Persistent Immune Dysregulation during Long COVID is Manifested in Antibodies Targeting Envelope and Nucleocapsid Proteins
Marcin Kwissa, Manikannan Mathayan, Satyajeet S. Salunkhe, Velavan Bakthavachalam, Zijing Ye, Mark A. Sanborn, Samantha Condo, Aditi Upadhye, Athulith Nemakal, Haoyang Wang, James Chan, Justin M. Richner, Sanjib Basu, Richard M. Novak, Jeffrey R. Jacobson, Balaji B Ganesh

TL;DR
The study finds that people with Long COVID have persistent immune issues, including high antibody levels against certain SARS-CoV-2 proteins and elevated immune cell activity.
Contribution
The study identifies specific immune markers and antibody patterns that persist in Long COVID patients over six months.
Findings
LC participants showed elevated IgG titers against SARS-CoV-2 Envelope and Nucleocapsid proteins for up to 6 months.
LC patients had higher levels of circulating T follicular helper cells and mucosa-associated invariant T cells.
LC participants exhibited elevated serum cytokines and higher rates of autoantibodies compared to convalescent individuals.
Abstract
Long COVID (LC) or Post-Acute Sequelae of SARS-CoV-2 infection (PASC) syndrome represents a widespread health challenge that necessitates the development of novel diagnostic approaches and targeted therapies that can be readily deployed. Immune dysregulation has been reported as one of the hallmarks of LC, but the extent of LC immune dysregulation in patients over time remains unclear. We therefore assessed SARS-CoV-2-specific antibody responses, peripheral immune cell profiles, autoantibody profiles and circulating cytokines for up to 6 months in participants with a SARS-CoV-2 infection who either convalesced or developed LC. Compared to convalescent, LC participants with a broad range of LC phenotypes exhibited persistently elevated IgG titers for SARS-CoV-2 Envelope and Nucleocapsid proteins over the 6 months of study duration. In contrast, the IgG responses to Spike protein were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Extended Data Fig. 1
Extended Data Fig. 1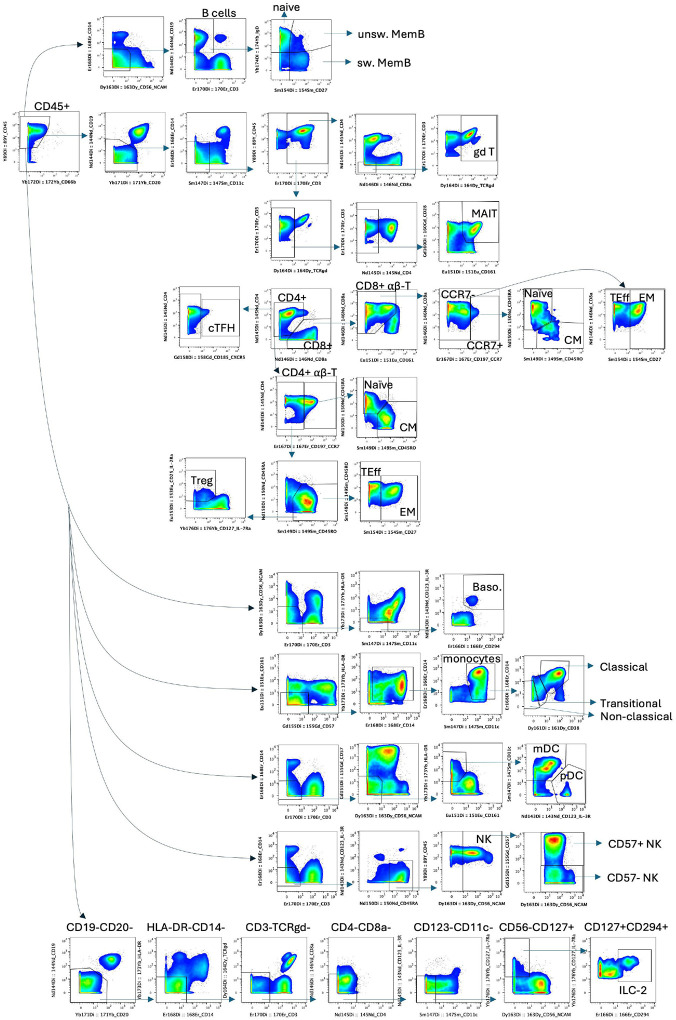 Extended Data Fig.2
Extended Data Fig.2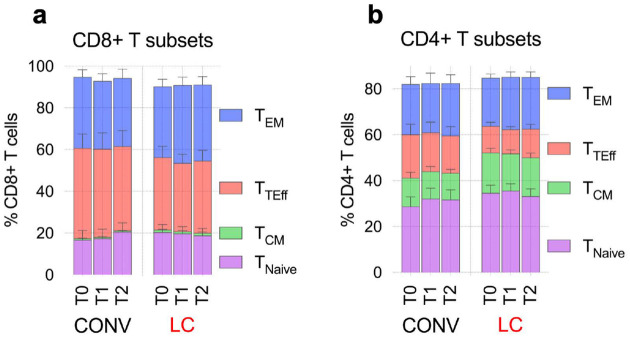 Extended Data Fig. 3
Extended Data Fig. 3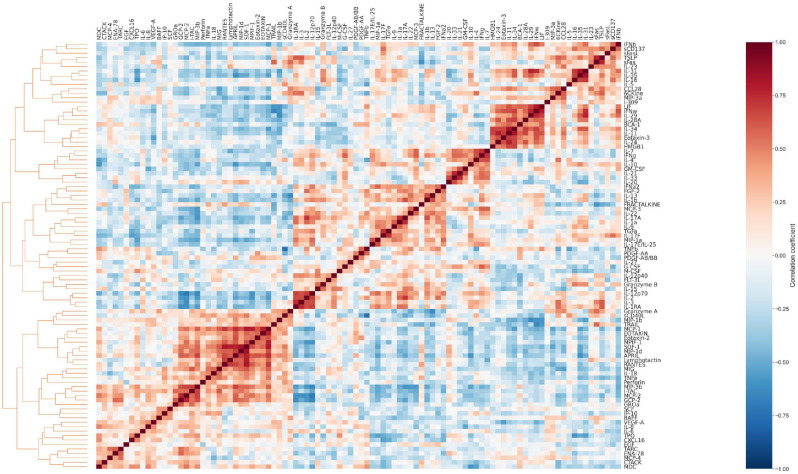 Extended Data Fig. 4
Extended Data Fig. 4 Extended Data Fig. 5
Extended Data Fig. 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLong-Term Effects of COVID-19 · Autoimmune Neurological Disorders and Treatments · Neuroinflammation and Neurodegeneration Mechanisms
Introduction
Long COVID (LC) or Post-Acute Sequelae of SARS-CoV-2 infection (PASC) syndrome is a debilitating chronic disease for which there are currently no targeted therapies available and which highlights the enduring impact of acute SARS-CoV-2 infection on human health^1,2^. LC is a syndrome comprised of multiple endotypes or lingering disease manifestations such as excessive fatigue, “brain fog”, dysautonomia, and additional neurological, cardiovascular, musculoskeletal or respiratory complications^3–6^. The clinical manifestations vary in duration and severity and affect multiple organ systems, with considerable inter-individual variability^7,8^. Due to the heterogeneity of LC manifestations and the breadth of the symptoms, diagnosing LC and also distinguishing it from other chronic diseases with congruent symptoms has emerged as a major challenge in clinical practice.
