Unravelling the triad of penicillin-binding proteins, β-lactamase activity, and mRNA dynamics in Pseudomonas aeruginosa AmpC induction
Maria Montaner, Mariella Montes, Francina Alajarin, Silvia López-Argüello, Antonio Oliver, Bartolome Moya

TL;DR
This study explores how different antibiotics affect gene expression and enzyme activity in Pseudomonas aeruginosa, revealing that gene activity doesn't always match enzyme levels.
Contribution
The study reveals that ampC transcription does not predict functional enzyme levels, especially for PBP3-binding drugs.
Findings
Carbapenems and cefoxitin induced strong ampC transcription and β-lactamase activity by targeting PBP4.
LMW-PBP-deficient mutants showed ampC overexpression but activity increases plateaued despite mRNA induction.
PBP4 targeting and CreBC activation appear necessary for full ampC induction.
Abstract
The main objective of the present work was to dissect the interplay between penicillin-binding protein (PBP) occupancy, ampC transcriptional dynamics, and β-lactamase activity in Pseudomonas aeruginosa. Using wild-type PAO1 and isogenic LMW-PBP knockout mutants (PAOΔdacB, PAOΔdacBdacC, and PAOΔdacBdacCpbpG), we assessed ampC induction following exposure to 18 β-lactams and β-lactamase inhibitors. PBP binding (IC50) was quantified via Bocillin-FL labelling. AmpC mRNA levels were measured by qRT-PCR, and β-lactamase activity was determined in crude extracts, periplasmic and extracellular fractions using nitrocefin and cefalotin substrates. Carbapenems and cefoxitin, the drugs that maximally inhibited PBP4, induced strong ampC transcription and β-lactamase activity, while ceftazidime, aztreonam, and penicillins (PBP3-binding) triggered mRNA upregulation without enzymatic output.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
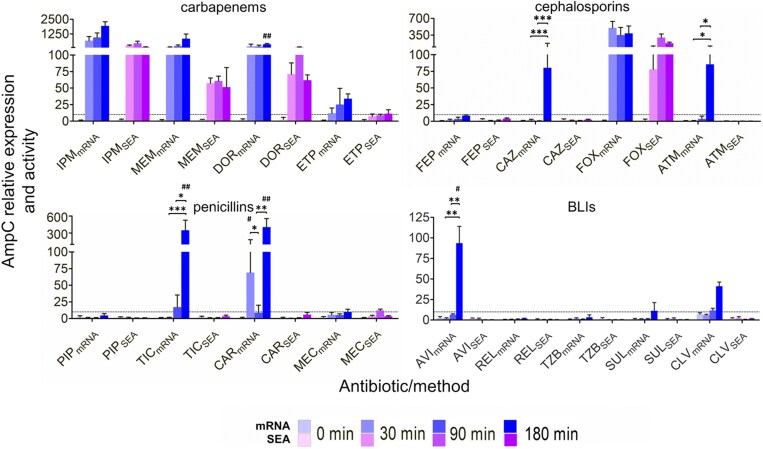 Figure 1
Figure 1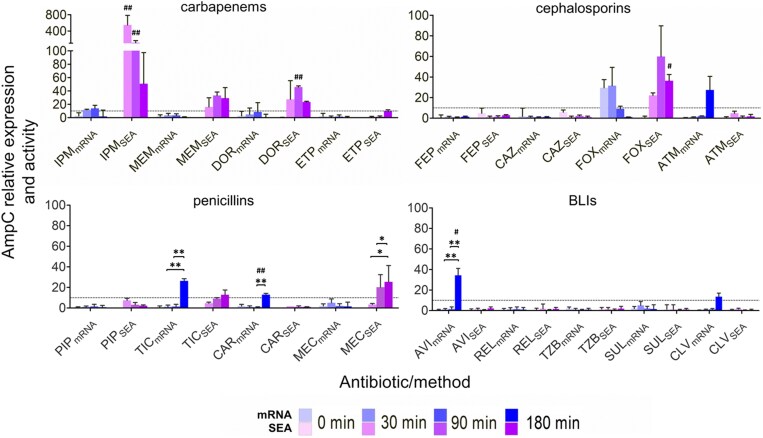 Figure 2
Figure 2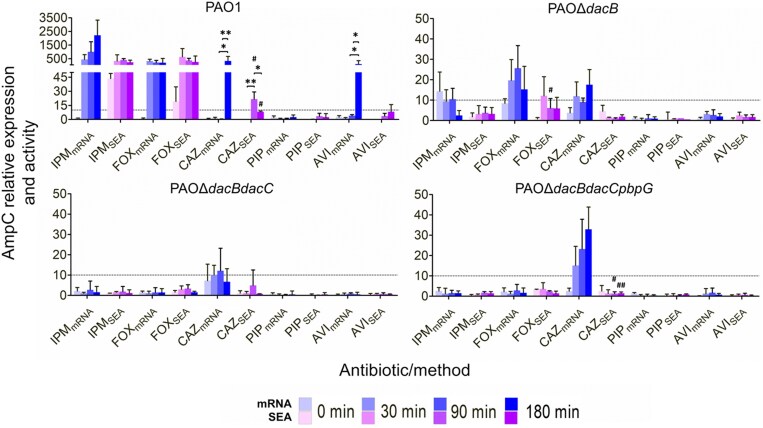 Figure 3
Figure 3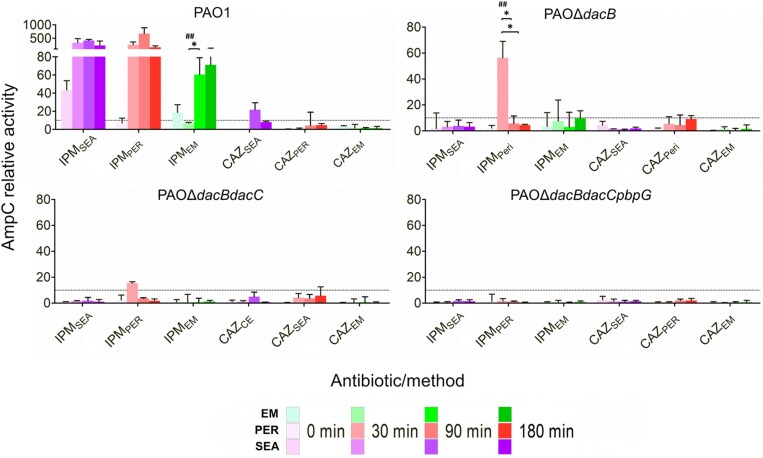 Figure 4
Figure 4| β-Lactam subclass | Drug | MIC (mg/L) | Studied drug concentration in 0.5 × MIC induction assays (mg/L) | Studied drug concentration in PBP430IC50 induction assays (mg/L) |
