predicts future memory decline. We analyzed 92 amyloid positive MCI participants from the Alzheimer’s…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
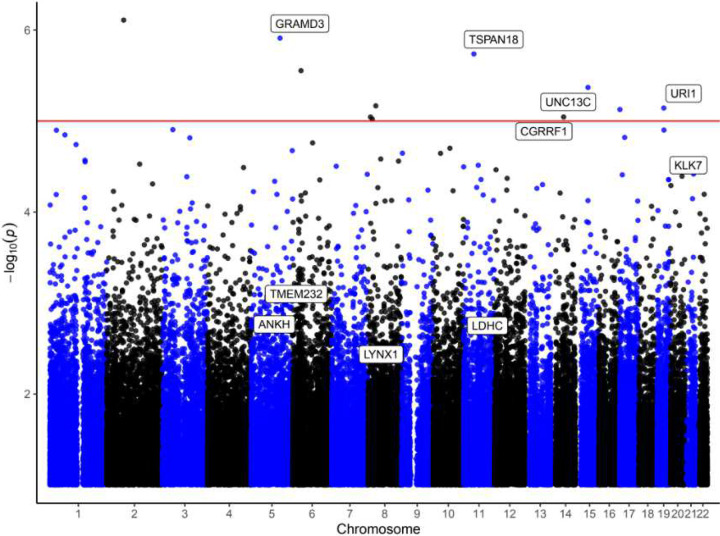 Figure 1
Figure 1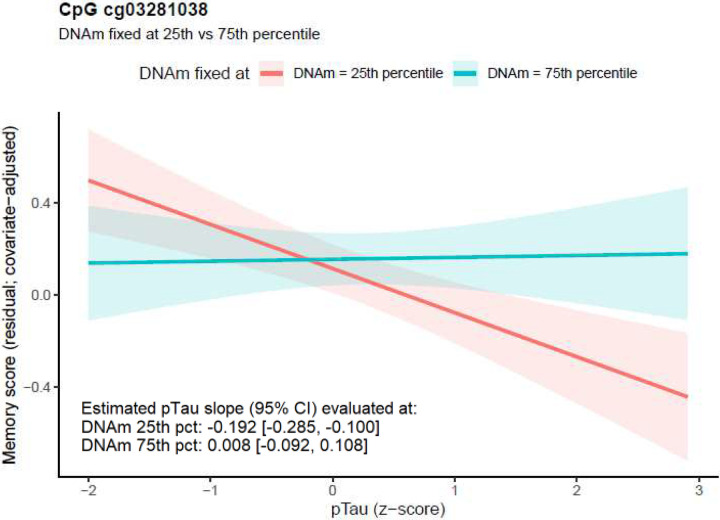 Figure 2
Figure 2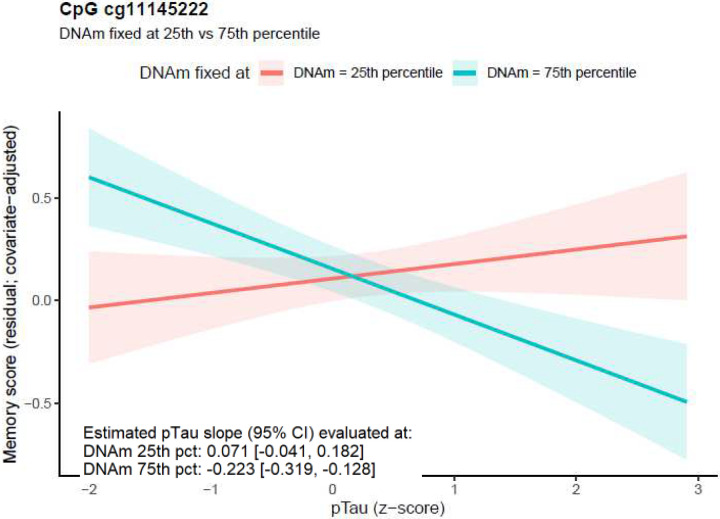 Figure 3
Figure 3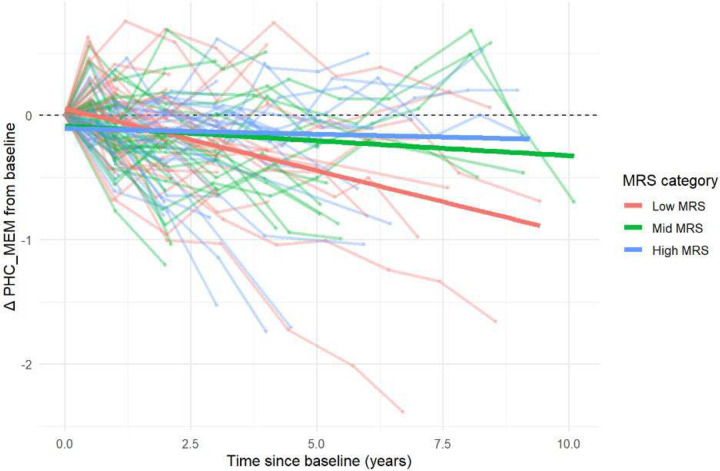 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Neuroinflammation and Neurodegeneration Mechanisms · Tryptophan and brain disorders
BACKGROUND
Alzheimer’s disease (AD) is a major public health challenge and one of the most financially costly diseases^1^. The pathological hallmark of AD is the accumulation of aggregated amyloid beta plaques and tau neurofibrillary tangles in the brain. However, there is considerable heterogeneity in clinical presentation that cannot be fully explained by the extent of AD pathology. Autopsy studies revealed that about one-quarter of cognitively normal older adults have sufficient amyloid pathology in the brain to meet neuropathological criteria for AD.^2,3^ It has been estimated that age-related neuropathological burden explains only about half of the variance in cognitive decline^4–6^, suggesting that additional factors are involved in the clinical manifestation of AD.