Persistent immune dysregulation is emerging as a fundamental pathogenic mechanism of LC, and characterizing such immune dysregulation holds great promise because it allows for the development of specific diagnostic biomarkers as well as targeted therapeutics. Several studies have reported sustained alterations in the innate immune system, including prolonged activation of the complement cascade^9,10^, distinct pro-inflammatory cytokine milieu^11,12^ and continued stimulation of innate immune cells of myeloid origin such as monocytes, macrophages^13–15^ and neutrophils^16–18^ as well as natural killer (NK) cells^19,20^. Additionally, dysregulation within the adaptive immune compartment has also been observed, characterized by exhausted T cell phenotypes and aberrant B cell activation^11,15,19,21,22^. Together, these immune abnormalities suggest that a failure to fully resolve the inflammatory response to the initial SARS-CoV-2 infection may underlie the persistent symptoms observed in individuals with LC. Additionally, autoimmune responses have also been associated with LC pathology, such as the presence of autoantibodies (auto-Abs) targeting a wide range of self-antigens, including components of key immunological pathways. Some studies have identified elevated levels of auto-Abs targeting interferons^23,24^, chemokines^25^ or nuclear antigens^26^ in subsets of LC patients. The observed heterogeneity suggests that such auto-Abs, when present, may reflect underlying predisposition, prior immune priming, or secondary consequences of sustained inflammation, rather than a unifying etiological feature of LC.
More recent studies suggest that the persistence of SARS-CoV-2 in various tissues may also contribute to the pathogenesis of LC because lingering SARS-CoV-2 RNA and proteins in tissues could potentially sustain immune activation. Multiple studies have detected viral RNA and proteins in tissues such as the lungs, gastrointestinal tract, and brain months after the acute phase of infection^27–31^. Additionally, disruption of mucosal barrier integrity, particularly in the gut and lungs, has been reported in LC, raising the possibility of viral persistence promoting microbial translocation that could further perpetuate systemic inflammation and immune imbalance^5,17,32^.
In addition to the complexity of the immune dysregulation and heterogeneity of LC, the severity of the symptoms can wax and wane over time, thus creating a challenge for developing diagnostic biomarker panels and choosing optimal temporal windows for therapeutic interventions. Longitudinal studies with serial sampling are thus required to determine whether immune dysregulation is transient, progressive, or persistent. To date, comprehensive multi-timepoint studies in well-characterized cohorts remain limited, leaving critical questions about temporal immune dynamics in LC unanswered. Documenting viral persistence in tissues would be valuable for diagnostics and for choosing which patients would benefit from antiviral therapies, but obtaining tissue samples from patients often requires invasive studies that are not feasible for longitudinal analysis. As a result, conclusions regarding viral persistence remain difficult to study non-invasively, and several studies have failed to detect SARS-CoV-2 RNA or protein in blood, stool^33^, or tissue samples from individuals with LC^21,34,35^. As an alternative approach, the adaptive immune system may serve as an indirect biosensor of persistent viral antigens and could be assessed by non-invasive means. Long-lived memory B and T cells can retain antigen specificity, and the detection of sustained antibody responses to viral antigens such as nucleocapsid or envelope proteins that are not used in COVID-19 vaccines may offer indirect evidence of ongoing viral antigen exposure and potential viral persistence.
We therefore studied immune dysregulation and antibody responses over time in participants with LC and those who recovered fully from COVID-19. We show a marked persistence in SARS-CoV-2–specific antibody responses, particularly elevated IgG to envelope and nucleocapsid proteins, along with altered immune cell profiles including increased MAIT and cTFH cells in LC participants. LC was also associated with a proinflammatory cytokine environment and the emergence of autoantibodies, suggesting persistent immune activation and dysregulation.
Results
Elevated IgG response against SARS-CoV-2 Envelope and Nucleocapsid in LC
We studied thirty participants who enrolled in the parent NIH RECOVER Adult Cohort Study during the post-acute phase of a SARS-CoV-2 infection. Details about the design of the Adult Cohort Study are available elsewhere^36^. This report examines peripheral blood samples from 30 participants (20 with LC, and 10 participants who were convalescent (CONV) without any persistent LC symptoms). Participants in the LC group presented with different symptoms, including cardio-pulmonary, musculoskeletal, neuropsychiatric, dysautonomia or a combination of other LC symptoms, which showcased the variety of global LC manifestations (Table 1 and Supplementary Table 1), mirroring the heterogeneity seen in a typical LC patient population^8,37,38^. To study possible longitudinal shifts in immune dysregulation, peripheral blood samples were obtained at baseline during enrollment into the parent study (Timepoint 0 or T0, on average 23 days post infection ±7.7 s.d.), approximately 3 months or 97 days later (±10.1 s.d., T1) and the final timepoint after an additional 3 months (at 6 months or 189 days post infection, ±13.8 s.d., T2). We conducted a comprehensive analysis of peripheral immunological signatures in the blood by testing serum antibody repertoire against multiple SARS-CoV-2 antigens, immunophenotyping peripheral blood mononuclear cells (PBMC) using multiparametric CyTOF analysis, analyzing circulating cytokines as well as autoantibodies and the plasma across all time points (Fig. 1a).
We quantified serum IgG titers of antibodies targeting key SARS-CoV-2 structural proteins such as the spike, envelope (Env), nucleocapsid (Nucl), and membrane (Mem) by ELISA (Fig. 1b–e). To validate the antibody specificity, pre-COVID-19 sera (n=3) were included as negative controls in all assays. The total IgG responses to Env protein were significantly (p=0.0022) elevated over time in LC with 1.98-fold higher mean OD450 value of anti-Env IgG when compared to CONV individuals across all timepoints (Fig. 1b). The IgG titers against the Nucl protein were also significantly higher (p=0.0419) in LC individuals. In contrast, the IgG response to Spike protein was significantly reduced (p=0.0025) in individuals with LC compared to the CONV cohort across all time points (Fig. 1b). We also observed a trend in the reduction of Mem-specific IgG titers among LC individuals over time, when compared to the CONV control cohort. Titers of total serum IgG response specific for Env protein correlated significantly with anti-Mem and anti-Spike IgG antibody response in the LC cohort (Fig. 1c). The viral Spike protein is the target antigen of the anti-SARS-CoV-2 humoral response commonly used in the majority of COVID-19 vaccines^39^. Therefore, to better characterize the immunological profile of Spike-specific IgG responses, we evaluated longitudinal levels of IgG1, IgG2, IgG3, and IgG4 subclasses in both LC and CONV cohorts (Fig. 1d and e). LC participants exhibited significantly higher titers of Spike-specific IgG1 compared to CONV (p = 0.0392), indicating a sustained Th1-biased response. We observed a similar trend for IgG3, with elevated levels in LC over time (Fig. 1d and e). In contrast, CONV participants demonstrated a higher titer of IgG4 (p = 0.0225), consistently across all time points, suggesting a more tolerogenic or regulatory profile following recovery from COVID-19 (Fig. 1d)^40–42^. In fact, in CONV the proportion of IgG4 within all IgG specific to Spike was on average at 54.5% (+/− 7%) among all IgG subclasses (Fig. 1e), showcasing a class-switch when compared to IgG4 in LC (IgG4=27.2%).