|---|---|---|---|---|
| Carbapenem | Imipenem | 1 | 0.5 | 0.01 |
| Carbapenem | Doripenem | 1 | 0.5 | 0.02 |
| Carbapenem | Meropenem | 0.5 | 0.25 | 0.02 |
| Carbapenem | Ertapenem | 4 | 2 | 0.05 |
| Penicillin | Piperacillin | 4 | 2 | 1.67 |
| Penicillin | Carbenicillin | 32 | 16 | 4.98 |
| Penicillin | Ticarcillin | 16 | 8 | 5.21 |
| Penicillin | Mecillinam | 16 | 8 | 211.78 |
| Monobactam | Aztreonam | 4 | 2 | 1.35 |
| Cephalosporin | Ceftazidime | 1 | 0.5 | 0.32 |
| Cephalosporin | Cefepime | 1 | 0.5 | 0.25 |
| Cephalosporin | Cefoxitin | 1024 | 512 | 20.42 |
| DBO-BLI | Avibactam | ND | 4 | 2.39 |
| DBO-BLI | Relebactam | ND | 4 | 8.49 |
| BLI | Tazobactam | ND | 4 | 4.78 |
| BLI | Sulbactam | ND | 4 | 2.89 |
| BLI | Clavulanic Acid | ND | 4 | 4.34 |
| Time (min) | PBP | Drug mg/L | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IPM | ETP | MEM | DOR | PIP | TIC | CAR | MEC | ATM | ||
| 30 | PBP2 | 0.06 ± 0.02 | 0.32 ± 0.04 | 1.53 ± 0.38 | 0.29 ± 0.05 | 2.18 ± 0.54 | 19.8 ± 3.25 | 32.3 ± 12.6 | 283 ± 65.2 | 7.42 ± 6.70 |
| PBP3 | 1.01 ± 0.64 | 0.54 ± 0.32 | 0.60 ± 0.26 | 0.44 ± 0.12 |
|
|
| 564 ± 71.3 |
| |
| PBP4 |
|
|
|
| 1.67 ± 0.67 | 4.98 ± 2.02 | 5.21 ± 5.13 |
| 1.35 ± 0.72 | |
| PBP5 | 0.06 ± 0.02 | 0.81 ± 0.12 | 1.40 ± 0.22 | 0.90 ± 0.25 | 3.15 ± 2.11 | 262 ± 66.1 | 48.8 ± 26.9 | 423 ± 226 | 4.55 ± 3.84 | |
| 90 | PBP2 | 0.07 ± 0.01 | 0.07 ± 0.04 | 0.14 ± 0.08 | 0.36 ± 0.17 | 1.54 ± 0.30 | 11.9 ± 7.85 | 24.4 ± 7.81 | 58.2 ± 11.8 | 6.52 ± 2.07 |
| PBP3 | 1.51 ± 0.63 | 0.25 ± 0.16 | 0.32 ± 0.12 | 0.22 ± 0.15 |
|
| 7.27 ± 3.57 | 95.1 ± 33.7 |
| |
| PBP4 |
|
|
|
| 0.64 ± 0.25 | 3.19 ± 0.22 |
|
| 1.41 ± 1.51 | |
| PBP5 | 0.14 ± 0.01 | 0.80 ± 0.24 | 0.68 ± 0.16 | 1.01 ± 0.19 | 6.35 ± 11.6 | 166 ± 89.4 | 48.8 ± 26.9 | 376 ± 118 | 7.03 ± 4.19 | |
| 180 | PBP2 | 0.15 ± 0.04 | 0.26 ± 0.11 | 0.18 ± 0.03 | 0.05 ± 0.04 | 2.13 ± 0.52 | 14.21 ± 4.72 | 13.67 |
| 1.32 ± 0.32 |
| PBP3 | 0.93 ± 0.22 | 0.10 ± 0.04 | 2.24 ± 0.76 | 0.64 ± 0.27 |
|
|
| 80.8 ± 32.8 |
| |
| PBP4 |
|
|
|
| 0.38 ± 0.14 | 3.93 ± 1.39 | 3.87 ± 0.73 | 39.2 ± 4.45 | 1.22 ± 0.62 | |
| PBP5 | 0.12 ± 0.07 | 0.73 ± 0.31 | 0.48 ± 0.27 | 0.33 ± 0.11 | 0.79 ± 0.87 | 68.7 ± 14.8 | 21.3 ± 18.2 | 254 ± 157 | 24.8 ± 1.37 | |