Cognitive reserve (CR) refers to differences in the adaptability of cognitive processes that modify the impact of AD pathology on cognitive performance^7,8^. In this study, we operationalized CR as memory reserve (i.e., memory-related CR), defined as attenuation of the CSF pTau181-memory association. CR has been studied using three broad approaches. First, the proxies approach estimates CR from activities thought to promote it, such as higher educational attainment, occupational complexity, or cognitively stimulating activities^9–12^. Second, the residuals approach estimates CR based on the amount of cognitive performance that remains unexplained after accounting for demographic factors and neuropathology burden^13,14^. Finally, the moderation approach, widely regarded as the most direct evidence of CR, tests whether a candidate CR factor attenuates the pathology-to-cognition association^7,15^.
CR is most informative at the prodromal stage of AD, where individuals with mild cognitive impairment (MCI) show substantial heterogeneity in disease progression. While many MCI patients experience rapid cognitive decline, a considerable proportion remain stable or even revert to normal cognition. It has been observed that higher CR is associated with preserved cognitive function, even after accounting for brain pathologies^16^.
Despite its recognized importance, the biological underpinnings of CR remain poorly understood. While a number of studies investigated CR using genetic and proteomic approaches^17–19^, few have examined its epigenetic basis. DNA methylation (DNAm) is the most extensively studied epigenetic mechanism, it is influenced by both genetic and environmental factors, including lifestyle factors such as smoking, diet, and exercise^20^, which have been shown to contribute to CR^21^.
In this study, we aimed to develop a blood DNAm signature of CR, by identifying CpGs that moderate the impact of AD pathology on memory. To this end, we analyzed 92 samples from the MCI subjects in Alzheimer’s Disease Neuroimaging Initiative (ADNI) study with matched blood-based DNA methylation, CSF pTau181 levels, and PHC_MEM harmonized memory scores measured at the same visit. Specifically, we modeled memory as a function of DNAm, pathology as quantified by CSF pTau181 levels, and their interaction, adjusting for age, sex, number of APOE4 alleles, and major immune cell type proportions in the blood samples. We used CSF pTau181 as our index of tau pathology because it is sensitive to early disease-related changes, significantly associated with clinical cognitive symptoms, and correlates with greater baseline tau-PET signal, as well as faster longitudinal tau accumulation, especially among amyloid-positive subjects^22,23^.
To better understand the functional roles of CR-related DNAm changes, we performed integrative analyses leveraging multiple external resources, including eQTM (methylation and gene expression associations), mQTL (methylation and genetic variant associations), GWAS summary statistics, brain-blood correlations, and biological pathway databases. Furthermore, we evaluated the feasibility of DNAm as a biomarker for individualized prognosis in MCI, a stage when disease trajectories may still be modifiable.
METHODS
Study participants
We analyzed blood DNA methylation data from 92 amyloid-positive, self-identified non-Hispanic White participants who were diagnosed with MCI in the ADNI cohort. In addition to DNAm data, each of these subjects also had available data on CSF p-tau181 (PHC_pTau), memory composite scores (PHC_MEM), and amyloid positivity status, all generated by the Alzheimer’s Disease Sequencing Project Phenotype Harmonization Consortium (ADSP-PHC)^24^. Data was obtained from the ADNI portal (adni.loni.usc.edu).
Cognitive measures and CSF biomarkers
PHC_MEM is a harmonized memory composite derived primarily from verbal learning and delayed-recall measures (e.g., WMS-R Logical Memory, Rey AVLT, and memory items from ADAS-Cog, and screening instruments), co-calibrated across ADNI, Adult Changes in Thought (ACT) and Religious Orders Study and Memory and Aging Project (ROS/MAP) cohorts using confirmatory factor analysis^24^.
The CSF AD biomarkers Aβ42, total tau, and pTau181 in ADNI were quantified using the Roche Elecsys platform^25^ and subsequently co-calibrated across ADSP-PHC cohorts by the ADSP-PHC consortium. Harmonization involved removing duplicate observations, excluding log10-scale outliers beyond the interquartile range (Q1−1.5 IQR or Q3+1.5 IQR), and standardizing the remaining values to z-scores to generate harmonized metrics comparable across ADNI, NACC, and Knight ADRC datasets^26^. Amyloid-positive (A+) status was then assigned using data-driven cut points from a two-component Gaussian mixture model applied to the post-QC z-scores of CSF Aβ42.
DNA methylation data pre-processing
The analysis pipeline followed our established workflow^12^. CpG methylation was measured using the Illumina Infinium MethylationEPIC v1.0 Beadchip arrays. Supplementary Table 1 shows detailed probe and sample counts at each quality control (QC) step. For the QC of samples, we removed samples with low bisulfite-conversion efficiency (< 85%) or had discrepancies between the recorded sex and DNAm-predicted sex status. For the QC of probes, we removed probes lacking annotations, mapping to sex chromosomes, or whose IDs did not start with “cg”. Missing probe values, as well as probes with detection P-values > 0.01, were imputed using methyLImp2 R package^27^. We also removed CpGs that are cross-reactive^28^ or located close to single nucleotide polymorphism (SNPs).