We also tested the Spike-, Env-, Nucl- and Mem–specific titers of IgA in plasma (Fig. 1f). We noted a trend toward increased levels of anti-Spike IgA responses in the LC cohort, however, there was no significant difference between experimental groups. None of the other tested SARS-CoV-2 antigens showed substantial differences between the experimental groups over time (Fig. 1f). Virus neutralization titers in plasma varied markedly among individuals with titers ranging from below detection levels up to 3×10^4^ of IC50 units (Fig. 1g). We observed a mild increase in the mean neutralization activity against the SARS-CoV-2 infection in sera from the LC group, however this trend was not significantly different from the mean neutralization titer in the CONV group. Our data thus indicate a distinct shift in SARS-CoV-2 antigen specificity of the humoral IgG response with augmented titers against Env and Nucl proteins in LC participants. Additionally, anti-Spike IgG titers indicated differential isotype skewing, favoring the pro-inflammatory IgG1/IgG3 subclasses in LC, in contrast to the IgG4-biased response in convalescent individuals.
Mucosal immunoglobulin signature in LC plasma
Next, we used mass spectrometry analysis to characterize the total immunoglobulin (Ig) content in the plasma proteome from LC and CONV individuals (Fig. 2). A heatmap of Z-scores representing the relative abundance of Ig proteins in plasma revealed a consistent increase in total Ig protein levels in individuals with LC compared to convalescent controls (Fig. 2a). Across all detected Ig peptides and at all longitudinal timepoints analyzed, the LC group exhibited higher normalized expression, suggesting sustained or dysregulated antibody production. Next, we compared quantities of the constant regions of heavy chains to evaluate total subsets of class-switched, circulating immunoglobulins and their subclasses. Individuals with LC showed persistently elevated levels of IGHM but also IGHA1 and IGHA2 compared to CONV (Fig. 2b). The total protein spectrum counts of each of the IgM, IgA1, and IgA2 were significantly more abundant (p=0.0009, p=0.0026, and p=0.004, respectively) in LC participants when compared to CONV over time. Additionally, J chain protein fragments, which link monomers of IgM and IgA to form polymers, were also highly expressed in LC (p=0.0028). In contrast, we did not observe differences in the abundance of IGHG1, IGHG2, IGHG3, or IGHG4 between the two groups at either time point. These findings suggest a selective enrichment of mucosal and early-phase antibody isotypes in LC, potentially indicative of ongoing or dysregulated immune activity, while class-switched IgG subtypes remain stable.
We further evaluated potential changes in the Ig heavy, kappa, and lambda variable chains in plasma of LC and CONV cohorts (Fig. 2c). Fold-change analysis revealed that multiple variable region peptides were significantly upregulated in individuals with LC compared to convalescent controls (CONV). As shown in the volcano plot (Fig. 2c), numerous IGHV, IGKV, and IGLV chains exhibited elevated expression, highlighting a broader shift in variable region usage or expansion of specific B cell clones in the LC group. This differential abundance suggests persistent or skewed antibody responses that may contribute to the immunological landscape of LC.
Overall, dysregulated plasma immunoglobulin patterns in LC may reflect an ongoing B cell activation or an altered humoral immune response at the mucosal sites in individuals with persistent post-viral symptoms.
LC participants exhibit elevated levels of cTFH and MAIT cells in the blood
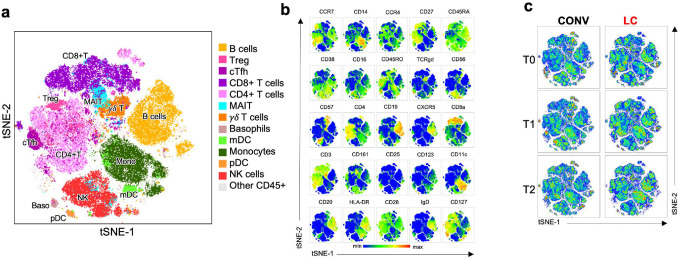
To further define dysregulation of the innate and adaptive immunological system in LC participants, we performed high-dimensional profiling of PBMCs using CyTOF with a comprehensive 26-antibody panel targeting key cell lineage markers. To visualize cellular subsets, we performed UMAP clustering of concatenated samples from all individuals across both groups and all timepoints (Fig. 3a). Within the CD45+ lymphocyte population, we identified clusters corresponding to canonical immune cell lineages, including total CD19+ B cells, CD3+CD8+ T cells and CD3+CD4+ T helper cells (including CD25hi CD127- T regulatory cells, Treg and CXCR5+ circulating T follicular helper cells, cTFH), unconventional T cells (mucosa associated invariant cells, MAIT and γδ-T cells), NK cells, innate-like lymphocytes type 2 (ILC-2), and myeloid cell subsets such as monocytes, myeloid dendritic cells (mDC), basophils, as well as plasmacytoid DCs (pDCs; Fig. 3a and b). A similar clustering pattern was observed using t-SNE (Extended Data Fig. 1a). As shown in Fig. 3b and Extended Data Fig. 1b, each cluster displayed characteristic marker expression consistent with lineage identities as defined in our gating strategy (Extended Data Fig. 2). When comparing the CD45+ immune cell subpopulations between the CONV and LC groups at T0, T1, and T2 (Fig. 3c, Extended Data Fig. 1c), we observed differences in cluster size and density for cTFH and MAIT cells between groups, though these cell groups did not exhibit marked temporal changes. This prompted a more detailed subtype-specific analysis based on traditional gating (Extended Data Fig. 2).
The frequency of CD3+CD4+ and CD3+CD8+ T cell populations within the total CD45+ PBMC did not vary significantly between the LC and CONV groups over time (Fig. 3d and f). Similarly, within the CD8+ T cells, the CCR7hiCD45RA-CD45RO+ central (TCM) memory, CCR7lo/-CD8+CD27+ effector (TEM) memory cells and CCR7lo/-CD8+CD27- terminally differentiated effector (TEff) were similarly distributed in both experimental groups (Fig. 3e and Extended Data Fig. 3a). However, in accordance with the clustering analysis in Fig. 3c, LC individuals showed a significant increase in the proportion of CD4+CCR7hiCD45RA-CD45RO+ central memory cells (TCM) within all CD45+ lymphocytes when compared to CONV (Fig. 3g and Extended Data Fig. 3b). The frequency of the CD4+CD25+CD127low/neg Treg cells remained unchanged between the LC and CONV cohorts throughout the study period (Fig. 3h). In parallel with the increase in CD4+ TCM observed in the LC cohort (Fig. 3g) we observed a substantial expansion in the population of the CD4+CXCR5+ circulating follicular helper cells (cTFH) in LC participants in contrast to CONV (p=0.0352; Fig. 3i) possibly indicative of an ongoing germinal center (GC) reaction and B cell activation, even though the total numbers of circulating B cells or B cell subsets were not different between LC and CONV (Fig. 3j). The fractions of CD4+ TCM correlated strongly with cTFH subsets in LC (p<0.0001, r=0.55; Fig. 3k).
To better characterize possible persistent, systemic inflammatory activation in LC we validated innate cell subsets in the blood including antigen presenting cells (APC), cells of myeloid lineage, and innate-like lymphocytes (Fig. 4). The frequency of total Lineage-HLA-DR+CD11c+ classical myeloid DC (mDC) or Lineage-HLA-DR+CD11c-CD123+ plasmacytoid DC (pDC) populations within CD45+ lymphocytes remained constant between the two experimental cohorts (Fig. 4a). Similarly, we did not detect significant changes in the magnitude of total CD14+ monocytes, nor any of the monocyte subpopulations (Fig. 4b) or basophils (Fig. 4c). Further, we characterized subpopulations of innate lymphocytes and invariant T cells (Fig. 4d–g), and did not observe any significant differences between LC and CONV cohorts in the circulating numbers of CD56+CD57+ mature, cytotoxic cells, circulating ILC-2 and the γδ-T cell populations (Fig. 4e, f). In contrast, there was a significant 1.73-fold increase (p=0.0461) in the total frequency of CD3+CD4-CD28+CD161hi MAIT cells in LC at all timepoints of specimen collection (Fig. 4g). Importantly, the extent of increase in circulating MAIT cells in LC participants was correlated with the high titers of anti-Env IgG at the group level (r=0.3, p<0.026; Fig. 4h).
In summary, these analyses show an augmented number of circulating TFH cells that likely reflect an ongoing germinal center reaction and B cell activation. Additionally, the increase in MAIT cells in LC and the positive correlation between circulating MAIT cell numbers and anti-Env IgG titers suggest ongoing mucosal inflammation in LC individuals that may reflect persistence of viral antigens in the mucosal compartments.
Inflammatory cytokine profile is associated with LC
We analyzed serum cytokine milieu in samples from LC and CONV individuals across all timepoints using a 96-plex cytokine panel (Fig. 5). As shown in Fig. 5a, a t-SNE analysis of cytokine profiles identified two distinct clusters corresponding to LC and CONV cohorts, although samples from the specific timepoints (indicated by distinct symbols) clustered closely within each individual among the two groups. This suggested marked between-group differences in cytokine expression patterns, with relatively stable differences over time.
To further explore co-expression patterns, we performed cytokine clustering analysis (Fig. 5b and Extended Data Fig. 4). This approach revealed three major cytokine expression modules that were consistently observed across all individuals from both groups (Fig. 5b and Extended Data Fig. 4). Each of the modules clustered a collection of cytokines with similar expression kinetics and values in both experimental groups over time. We calculated the odds ratios of cytokines in each of the modules to define the strongest association with LC diagnosis (Fig. 5c). A cluster of cytokines grouped in module 2 showed the highest odds ratio of positive association with LC, with significant enrichment of cytokines such as IL-11, LIF, Eotaxin-3, IL-23, IFN-β, and HMGB-1, which were closely correlated with LC outcome (Fig. 5c). In module 1, expression of ENA-78 (CXCL5) was also positively associated with LC whereas IP-10 (CXCL-10) correlated negatively with LC diagnosis (Fig. 5c). Finally, we compared changes in total cytokine values between LC and convalescent cohorts over time (Fig. 5d). Overall fold increase in circulating cytokine levels of LC participants over the convalescent cohort over time was most significant for IL-11 and LIF, but also IFN-β, IL-24, ENA-78 (CXCL5), and TNF-β, resembling cytokine modules defined by clustering analysis (Fig. 5b–d).
The LC diagnosis was strongly associated with a long-lasting systemic immune activation represented by overexpression of a several cytokines and chemokines which signal tissue damage and can potentially exacerbate chronic inflammation.
Auto-IgG and -IgM antibodies in LC highlight autoreactive response
Acute COVID-19 disease as well as the post-acute phase are often accompanied by an autoimmune reaction^22,43–45^. To assess for the presence of autoantibodies in LC, we tested participant plasma for IgG and IgM reactivity against an array of over twenty-six thousand human proteins using HuProt technology. Global analysis of the protein array data showed an increase in total autoantigen reactivity in LC participants when compared with convalescent (Figures 6 and Extended Data Fig. 5). We identified 16 proteins targeted by auto-IgG in plasma that showed significantly elevated titers in LC compared to CONV serum, as illustrated in the heatmap of Z-scores representing the relative abundance of target-specific autoreactive IgG (Fig. 6a) or top 20 proteins targeted by autoreactive IgM (Extended Data Fig. 5). Mean absolute values of each of the auto-IgG titers remained stable with significantly higher relative intensity values in LC individuals at least until the T2 timepoint (Fig. 6b). Fig. 6c visualizes the correlation matrix of the top 16 autoantigens targeted by plasma IgG autoantibodies in individuals with LC identified in Figs. 6a and b. Pearson analysis revealed that auto-IgG intensity values against 9 of the 16 autoantigens were strongly correlated (r > 0.7; p < 0.0001), suggesting that a subset of LC individuals developed simultaneous autoreactive responses against multiple self-proteins. This pattern implied broad autoreactivity in a portion of the cohort, whereas other individuals exhibited more limited auto-IgG responses, targeting only one or a few specific autoantigens. The heterogeneity in autoantibody profiles highlights the diverse immunological manifestations of LC and suggests multiple underlying mechanisms of autoreactivity. Interestingly, no single autoantibody was pathognomonic for LC, and the targets of the autoantibody comprised a broad range of cellular targets, likely derived from different tissues.
Taken together, we detected a series of autoantibody titers upregulated in LC participants and observed that they had developed IgG against multiple autoantigens.
Effect of the Time Post-Infection on Antibody Responses in CONV and LC
Finally, we evaluated whether the interval between the SARS-CoV-2 infection and the subsequent timepoints of specimen collection influenced the distinct dynamics of anti-SARS-CoV-2 antibody response defined by our analysis in both LC and CONV participants. Participants were enrolled in the NIH RECOVER cohort over a period of 10 months between December 2021 and October 2022, as illustrated with the chronological timeline for each participant, indicating the date of infection, study enrollment, and each blood draw (Fig. 7a). To standardize the timing gap across all individuals in both groups, we normalized all blood collection timepoints to the initial date of SARS-CoV-2 infection (Fig. 7b). Despite the intra-cohort variability, there were no significant differences in the timing of specimen collection between LC and CONV groups at any of the timepoints (T0, T1, and T2). To further investigate the dynamics of the antibody response, we stratified antigen-specific IgG titers by days post-infection for each individual in our experimental cohort. Spaghetti plot analysis exposed alterations in the temporal profiles of IgG responses to Spike, Envelope, Nucleocapsid, and Membrane proteins which varied overtime (Fig. 7c). We performed Pearson correlation analysis between IgG titers against all SARS-CoV-2 antigens and the time interval from infection to specimen collection at each timepoint (T0, T1, and T2). This analysis revealed significant correlation between the time gap and anti-Membrane IgG titers at timepoint T2 with reduced Ab response in samples collected later post infection but not for any of the other measured antibody titers and timepoints. This indicated that the interval between infection and blood draw did not significantly influence the magnitude of the antibody response (Fig. 7d).