Methylation beta values were normalized using β-mixture quantile normalization (BMIQ) as implemented in the watermelon R package^29^. Batch effects were corrected with Harman R package^30^. We removed outlier samples identified in principal component analysis (PCA), defined as those falling outside –3 standard deviations from the mean of PC1 and PC2. Restricting to samples from MCI subjects, our final DNAm dataset included 92 samples.
Next, using the quality controlled DNAm data, we estimated the immune cell-type proportions (B lymphocytes, natural killer cells, CD4+ T cells, CD8+ T cells, monocytes, neutrophils, and eosinophils) using the EpiDISH R package. Consistent with previous blood-based DNAm studies^31–33^, granulocyte proportions were computed as the sum of neutrophils and eosinophils proportions, as both cell types are classified as granular leukocytes.
Statistical Models for identifying DNAm involved in memory reserve
We first regressed PHC_MEM scores on covariates (age, sex, number of APOE4 alleles, estimated major immune cell type proportions) and used the residuals as covariate-adjusted memory scores. For each CpG, we then fit
where DNAm is DNA methylation beta value, pTau181 is CSF pTau181 level standardized to z-scores, and ε is residual error term.
A significant DNAm×pTau181 interaction (γ≠0) indicates that the association between pTau181 and memory depends on DNAm at that CpG. Assuming higher pTau181 is associated with worse memory (β<0), a positive γ indicates attenuation of the negative pTau181-memory association at higher DNAm, consistent with greater memory reserve. On the other hand, a negative γ indicates more negative pTau181-memory association at higher DNAm (i.e., less negative association at lower DNAm), where greater memory reserve corresponds to lower DNAm. The pTau181 slope at a given DNAm level is estimated by β + γDNAm.
Inflation assessment and correction
We estimated genomic inflation factor (lambda value) using the bacon method, which was specifically designed for EWAS^34^. The estimated inflation factor was 1.06 and the estimated bias was −0.058. After genomic correction using the bacon method, the estimated inflation factor and bias were 1 and 9.90×10^−7^, respectively. The bacon method was then used to compute bacon-corrected effect sizes, standard errors, and P-values for the DNAm × pTau181 interaction. To exclude CpGs with minimal variability, we required an interquartile range (IQR) greater than 0.02 in beta values across all samples. We considered CpGs with P-values less than 1 × 10^−5^ to have suggestive significance. To account for multiple comparisons, we computed False Discovery Rate (FDR).
Differentially methylated regions analysis
For region-based analysis, we used the comb-p method^35^, which scans genome-wide CpG locations and P-values to identify regions enriched with clusters of low P-values. We used P-values of the DNAm × pTau181 interaction effect as input for comb-p and parameter settings with --seed 0.05 and --dist 750 (a P-value of 0.05 is required to start a region and extend the region if another P-value was within 750 base pairs). These parameters were shown to have optimal statistical properties in our previous comprehensive assessment of the comb-p software^36^. As comb-p uses the Sidak method to account for multiple comparisons, we selected differentially methylated regions (DMRs) with Sidak P-values less than 0.05. To further reduce false positives, we also required all the CpGs within the DMR to have a consistent direction of change.
Functional annotation and pathway analysis
Significant individual CpG methylation signals and DMRs were annotated using gene annotations from Illumina and the Genomic Regions Enrichment of Annotations Tool (GREAT) software^37^, which associates genomic regions with target genes.
To identify biological pathways enriched for CR-related DNAm, we used the methylRRA function from the methylGSA R package^38^, which analyzes single CpG P-values as input. Briefly, methylGSA computes a gene-wise ρ value by aggregating P-values from multiple CpGs mapped to each gene, adjusts for the different numbers of CpGs per gene using Bonferroni correction, and then performs Gene Set Enrichment Analysis^39^ in pre-ranked mode to identify pathways enriched with significant CpGs. We analyzed GO terms from the KEGG and REACTOME databases, restricting analyses to pathways containing between 5 and 200 genes. To avoid gene sets where the enrichment signal is driven by only one or two genes, we additionally required that significant gene sets include at least three genes in the “core enrichment” subset.
Integrative analyses with gene expression, genetic variants, and brain-to-blood correlations
To evaluate the effect of the significant DNAm on the expression of nearby genes in blood samples, we overlapped our CR-related DNAm, including both significant individual CpGs and those located within DMRs, with eQTm analysis results in Supplementary Tables 2 and 3 of Yao et al. (2021)^40^.
To assess the correlation of DNAm levels between blood and brain samples at CR-related CpGs, we used the London dataset, which includes 69 matched samples from blood and brain prefrontal cortex tissues^41^. Brain-blood correlations were assessed using two approaches: (1) unadjusted correlations based on methylation beta values and (2) adjusted correlations using residuals from regression models accounting for covariates. In unadjusted analysis, we calculated Spearman rank correlations between DNA methylation beta values measured in brain and blood samples. For adjusted analysis, we accounted for potential confounders by removing the effects of covariates. Specifically, we removed effects from estimated neuron proportions in brain samples (or estimated immune cell-type proportions in blood samples), batch, age at death (for brain samples) or age at blood draw (for blood samples), and sex, by fitting linear models separately for brain and blood samples and extracting residuals. Spearman correlations were then calculated using these residual values to assess the relationship between brain and blood methylation levels independent of confounders.
For correlation and overlap with genetic loci, we searched blood mQTLs using the GoDMC database (http://mqtldb.godmc.org.uk/downloads).^42^ Using the same criteria from the original GoDMC study,^42^ we considered a cis P-value smaller than 10^−8^ and a trans P-value smaller than 10^−14^ as significant. The genome-wide summary statistics for genetic variants associated with dementia described in Bellenguez et al. (2022)^43^ were obtained from the European Bioinformatics Institute GWAS Catalog (https://www.ebi.ac.uk/gwas/) under accession no. GCST90027158.