Discussion
Here, we studied the humoral, cellular and molecular indicators of a potential immune dysregulation over time in participants suffering from LC and those who did not exhibit any persistent symptoms after a resolved SARS-CoV-2 infection. Our study shows a distinct shift in the SARS-CoV-2-specific antibody profile with dominant IgG responses to Envelope and Nucleocapsid viral proteins in individuals with LC over the period of at least 6 months after the initial infection. Further analysis determined a close correlation of high Envelope-specific IgG titers with elevated proportions of circulating MAIT cells as well as an increase in the cTFH during LC. Additionally, LC participants exhibited an inflammatory cytokine signature and developed distinct autoreactive antibody responses.
Among the many hypotheses regarding the underlying mechanisms of LC, a dysregulation of the immune system has emerged as a prominent pathogenic mechanism^3,7^. In our study the elevated anti-Envelope and anti-Nucleocapsid IgG response in LC suggest an ongoing antigenic stimulation and suggests persistence of viral proteins or protein fragments in chronically ill individuals^31,46^. Although all participants in both study cohorts reported a positive vaccination status (Table 1), the higher anti-Spike IgG titers observed in the CONV cohort may suggest a more efficient vaccine-induced response in this group^39,47^. This interpretation is further supported by our data of predominant Spike-specific IgG4 levels in CONV participants (Fig. 1d), consistent with previous reports demonstrating high IgG4 class-switching of Spike-specific IgG response in individuals who received multiple doses of a mRNA vaccine^40–42^. IgG4 inherently has an altered capacity to efficiently bind Fc receptors leading to reduced antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) related events^48^. In contrast, IgG3 can effectively bind IgG-Fc-receptor and induce NK cell mediated ADCC^49^. Therefore, it is plausible that LC patients experience more inflammation-prone events due to the persistence of elevated IgG3, while the control group having higher levels of the less inflammatory IgG4 is comparatively protected.
The overall IgG titer against Spike, albeit lower in the LC group, remained stable over time for up to 6 months after acute infection in contrast to the convalescent group where Spike-specific IgG response diminished at later timepoints. Such a pattern may suggest a sustained immunogenic stimulus in LC, potentially driven by persistent presence of viral antigens or delayed resolution of immune activation. The exact reason of high immunogenicity of Envelope and Nucleocapsid proteins (but not Spike or Membrane) in LC remains unclear, but the consistency of IgG responses within our cohort supports the hypothesis of a prolonged Envelope and Nucleocapsid viral antigen exposure. The precise location of the latent residual SARS-CoV-2 virus or viral RNA and the tissue serving as a reservoir for ongoing antigenic shedding cannot be ascertained from the analysis of circulating peripheral blood. Our mass spectrometry analysis of plasma proteome revealed elevated IgA and IgM immunoglobulin and J-chain peptides in LC participants, along with elevated levels of Ig variable region peptides but none of the IgG subclasses (Fig. 2). This supports the notion of de novo immune activation, especially at mucosal sites, and may suggest that intestinal and lung mucosa may serve as a location for viral persistence and sustained immune stimulation^21,50^.
Reactivation of other latent viruses such as Epstein-Barr virus (EBV) during or following SARS-CoV-2 infection has been increasingly recognized as a putative contributor to the immunopathogenesis of LC^22,51,52^. Though our analysis focused on SARS-CoV-2-specific immune response, our proteomic analysis showing elevated plasma levels of IGHA, IGHM, and immunoglobulin variable region peptides may reflect mucosal antibody activity which is consistent with EBV’s tropism for the oropharyngeal and gastrointestinal epithelium^53^. Lytic EBV replication in epithelial cells induces the expression of pro-inflammatory mediators, including IL-6 family cytokines such as LIF and IL-11 (Fig. 5), as well as BAFF, which may collectively promote local B cell activation and polyclonal expansion of mucosal antibody-secreting cells. These findings are consistent with the possibility that EBV or other herpesvirus reactivation, together with ongoing SARS-CoV-2 antigenic exposure, may contribute to the systemic immune alterations observed in LC.^51^.
Our immunophenotyping of PBMCs revealed significant alterations in individuals with LC compared to convalescent controls, specifically in the frequencies of circulating TFH, CD4+ TCM and MAIT cells. Elevated proportions of circulating TFH likely indicate their critical role in B cell activation, and the generation of high-affinity antibodies during the acute but also post-acute phases of the infection. The surge of cTFH and CD4+ TCM cells also suggests a persistent ongoing T-cell driven immune response and dysregulated B cell activity^19,54^. One possibility is that persistent antigen exposure, potentially due to viral protein reservoir or latent viral reactivation, sustains TFH cell differentiation and circulation. Alternatively, this could indicate altered trafficking or retention of TFH-like cells outside of secondary lymphoid tissues. Elevated frequencies of cTFH could contribute to prolonged or atypical antibody responses, including those observed in our proteomic analyses showing sustained Envelope- and Nucleocapsid-specific IgG titers in LC, as well as elevated mucosal immunoglobulin signature in the plasma proteome.
Elevated presence of MAIT cells in peripheral blood during LC is indicative of systemic immune activation, altered tissue trafficking, or an ongoing mucosal immune stimulation^55,56^. During acute SARS-CoV-2 infection, MAIT cells undergo significant perturbation, exhibiting activation during acute disease followed by numerical alterations and functional impairments^57–59^. Interestingly, in our study, LC participants demonstrate sustained elevation of MAIT cell frequencies for at least 6 months post-acute infection. This persistence strongly correlates with heightened IgG titers against the SARS-CoV-2 Envelope protein, suggesting a prolonged immune activation state that is absent in convalescent individuals. In fact, in an animal model of SARS-CoV-2 infection, MAIT cells have been shown to provide help to B cells and enhance Ab response^60^. This raises important questions regarding MAIT cell involvement in the pathogenesis of LC^61^. Their continued presence could indicate chronic inflammatory signaling, potentially mediated by IL-18 and IL-12 or other combination of inflammatory cytokines, leading to dysregulated immune responses. Moreover, the sustained anti-Envelope IgG levels point to ongoing B cell activity, which may be influenced by MAIT cell-derived cytokines such as IFN-γ and IL-17. In chronic viral infections like HIV and hepatitis, MAIT cells frequently exhibit exhaustion phenotypes, contributing to disease persistence rather than resolution^55,62–64^. A similar mechanism may be at play in LC, where sustained MAIT activation perpetuates inflammatory cascades, leading to lingering symptoms. The interplay between prolonged MAIT activity and persistent B cell activation necessitates further exploration, as it could provide valuable insights into the immune dysregulation characteristic of LC. Investigating exhaustion markers and functional capacity of these cells in LC patients may help clarify whether they are contributing to pathogenesis or attempting to resolve residual viral presence. Given their known roles in chronic viral infection, it is plausible that MAIT cells serve as mediators of long-term immune alterations in SARS-CoV-2 infection, warranting deeper mechanistic studies.