Computation of Methylation Reserve Scores (MRS) and assessment of its utility in predicting future memory decline
The MRS was computed based on the 6 significant individual CpGs and 88 CpGs located within DMRs. In Model 1 described above, for each subject and at each CpG, pTau effect on memory at a given DNAm level is estimated as βpTau + γ_pTau× DNAm_M, where βpTau is the estimated main effect of pTau, γpTau× DNAm is the interaction effect, and M is methylation beta value. We then averaged the CpG-specific values across the 94 CpGs to obtain a subject-specific raw score (MRS_raw_), so that lower values indicate greater pTau-associated memory decline. It was then rescaled as a z-score using the scale () function.
To evaluate if the MRS associates with future memory decline, we fitted the mixed-effects model below (Model 2), using the lmerTest R package, to the 88 ADNI subjects with longitudinal follow up information.
RESULTS
Study cohort
To identify DNAm signatures for memory reserve, we analyzed a cross-sectional dataset that included CSF pTau181 biomarker, memory assessments, and blood DNAm measured at the same visit. Our analysis included 92 amyloid positive individuals diagnosed with MCI from the ADNI cohort. Participants had a mean age of 74.67 ± 7.51 years; 44.57% (41 subjects) were women, and the mean duration of education was 16.21 ± 2.78 years. Approximately half (52%, 48 subjects) carried at least one APOE ε4 allele, and 43% (40 subjects) reported a history of smoking. Baseline CSF pTau181 biomarker concentration was 46.46 ± 27.32 pg/mL (Table 1).
The CSF pTau181-memory association are dependent on DNA methylation at individual CpGs and DMRs
After regressing PHC_MEM on covariate (age, sex, APOE ε4 allele count, and immune cell type compositions), we used the residuals as covariate-adjusted memory scores and fit Model 1 at each CpG, testing DNAm×pTau181 interaction (with corresponding main effects of DNAm and pTau181). After correcting for genomic inflation, we identified 11 CpGs with a DNAm×pTau181 interaction effect meeting the suggestive significance threshold (P < 1×10^−5^); none reached the 5% FDR significance. After excluding low-variability CpGs (IQR < 0.02 in beta values across all samples), 6 CpGs remained. Among them, 1 CpG (cg03281038) was located in the promoter region of UNC13C gene. Four CpGs were located in the gene bodies of GRAMD3, TSPAN18, CGRRF1, and URI1 genes, and 1 CpG was located in an intergenic region (Table 2, Figure 1, Supplementary Figure 1).
Across the 6 CpGs with suggestive DNAm × pTau181 interaction effects, DNAm moderated the association between pTau and memory, with the strength of the association varying by CpG-specific methylation level. Four CpGs showed positive interaction estimates, such that higher DNAm was associated with a weaker (less negative) pTau-memory relationship. In contrast, for two CpGs (cg01602139 and cg11145222), the interaction estimates were negative, indicating that lower DNAm was associated with a weaker pTau181- memroy relationship (i.e., greater memory reserve).
For example, at cg03281038 in the UNC13C promoter region, the pTau memory association varied by DNAm level. When DNAm was fixed at the 25^th^ percentile, higher pTau was significantly associated with poorer memory performance (β = −0.192, 95% CI −0.285 to −0.100). In contrast, when DNAm was fixed at the 75^th^ percentile, the pTau-memory slope was near 0 (β = 0.008, 95% CI: −0.092 to 0.108), indicating attenuation of pTau-memory association and supporting a moderation effect (Figure 2). UNC13C encodes a presynaptic protein that primes synaptic vesicles and is essential for neurotransmitter release, a process supporting synaptic function and neural communication that has been repeatedly implicated in cognitive resilience^44^.
Figure 3 shows a CpG with a negative DNAm × pTau181 interaction estimate. At cg11145222 located in TSPAN18, when DNAm was fixed at the 75^th^ percentile, higher pTau181 was associated with poorer memory (β = −0.223, 95% CI: −0.319 to −0.128). In contrast, when DNAm was fixed at the 25^th^ percentile, the pTau-memory association was not significant (β = 0.071, 95% CI: −0.041 to 0.182), indicating attenuation at lower DNAm. Biologically, TSPAN18 has been implicated in endothelial calcium signaling and thrombo-inflammatory responses, processes contributing to vascular and blood-brain barrier (BBB) integrity^45^. Impairment of vascular and BBB integrity has been repeatedly associated with cognitive decline, aging, and AD pathology^46^.
Interestingly, we found that none of these CpGs showing a significant DNAm × pTau interaction had marginal associations with CSF pTau181 or memory that survived Bonferroni correction; the only nominal signal was for cg26869675 with memory (P-value = 0.038), which did not remain significant after multiple-testing correction (Supplementary Figure 2).
Using P-values for individual CpGs as input, comb-p software^35^ identified 11 DMRs, which achieved both a nominal P-value < 1×10^−5^ and a Sidak multiple comparison-adjusted P-value < 0.05. We also required all the CpGs within each DMR have a consistent direction of change in estimated effect sizes in the individual CpG analysis (Table 3). The number of CpGs in these DMRs ranged from 3 to 17. Among these DMRs, the majority (7 DMRs, 64%) showed hypermethylation associated with greater memory reserve. A total of 5 DMRs (45%) are in promoter regions of the genes KLK7, TMEM232, ANKH, LYNX1, and LDHC. As with the significant individual CpGs, we found that all DMRs, except one located in gene body of JAKMIP3, showed no significant association with either CSF pTau181 or memory (Supplementary Figure 3).
Because 43% of participants reported a history of smoking, we refitted the models with additional adjustment for smoking status. Effect estimates and P-values were very similar, and all CR-related CpGs and DMRs remained highly significant (Supplementary Table 2), indicating that these associations are unlikely to be explained by smoking history.
Pathway analysis revealed memory reserve-related DNA methylation are enriched in biological pathways involved in metabolic health and inflammation
To further understand the biological processes underlying cognitive reserve, we performed pathway analysis using the methylGSA software which used P-values from all the CpGs as input.^38^ We applied a 25% false discovery rate (FDR) threshold, an alternative to the conventional 5% FDR that has been suggested for GSEA.^47^ At this threshold, we identified 2 KEGG and 20 Reactome pathways significantly enriched for memory reserve-related DNA methylation (Supplementary Table 3). The most significant pathway was the KEGG adipocytokine signaling pathway (P-value = 2.6 × 10^−4^, FDR = 0.056), which represents the network of hormones and cytokines released by adipose tissue, most prominently leptin and adiponectin. Leptin enhances synaptic plasticity in the hippocampus, promotes β-amyloid clearance, and improves memory performance in animal models of aging and AD.^48^ In the Framingham Heart Study, higher plasma leptin levels were associated with a lower risk of incident dementia and with favorable brain health indices.^49^ Adiponectin, on the other hand, exerts insulin-sensitizing, anti-inflammatory, and antioxidant effects in peripheral tissues, functions that may also provide protection against neurodegenerative processes such as AD.^50^
Similarly, several significant Reactome pathways were also related to adipose and metabolic health. These included Regulation of lipid metabolism by PPARα (a lipid-sensing signaling cascade that enhances fatty acid oxidation and energy metabolism), Triglyceride catabolism (breakdown of stored fat to provide energy), Adipogenesis (formation of mature fat cells capable of safely storing and releasing lipids), Transcriptional regulation of white adipocyte differentiation (activation of gene programs that produce insulin-sensitive white adipocytes), and Epigenetic regulation of adipogenesis genes by MLL3 and MLL4 complexes (histone-modifying complexes that epigenetically control metabolic and inflammatory gene expression).
Taken together, these results highlighted the importance of maintaining metabolic balance, preventing lipid accumulation in the brain and peripheral tissues where it can trigger inflammation, preserving insulin sensitivity, and reducing systemic metabolic stress that may worsen neuropathology.^51^ Balanced adipose and lipid signaling pathways support healthy lipid handling and anti-inflammatory responses, which in turn may help preserve brain function and promote cognitive resilience in the presence of neurodegenerative pathology.
Correlation of significant DNAm with expression of nearby genes in blood samples
To evaluate the functional relevance of our significant DMRs and CpGs involved in memory reserve, we compared them with established DNAm to gene-expression associations (i.e., eQTMs) computed from matched blood DNAm and gene-expression data in over 4000 participants from the Framingham Heart Study^40^. Among the 6 significant individual CpGs and 88 CpGs within the 11 DMRs, 3 CpGs in the promoter region of ANKH and 6 CpGs in the promoter region of LDHC, all of which were located in DMRs, showed significant cis (within 500 kb) associations with gene expression levels (Supplementary Table 4).
Notably, the ANKH gene encodes a transmembrane protein that regulates extracellular mineralization by controlling the export of key metabolites such as ATP and citrate. A recent genome-wide association study (GWAS) identified a protective allele (rs112403360) in ANKH that is strongly associated with cognitive resilience and a reduced risk of AD among cognitively healthy centenarians^52^. Impaired ANKH function can lead to pathological calcification, including arterial and vascular mineralization. The protective allele, as well as the hypomethylation we observed in the promoter of this gene, may help preserve vascular integrity and reduce neuropathological burden, thereby supporting sustained cognitive function.
At the other eQTM locus, the LDHC gene is mainly expressed in testis. However, its paralog LDHA, which shares high sequence similarity and is located in proximity (< 7 kb), is ubiquitously expressed in somatic tissues, including blood and brain. Because both DNAm and gene expression in the eQTM resource were measured using expression arrays, which are susceptible to cross-hybridization between paralogous genes, the observed LDHC eQTM likely reflects transcriptional activity at the broader LDHC/LDHA locus, and may be driven predominantly by LDHA expression. LDHA encodes lactate dehydrogenase A, which produces lactate, a key neuronal energy substrate essential for long-term memory formation^53^. Although LDHA-associated lactate production is required for normal neuronal plasticity, studies in animal models of aging suggest that sustained alterations in neuronal LDH/LDHA activity and a shift toward higher brain lactate are associated with shortened lifespan, neurodegeneration, and age-related memory impairment, suggesting that tight regulation of this locus is important for cognitive resilience^54^.
Methylation quantitative trait loci and intersection with genetic risk loci in dementia
To identify methylation quantitative trait loci (mQTLs) for the significant DMRs and CpGs, we next performed look-up analyses using the GoDMC database^42^ for mQTLs. Among the 6 significant CpGs and 88 CpGs located in the DMRs, 64 and 21 CpGs had mQTLs in cis and in trans, respectively (Supplementary Table 5).