The presence and persistence of autoantibodies in a subset of LC participants further underscores the immune dysregulation that we observed at the cellular level. Interestingly, the pattern of autoantibodies varied between the affected LC participants, suggesting that there is no single autoantigen that promotes a state of persistent autoimmunity and immune dysregulation^22,44,65^. This heterogeneity in autoantibody responses may reflect the heterogeneity of endotypes in our LC population as well as the fact that the tissue injury observed during the acute and post-acute phases of the SARS-CoV-2 infection can vary widely between individuals. The location of the tissue injury and the types of cells involved in the various manifestations of tissue injury may dictate which autoantibodies are generated, thus resulting in substantial variability between individual LC participants.
One limitation of our study is the relatively small cohort sizes and the heterogeneity within the LC group, both in terms of demographics and clinical presentation. LC encompasses a wide spectrum of symptoms, and a larger-scale immunophenotyping study may be required to ascertain differences between distinct LC endotypes. The divergency of LC symptoms remains a central issue for many studies and addressing this complexity will require large, more stratified studies with sufficient power to explore associations between immune features and clinical phenotypes.
Our analysis focused on immune cell populations in peripheral blood as the primary source of study specimens. However, both our findings and other studies^19,21,27,28,30,57,66^ suggest that the most significant immune activation, dysregulation, and potential viral reservoirs likely reside in tissues, particularly mucosal sites. This limitation poses a challenge, as human tissue samples are difficult to access, making blood-based studies the most feasible approach despite their inability to fully capture tissue-specific immune dynamics. While peripheral blood provides valuable insights into systemic immune responses, it may not reflect the localized inflammation and persistence of viral antigens in tissues.
While our approach is a limitation for the purpose of mechanistic analyses and determination of possible tissue reservoirs of viral persistence, the detection of persistent antiviral IgG and of key markers of immune dysregulation in peripheral blood has significant value for its implementation in the clinical setting because blood is readily accessible during routine outpatient visits. Our work highlights the potential value of obtaining longitudinal samples over a period of several months to aid in the diagnosis and monitoring of LC in clinical settings and intervention studies.
In summary, our longitudinal immunoprofiling reveals persistent humoral, cellular, and molecular immune dysregulation in individuals with LC. We demonstrate that IgG antibodies in LC participants exhibit persistent skewing in SARS-CoV-2 antigen recognition, a pattern that correlates with long-lasting activation of circulating T follicular helper (cTFH) cells and mucosal-associated invariant T (MAIT) cells. Importantly, these features persist for at least six months following LC diagnosis, consistent with a model of ongoing viral or antigenic persistence, possibly in the mucosa. This immunological imprint is further characterized by continued systemic elevation of inflammatory cytokines and the presence of autoreactive antibody responses, suggesting sustained immune activation and dysregulation. Taken together, our findings provide new insights into the immunopathology of LC and support the hypothesis that unresolved antigen exposure may underlie the chronic immune perturbations that define this syndrome. These findings highlight the importance of longitudinal profiling in LC patients for diagnostic purposes, and they also suggest that such longitudinal profiling could be invaluable for the monitoring of the efficacy of novel therapeutics in LC or Long COVID.
Methods
Study design and specimens
This retrospective longitudinal study included samples collected from n=30 adult participants already enrolled in the parent Researching COVID-19 to Enhance Recovery (RECOVER) Adult Cohort Study^36,38^. Participants in the parent RECOVER Adult Cohort Study were enrolled at 83 sites across the United States recruited through clinician referrals, mailings to participants who tested positive for SARS-CoV-2, or responses to medical center paper postings, websites, and advertisements. Adult participants were enrolled in this cohort approximately 3 weeks after symptomatic, polymerase chain reaction (PCR)-confirmed SARS-CoV-2 infection between December 2021 and October 2022. The LC group definition was: SARS-CoV-2 infected and RECOVER Long COVID Research Index 2023 (LCRI)^67^ ≥ 12 at T2 (6 months post infection) with only a single infection recorded and vaccinated. The CONV group were defined to have 2023 LCRI < 12 at T2 (6 months post infection). The parent RECOVER-Adult protocol was approved by the IRB at NYU Grossman School of Medicine and collaborating sites, including the University of Illinois at Chicago (UIC, IRB study #2021–1287). Cryopreserved PBMC, plasma and serum specimens were received from the RECOVER cohort at Mayo Clinic, Rochester. Cryopreserved PBMC were stored in liquid nitrogen, and plasma and serum were stored at −80 °C until use. Trained personnel processed and tested frozen specimens at UIC under BSL-2 or BSL-3 conditions. Details about participant cohorts are summarized in Table 1 and Suppelementary Table 1. Specific dates for the participant’s infection (self-reported), study enrollment, and each of the 3 collections are summarized in Table 1, Fig. 7a; the timepoints of specimen collections are summarized in Figs. 1a and 7b,c as days post-infection.
Antibody ELISA
We used 96-well flat bottom microtiter ELISA plates (MaxiSorp Thermo Scientific Nunc Cat. No. 442404) pre-coated with 0.1 μg/ml of SARS CoV-2 B.1.1.529/Omicron recombinant Spike trimer protein (His Tagged, Acro Biosystems, Cat.No. SPNC52Hz), SARS-CoV-2 B.1.1.529/Omicron Membrane Glycoprotein (RayBiotech Code: 230-01124-100) at 0.2 μg/ml, SARS-CoV-2 B.1.1.529/Omicron Envelope protein (ABclonal Cat.No. RP01263LQ) at 0.1 μg/ml, SARS-CoV-2 B.1.1.529/Omicron Nucleocapsid Protein (Ray Biotech Cat.No, 230-30164-100) at 0.1 μg/ml. The coating buffer consisted of 15 mmol/L Na_2_CO_3_, 35 mmol/L NaHCO_3_, 7.7 mmol/L NaN_3_ at pH 9.6, and the plates were incubated overnight at 4°C. The wells were washed three times with 300 μL/well of washing buffer (0.05% Tween-20 in PBS, pH 7.4) and dried before blocking. For blocking, each well was treated with 300 μl Blocking Buffer (2% BSA in Washing Buffer, pH 7.4) at room temperature for 2 hours. After blocking, 100 μl of heat-inactivated serum were added to the respective wells at dilutions: 1:3200 for Spike, 1:200 for Envelope, 1:2000 for Nucleocapsid and 1:200 for Membrane. The samples were diluted in sample dilution buffer (0.5% BSA in Washing Buffer, pH 7.4) and incubated at 37 °C for 1 hour. Following the serum sample incubation, 100 μl of Goat anti-human IgG polyclonal secondary antibody conjugated with HRP (Invitrogen Cat. No. A18805) Goat anti-Human IgA Polyclonal Secondary Antibody, HRP from (Invitrogen Catalog # PA1–74395) or Goat anti-human IgG1, IgG2, IgG3 or IgG4 (all Invitrogen) was added to the respective wells and incubated at 37 °C for 1 hour. The antibody was diluted to 1:5000 in Antibody dilution Buffer. Next, 100 μl of the Substrate Solution was added to each well and incubated at 37 °C for 10 min. The Substrate Solution consists of 8 μl 3% H_2_O_2_ and 100 μl 10 mg/mL TMB in 10 mL of Substrate Solution A (50 mmol/L Na_2_ HPO_4_·12H_2_O, 25 mmol/L Citric acid, pH5.5). Finally, 50 μl of stop solution one mol/L sulfuric acid was added to each well, and the plate was read in a microtiter plate reader at OD 450 nm to obtain the OD values for calculations.