Next, to evaluate if the significant mQTLs overlapped with genetic risk loci implicated in AD, we compared the mQTLs with genetic variants nominated by a recent Alzheimer’s Disease and Related Dementias (ADRD) GWAS meta-analysis.^43^ We found 237 mQTLs associated with DNAm at 16 significant CpGs (all located within DMRs) overlapped with genetic variants reaching suggestive significance (P-value < 1 × 10^−5^). Among them, 65 mQTLs associated with DNAm at the same 16 significant CpGs, are in high LD with genome-wide significant loci rs6605556 (P-value = 7.1 × 10^−20^) within the HLA region on chr6:32,395,036–32,636,434, which included HLA-DRA, HLA-DRB5, HLA-DRB1, HLA-DQA1, and HLA-DQB1 genes (Supplementary Table 6).
Correlation of DNAm levels in blood and brain samples at memory reserve related CpGs
We next evaluated cross-tissue concordance of DNAm levels at significant CpGs using the London dataset, which includes matched pre-mortem blood and post-mortem prefrontal cortex DNAm profiles from 69 subjects^41^. At 5% FDR, among the 6 significant individual CpGs and 88 CpGs mapped within the identified DMRs, 46 CpGs (48.9%) showed significant brain-blood correlations in both unadjusted and adjusted analyses that accounted for covariate variables (Supplementary Table 7). These CpGs are located in promoter regions of the LDHC, KLK7, ANKH, TMEM232, LYNX1 genes and intergenic regions. All CpGs showed positive brain-blood methylation correlations.
Methylation-based memory reserve biomarker is significantly associated with future memory decline in MCI subjects
To evaluate feasibility of leveraging cognitive reserved-related DNAm to predict future memory decline, we developed a methylation reserve score (MRS) from CpGs with significant DNAm × pTau interactions, including 6 individual CpGs and 88 CpGs located within DMRs. For each subject and at each CpG, the pTau effect on memory is computed as βpTau + γpTau× DNAmM, where βpTau is the estimated main effect of pTau, and γpTau× DNAm is the estimated interaction effect, and M is methylation beta values in Model 1 (Methods). For each subject, the MRS was then computed by averaging these CpG-specific values across all 94 selected CpGs. Note that the MRS reflects the DNAm-dependent effect of pTau for each subject, and lower MRS values indicate greater pTau-associated memory decline (i.e., a more negative pTau effect on memory), while higher values indicate attenuation of pTau’s impact (i.e., less negative pTau effect on memory, thus more memory reserve).
Among 92 MCI subjects with DNA methylation, PHC_MEM, and CSF pTau measured at the same visit (defined as baseline visit), 88 had at least one follow-up memory assessment. We restricted the longitudinal dataset to follow-up visits occurring after the baseline visit and fitted linear mixed-effects models to assess the association between baseline MRS and longitudinal memory (PHC_MEM). Random intercept effects for each subject were included to account for within-subject correlations among repeated measures. We adjusted for baseline pTau, age, sex, education, APOE4, and included interaction effects MRS × time, pTau × time, and pTau × MRS. The interpretations of the mixed effects model coefficients are presented in Table 4. Note that baseline age, pTau and MRS were standardized (z-scored), so that their mean baseline values are 0.
Our results showed that memory declined significantly over time (β_time_ = −0.050 per year, P-value = 1.8 × 10^−9^) (Table 5). At baseline (time = 0, average MRS and pTau), older age and APOE4 carrier status are significantly associated with lower memory (β_age_ = −0.308, P-value = 6.1 10^−6^; β_APOE4_ = −0.480, P-value = 2.2×10^−5^), while sex, education and the main effect of MRS were not significant. Higher baseline pTau is associated with lower baseline memory (β = −0.138, P-value = 0.025); it also predicted faster decline (β_time × pTau_ = −0.026 per year per 1 unit increase in pTau, P-value = 3.0 × 10^−4^). At baseline, MRS attenuates the harmful effect of pTau on memory (β_MRS × pTau_ = 0.217, P-value = 2.4 × 10^−4^). Importantly, higher MRS at baseline is significantly associated with slower decline (β_MRS × time_ = 0.034, P-value = 9.5 × 10^−5^). Taken together, these results indicate that higher MRS corresponds to greater memory reserve, reflected by slower memory decline and reduced pTau-related memory impairment at baseline.
Figure 4 shows changes from baseline in harmonized memory scores for the 88 MCI subjects, grouped by tertiles of their baseline MRS. Notably, subjects with higher MRS showed slower memory decline over time. To formally test group differences, we refit the mixed model described above, except by replacing MRS scores with MRS tertile indicators. Our results showed that compared to the high-MRS group, the low-MRS group declined significantly faster (β_low MRS × time_ = −0.053, P-value = 0.008) and showed a stronger harmful baseline pTau-to-memory association (β_low MRS×pTau_ = −0.520, P-value = 4.1×10^−4^); the mid- vs. high-MRS comparison was not significant (Supplementary Table 8). Overall, higher baseline MRS is associated with more attenuation in tau-related effects on memory at baseline, as well as slower longitudinal memory decline, consistent with greater memory reserve.
DISCUSSION
Consistent with the consensus that conceptualizes CR as a moderator of pathology-cognition relationships^7,8^, we operationalized CR specifically as memory reserve and performed comprehensive interaction-based analyses to identify a blood DNAm signature of CR that modifies the association between CSF pTau181 and memory in amyloid positive ADNI participants with MCI. Across the epigenome, we identified 11 CpGs showing a DNAm×pTau181 interaction at a suggestive significance threshold (P-value < 1×10^−5^), and 11 DMRs significant after multiple comparison correction. After excluding low-variability CpGs (IQR < 0.02), 6 CpGs remained.