Milliplex cytokine detection
To track and correlate the progression of 96 different analytes, serum samples from 30 participants were collected across three time points, heat inactivated at 56°C for 60 minutes in a water bath, and frozen for storage. The samples were then thawed for use in a multiplex assay using a Magpix platform (Millipore Sigma) to quantify the concentrations of 96 different analytes using 2 different kits. Samples were processed and loaded onto multiple 96-well plates, with procedures in doing so adhering to the protocol prescribed in the “MILLIPLEX^®^ Human Cytokine/Chemokine/Growth Factor Panel A Magnetic Bead Panel - 96-Well Plate Assay” and the “MILLIPLEX^®^ Human Cytokine/Chemokine/Growth Factor Panel B Magnetic Bead Panel - 96-Well Plate Assay” guides packaged with the kits used in the assay. Kits used in the assay were provided by MilliporeSigma (HCYTA-60K-PXBK48 and HCYTB-60K-PXBK48). The reagents used in this assay, including standards, quality controls, and detection antibodies were included in the respective kits. To measure the concentrations of each analyte in the samples after well plate preparation, samples were run on the Luminex^®^ MAGPIX^®^ instrument, an instrument used for multiplexing assays, using the xPONENT^®^ software. Calibration and performance verification were conducted, adhering to instructions in the kit guides, prior to running the samples. After running the samples, data was outputted onto a Microsoft^®^ Excel^®^ spreadsheet, which included average concentration and median fluorescence intensity measurements in each well. Cytokine expression clustering analysis was done using Cytomod as described^68^. Briefly, cytokine concentrations were first normalized within each participant from LC and CONV groups using a regression-based background adjustment. Pairwise Spearman correlations were then computed on adjusted values to select cytokines with similar expression kinetics. Unsupervised hierarchical clustering of the correlation matrix was performed to define cytokine modules, with the optimal number of clusters determined by the Tibshirani gap statistic. Cluster stability was assessed by 1,000 bootstrap iterations, and the final modules were assigned based on the probability that cytokine pairs consistently co-clustered. Associations between cytokine levels and long covid outcome variable were evaluated using linear regression models using GraphPadPrism v.10.2.
CyTOF
PBMC samples were retrieved from liquid nitrogen and thawed in a 37°C water bath for 1 minute or until the frozen media had dissolved. Subsequently, 1 ml of thawed PBMCs was transferred to 9 ml of cold RPMI-10% medium in conical tubes, and the mixture was allowed to equilibrate for 10 minutes. The PBMC-containing conical tubes were centrifuged at 1500 rpm at 4°C for 5 minutes. The supernatants were carefully removed, the cells were washed twice with RPMI-10% medium for 5 minutes each time, and the cells were suspended in 1 ml of RPMI-10% medium. PBMC viability was assessed using 0.4% Trypan blue. Following this, 2–3 ×10^6 PBMC were mixed with 5 mL of Maxpar Cell Staining Buffer (CSB 201068) and centrifuged at 300 × g for 5 minutes. The cells were then treated with 5μL of Human TruStain FcX^™^ FcR (Biolegend Cat. No. 422301) blocking agent for 10 minutes, and 215μL of staining buffer was added at room temperature. Then, 270μL of the FcR-blocked PBMCs were placed into a 5 mL tube containing the dry antibody pellet containing metal-tagged antibodies from a 30-marker panel and incubated at room temperature for 30 minutes. Cells were washed with staining buffer, then fixed with 1ml of fixed solution (1.6% formaldehyde) for 10 minutes at room temperature. After another wash with staining buffer, cells were suspended in 1ml of intercalator Ir solution (Fluidigm Cat. No. 201192A) and incubated overnight at 4°C. The cells were washed twice with staining buffer and twice with 2ml of cell acquisition solution (CAS Plus 201248). Labeled PBMCs were loaded into the Fluidigm CyTOF2^®^ instrument for data acquisition. FCS files were analyzed using FlowJo v10 software. Gating strategy is presented in Extended Data Fig. 2. For clustering analysis, 1×10^5^ CD45+ cells from each fcs file from each participant in the LC and CONV cohort, at each collection timpoint were concatenated in a single fcs file for further analysis. We used FlowJo v.10.10.0 built-in tSNE and UMAP v4.1.1 for cell clustering analysis.
Virus neutralization assay
The serum from each sample was diluted at a 1:50 ratio to achieve a 3-fold dilution in DMEM Medium. 60 ul of SARS CoV-2 omicron virus (B.1.1.529) at 5.2×10^5 FFU/mL was mixed with the DMEM and then incubated at 37°C for 1 hour. The mixture was then added to the Vero E6 cells and incubated for another hour at 37°C. Subsequently, 2% Methylcellulose medium was added to each well, and the plates were incubated for 36 hours. The overlay medium was removed, and the plates were treated with 4% paraformaldehyde for 30 minutes, followed by 10% formaldehyde for 15 minutes. Virus-inactivated plates were taken out of the BSL3 facility and washed with PBS. Later, the plates were incubated with Polyclonal Anti-SARS coronavirus, guinea pig (BEI – NR0361) primary antibody mixed in perm wash at a concentration of 1:15,000 for 3 hours at room temperature. Subsequently, the plates were washed with PBS and incubated with Goat anti-guinea HRP-labeled antibody (Sigma - A-7289) at a 1:5000 dilution for 3 hours. After being rewashed, the plates were stained with 30ul of TrueBlue stain for 15 minutes at room temperature. Finally, the plates were read by an Immunospot plate reader, and 20–200 spots/foci were counted and calculated.
Mass spectrometry
Sample Preparation:10μl of each were initially diluted with an equal amount of PBS and then mixed with 40μl of urea. From this dilution, 25μl of the sample was mixed with 5μl of urea and 200mM DTT. The mixture was then incubated at 56°C for 30 minutes. Following this, 3.3μl of 500 IAA was added to the sample and then incubated in a dark room for 40 minutes. Subsequently, 170μl of 100mM ABC and 8μl of Trypsin (2μg) were added to each sample, and the samples were incubated at 37°C overnight. The digestion process was stopped by adding 10% formic acid (FA) to reach a final concentration of 1% FA. The dried protein digests were resuspended in 400μl of 5% acetonitrile and 0.1% formic acid buffer for LC-MS analysis. The criteria for analysis included a 1% false discovery rate (FDR) for proteins and peptides, with a minimum of 1 peptide required for identification. A 1uL sample was analyzed using a Q Exactive HF mass spectrometer linked to an UltiMate 3000 RSLC nanosystem equipped with a Nanospray Flex Ion Source (Thermo Fisher Scientific). Digested peptides were loaded into a Waters nanoEase M/Z C18 trap column (100Å, 5um, 180um × 20mm) and then a 75 μm × 150mm Waters BEH C18 column (130A, 1.7um, 75μm × 15cm) and separated at a flow rate of 300nL/min. The LC solvent gradient was as follows: 5% B from 0–3 min, 8% B at 3.2min, 8–35% B at 110 min, 35–95% B at 119 min, washed 95% at 129 min, followed by 5% B equilibration until 140 min. Full MS scans were taken in the Q-Exactive mass spectrometer over the 350–1400 m/z range with a resolution of 60,000 (at 200 m/z) from 10 min to 130 min. The AGC target value for the full scan was 3.00E+06. The 15 most intense peaks with charge states 2, 3, 4, 5 were fragmented in the HCD collision cell with a normalized collision energy of 28%, and these peaks were then excluded for 30s within a mass window of 1.4 m/z. A tandem mass spectrum was acquired with a resolution of 30,000, and the AGC target value was 1.00E+05. The ion selection threshold was 1.00E+04 counts, and the maximum allowed ion injection time was 50 ms for both full and fragment ion scans. The spectra were searched against the UniProt human database using Mascot Daemon (2.6.0, updated on 09/28/21) with specific search parameters. The search results were then entered into Scaffold DDA software (v6.0.1, Proteome Software, Portland, OR) for compilation, normalization, comparison of spectral counts. The filtering criteria for protein identification were a 1% false discovery rate (FDR) for both proteins and peptides with a minimum peptide count of 1.
HuProt assay for autoantibodies
HuProt^™^ arrays were used for the ImmuneProfiler Assay by CDI Labs. Both, arrays and serum samples were blocked for 1 hour prior to the assay. For blocking, serum samples were diluted 1:1000 into CDISampleBuffer while arrays were incubated with CDIArrayBuffer at room temperature with gentle shaking. Each blocked and diluted sample was then probed onto a HuProt^™^ microarray at room temperature for 1 hour with gentle shaking. After probing, the arrays were washed with TBST (1xTBS / 0.1% Tween 20) for 10 min X 3, and then probed with Alexa 647-anti-human IgG, Fc specific and Cy3-anti-human IgM at room temperature for 1 hour in a light-proof box with gentle shaking, followed by 3 washes with TBST for 10 min each and 3 rinses with ddH2O. The arrays were then dried with an air duster and scanned using a GenePix^®^ 4000B scanner for data collection. Consistent reproducibility was noted among hits in the raw data obtained from the GenePix software within samples. Non-specific hits that directly bind to the secondary antibodies were eliminated from the analysis of the sample. CDI software was used to quantify the specificity of each individual sample to specific proteins on the array based on Z Scores. Further analysis on the HuProt data was performed in R. After background probes were removed the remaining 23,058 proteins were tested. Each time point was treated independently with the comparison of CONV (n=30) vs. LC (n=60) between groups. T-tests for each protein were performed with the comparison between CONV vs. LC. To control for multiple hypothesis testing, we adjusted the p-value to reduce the false discovery rate using the Benjamini Hochberg method. We filtered significant proteins with the adjusted p−value < .05 and (log fold change) > .5.
Analysis and Statistics
Statistical analysis of data was performed using GraphPadPrism v.10.2 software. Where indicated, datasets between LC and CONV cohort were analyzed using 2-way ANOVA, p values between experimental groups were calculated for all timepoints. For Milliplex multiplexed cytokine profiles modular clustering analysis in Fig. 5 we used R software with Cytomod^68^. We used Orange Datamining software v.3.3.8 for heatmap clustering analysis in Fig. 2a. Data visualization in Fig. 7a was done using Datawrapper. Fig. 1a was created with help of BioRender.
Extended Data
CyTOF analysis of PBMC by t-SNE.(a) A t-distributed stochastic neighbor embedding (t-SNE) analysis and map of all CD45+ cell subsets within concatenated samples from all participants in LC and CONV cohorts at all timepoints. Distinct, color-coded cell subsets within the PBMC are overlayed on the total CD45+ population (grey) on the t-SNE map. (b) Multigraph color maps of all markers used in cell phenotyping analyses; colors scaled from blue to red show min to max expression intensity, (c) Pseudo-color density t-SNE maps of CD45+ cells from concatenated CONV (left) or LC (right) cohorts at TO, T1 and T2 collection timepoint. tSNE clustering and cell types in b-c correspond to (a).
Gating strategy.Representative gating strategy of cell types and subtypes defined within the total CD45+ population in PBMC. Data was analyzed using FlowJo v10.
CyTOF analysis of T cell subsets.(a and b) Graphs represent frequency of CD8+ TCM, TTEff, TEM and Tnaive cell subsets within total CD8+ T cells (a) or CD4+ T cells (b). Cell subsets were defined as presented in the Extended Data Fig. 3.
Cytomod analysis of cytokine signatures associated with PASC.Pairwise Pearson’s correlations between cytokines following adjustment to the mean cytokine level. Cytokines were sorted along both axes using complete-linkage.
Autoreactive IgM repertoire in LC.Autoreactive IgG titers were measured by HuProt protein array. The heat map displays Z-scores representing the abundance of each top 20 target proteins of autoreactive IgM in LC. Columns indicate individuals from LC (red) and CONV (cyan) at each timepoint, rows represent HuProt array protein targets.
Supplementary Material
1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization, W. Post COVID-19 condition (long COVID). (2025).
- 2Eisinger R. W. Progress on the pathway to long COVID treatments. Nat Immunol 26, 2126–2129 (2025). 10.1038/s 41590-025-02336-y 41249484 · doi ↗ · pubmed ↗
- 3Peluso M. J. & Deeks S. G. Mechanisms of long COVID and the path toward therapeutics. Cell 187, 5500–5529 (2024). 10.1016/j.cell.2024.07.05439326415 PMC 11455603 · doi ↗ · pubmed ↗
- 4Greenhalgh T., Sivan M., Perlowski A. & Nikolich J. Z. Long COVID: a clinical update. Lancet 404, 707–724 (2024). 10.1016/S 0140-6736(24)01136-X 39096925 · doi ↗ · pubmed ↗
- 5An A. Y. Post-COVID symptoms are associated with endotypes reflecting poor inflammatory and hemostatic modulation. Front Immunol 14, 1243689 (2023). 10.3389/fimmu.2023.124368937680625 PMC 10482103 · doi ↗ · pubmed ↗
- 6Al-Aly Z. & Topol E. Solving the puzzle of Long Covid. Science 383, 830–832 (2024). 10.1126/science.adl 086738386747 · doi ↗ · pubmed ↗
- 7Davis H. E., Mc Corkell L., Vogel J. M. & Topol E. J. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol 21, 408 (2023). 10.1038/s 41579-023-00896-037069455 PMC 10408714 · doi ↗ · pubmed ↗
- 8Mehandru S. & Merad M. Pathological sequelae of long-haul COVID. Nat Immunol 23, 194–202 (2022). 10.1038/s 41590-021-01104-y 35105985 PMC 9127978 · doi ↗ · pubmed ↗