Notably, the direction of memory reserve, defined as attenuation of the pTau181-memory association, was locus-specific. At some sites, higher methylation was associated with weaker pTau181-memory association, while at others, lower methylation was protective. This heterogeneity is consistent with context-dependent DNAm regulation, in which hyper- and hypomethylation may have beneficial or adverse effects depending on genomic and regulatory context. Moreover, a composite methylation reserve score derived from these loci was significantly associated with slower longitudinal memory decline in MCI, independent of baseline pTau181, age, sex, APOE ε4, and education. Together, these findings support blood DNAm as a promising biological index of CR with potential clinical utility in the prodromal phase of AD.
A key feature of these CR-related loci is that, despite showing robust DNAm×pTau181 interaction effects, nearly all lacked strong marginal associations with pTau or memory (Supplementary Figures 2–3). Therefore, these DNAm do not simply reflect tau burden, nor do they serve primarily as biomarkers of current memory performance. Instead, they mark additional variations that determine how cognition varies about a given level of tau pathology. The lack of strong marginal effects further argues against a mediating role of DNAm in the pTau-memory relationship, but instead supports a moderating role, in which DNAm at these loci, or the biological mechanisms they mark (e.g., transcription factor binding, chromatin accessibility, histone modifications), influences the impact of tau pathology on memory. Future mechanistic studies are needed to determine how DNAm, or the regulatory elements it marks, modulates CR-related biological processes.
Several of the most significant loci point to biologically plausible mechanisms for cognitive reserve. The most significant DMR is located in the promoter of the KLK7 gene, which encodes an astrocyte-derived protease, an enzyme helps break down and clear deposited amyloid beta in the brain. Loss of KLK7 activity has been shown to exacerbate amyloid pathology in mouse models of AD, suggesting it plays a protective role in maintaining brain health^55^. Consistent with these findings, we observed hypomethylation in in the promoter of the KLK7 gene associated with greater memory reserve.
The second most significant promoter-associated DMR is located in the TMEM232 gene, which encodes a transmembrane protein that promotes inflammatory responses to atopic dermatitis, and has been implicated by genetic studies in allergic and atopic diseases characterized by immune dysregulation^56^. Importantly, recent DNAm studies in blood have also identified DMRs at TMEM232 associated with MCI, AD, as well as CSF AD biomarkers^57–59^. Consistent with these findings, the hypermethylation we observed at TMEM232 associated with greater CR may suggest that reducing TMEM232-mediated inflammatory signaling could contribute to a peripheral immune environment that mitigates the impact of AD pathology.
Finally, the DMR annotated to the LYNX1-SLURP2 read-through transcript, which spans the adjacent LYNX1 and SLURP2 genes, is involved in regulation of nicotinic acetylcholine receptor (nAChR) activity within the cholinergic system. In animal models, loss of LYNX1 leads to age-related neurodegeneration, highlighting its critical role in maintaining neuronal health during aging^60^. Moreover, treatment with a water-soluble LYNX1 analogue was shown to prevent amyloid beta-induced blockade of memory-related synaptic plasticity, indicating LYNX1 may be a potential therapeutic target for improving cognitive deficits in neurodegenerative diseases^61^. These findings are consistent with our observed hypomethylation at this locus associated with greater CR.
To help prioritize the most biologically relevant loci involved in CR, we next performed integrative analyses incorporating several external resources, to better understand the functional roles of the CR-related DNAm changes. These analyses nominated the ANKH and LDHA/LDHC loci, where DNAm was associated with nearby gene expression (Supplementary Table 4). Recent genetic studies have implicated ANKH in cognitive resilience and reduced AD risk, and impaired ANKH function is associated with pathological vascular mineralization^52^. The observed hypomethylation at ANKH promoter associated with greater CR may therefore reflect epigenetic upregulation of a protective vascular pathway, which helps to limit microvascular damage and neuropathological burden.
At the LDHC locus, the proximity and high sequence similarity between LDHC and its paralog LDHA suggest that the observed eQTM likely reflects transcriptional activity at the broader LDHC/LDHA locus. LDHA is a key enzyme in lactate production, and lactate serves as an important neuronal energy substrate that is critical for memory formation but may become maladaptive when chronically elevated in aging and neurodegeneration^53^. The association between CR-related methylation and LDH/LDHA activity is therefore consistent with the idea that tight regulation of neuronal energy metabolism and lactate homeostasis is important for preserving cognitive function in the presence of tau pathology. Intriguingly, our blood-to-brain correlation analysis also showed several CpGs in the LDHC/LDHA DMR showed strong positive correlations between blood and brain methylation, suggesting this locus may be a promising candidate for a blood-based biomarker of memory reserve.
Our pathway analysis of genes mapped to CR-related CpGs extended these gene-level observations and highlighted a strong enrichment of metabolic pathways, particularly those involving adipocytokine signaling, lipid handling, and adipose tissue function (Supplementary Table 3). The identification of the KEGG adipocytokine signaling pathway, along with Reactome pathways related to PPARα activation, triglyceride catabolism, adipogenesis, and epigenetic regulation of adipocyte differentiation, suggests that systemic metabolic health and lipid homeostasis are key biological processes involved in CR. Our findings support a model in which CR-related DNAm captures, at least in part, long-term exposure to metabolic states that maintain insulin sensitivity, prevent lipid accumulation, and reduce chronic inflammation. In turn, these favorable metabolic profiles may help preserve neural networks and reduce impact of accumulating tau pathology on memory.
A major strength of this study is the construction and evaluation of an MRS that aggregates information across multiple CR-related CpGs and DMRs into a single, interpretable measure. By definition, this score quantifies the DNAm-dependent effect of pTau on memory, such that higher values represent greater attenuation of tau’s harmful impact. In longitudinal mixed-effects models, a higher MRS at baseline was significantly associated with slower subsequent decline in harmonized memory scores among MCI participants, even after adjusting for age, sex, education, APOE ε4, and baseline pTau. Moreover, individuals in the lowest MRS tertile experienced both a stronger pTau-memory association at baseline and a faster rate of memory decline compared with those in the highest tertile (Supplementary Table 8). These findings demonstrate that CR-related DNAm patterns are not merely cross-sectional correlates but provide prognostic information about future clinical trajectories in prodromal AD. If replicated in larger and more diverse cohorts, MRS or similar DNAm-based metrics could be used to stratify individuals at baseline, and refine prognosis beyond traditional biomarkers, and identify those most likely to experience rapid decline.
Our results are consistent with prior work associating CR with modifiable lifestyle and cardiometabolic factors^21 ,62^. A number of the pathways enriched with CR-related DNAm in our study, including those involved in adipocytokine signaling, lipid metabolism, and vascular function, have previously been associated with smoking, obesity, physical inactivity, and other lifestyle factors in large epigenome-wide association studies^63,64^. This suggests that DNAm may serve as a biomarker capturing, at least in part, the cumulative impact of life-course exposures that build or erode reserve. Moreover, emerging evidence shows that lifestyle-based interventions can modify DNAm at cardiometabolic loci^65,66^, further supports the idea that CR is not a fixed trait determined only by early-life education or genetics, but a dynamic, biologically embedded property shaped by experiences and behaviors across the lifespan^7^. In this context, DNAm represents a dynamic and potentially reversible biomarker that not only reflects prior exposures but may also track response to lifestyle interventions.
Several limitations should be considered when interpreting these findings. First, because we operationalized reserve using PHC_MEM (a harmonized memory composite), our findings pertain to memory reserve and may not generalize to reserve in other cognitive domains (e.g., executive function, language, global cognition). Second, our analysis was conducted in a relatively small sample of ADNI MCI participants with concurrent CSF, DNAm, and memory data, which limits statistical power to detect small effects and may contribute to the fact that no CpG passed a stringent FDR threshold at the single-site level. We therefore focused on loci meeting a suggestive P-value threshold and on DMRs, which leverage spatial correlation across neighboring CpGs; nonetheless, replication in independent cohorts is essential. Third, the ADNI cohort is highly educated and predominantly of European ancestry, and our results may not generalize to more diverse populations or to individuals at different stages of the AD disease course. Fourth, although we adjusted for major immune cell proportions and key demographic (age, sex) and genetic covariates, residual confounding by unmeasured lifestyle factors or additional neuropathologies cannot be excluded. To help mitigate inflation and bias related to unmeasured confounding, we used the bacon method to empirically calibrate test statistics^34^. Finally, the use of CSF pTau181 as the pathology term in the interaction model focuses the analysis on tau-related processes that are closely associated with amyloid-β deposition, and may not capture reserve mechanisms that primarily operate through vascular lesions, TDP-43, or other non-AD pathologies.
In conclusion, this study provides convergent evidence that blood DNAm captures biologically meaningful variation in memory reserve among individuals with MCI. By focusing on DNAm×pTau interactions, we identified CpGs and DMRs that moderate the impact of CSF pTau181 on memory, mapped these loci to vascular, synaptic, metabolic, and immune pathways, and integrated them into an MRS that predicts future memory decline. These findings highlight the promise of blood DNAm as a non-invasive biomarker for resilience to AD pathology and suggest that interventions targeting vascular and metabolic health, along with traditional risk-reduction strategies, may enhance reserve and delay clinical progression in at-risk older adults.
Supplementary Material
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hurd M.D., Martorell P., Delavande A., Mullen K.J. & Langa K.M. Monetary costs of dementia in the United States. N Engl J Med 368, 1326–34 (2013).23550670 10.1056/NEJ Msa 1204629 PMC 3959992 · doi ↗ · pubmed ↗
- 2Jansen W.J. Prevalence Estimates of Amyloid Abnormality Across the Alzheimer Disease Clinical Spectrum. JAMA Neurol (2022).
- 3Ossenkoppele R. Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313, 1939–49 (2015).25988463 10.1001/jama.2015.4669 PMC 4517678 · doi ↗ · pubmed ↗
- 4Boyle P.A. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol 74, 478–89 (2013).23798485 10.1002/ana.23964 PMC 3845973 · doi ↗ · pubmed ↗
- 5Boyle P.A. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 83, 74–83 (2018).29244218 10.1002/ana.25123 PMC 5876116 · doi ↗ · pubmed ↗
- 6Tosun D. Contribution of Alzheimer’s biomarkers and risk factors to cognitive impairment and decline across the Alzheimer’s disease continuum. Alzheimers Dement 18, 1370–1382 (2022).34647694 10.1002/alz.12480 PMC 9014819 · doi ↗ · pubmed ↗
- 7Stern Y. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimers Dement 16, 1305–1311 (2020).30222945 10.1016/j.jalz.2018.07.219PMC 6417987 · doi ↗ · pubmed ↗
- 8Stern Y. A framework for concepts of reserve and resilience in aging. Neurobiol Aging 124, 100–103 (2023).36653245 10.1016/j.neurobiolaging.2022.10.015PMC 10424718 · doi ↗ · pubmed ↗