- —IdISBa Research Institute of Health Sciences of the Balearic Islands
- —Hospital Universitario Son Espases
- —Sustainable Tourism Tax
- —Govern de les Illes Balears10.13039/501100022397
- —National Institute of Health Carlos III
- —Agencia Estatal de Investigación10.13039/501100011033
- —Plan Estatal de Investigación Científica
- —2024 Research Consolidation Program
- —State Plan for Scientific, Technical and Innovation Research 2024–27
- —National Institute of Allergy and Infectious Diseases10.13039/100000060
- —National Institutes of Health10.13039/100000002
- —Ministerio de Economía y Competitividad of Spain10.13039/501100003329
- —Instituto de Salud Carlos III10.13039/501100004587
- —European Regional Development Fund10.13039/501100008530
- —Spanish Network for the Research in Infectious Diseases
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntibiotic Resistance in Bacteria · Bacterial Genetics and Biotechnology · Microbial Natural Products and Biosynthesis
Introduction
Pseudomonas aeruginosa remains a formidable clinical challenge due to its intrinsic resistance mechanisms, among which the upregulation of the chromosomal inducible cephalosporinase-type AmpC β-lactamase [Pseudomonas-derived β-lactamase (PDC)] stands as a pivotal contributor to multidrug resistance.^1^ The dynamic interplay between beta-lactam antibiotics and AmpC expression underscores the complexity of bacterial adaptive responses, highlighting the need for a comprehensive understanding of the molecular mechanisms driving this phenomenon.^2^
Additionally, most P. aeruginosa strains possess the genes for OXA-50 and PIB-1 β-lactamases. Despite the widespread distribution of PIB-1, its expression in the laboratory strain PAO1 and in clinical isolates is negligible. OXA-50, also known as PoxB, has been acknowledged as a naturally occurring oxacillinase in this bacterial species. However, neither of these enzymes has been experimentally linked to β-lactam resistance, which is primarily attributed to the presence of the AmpC cephalosporinase.^3–5^
In P. aeruginosa, the regulation of ampC is tightly controlled and inducible in response to certain β-lactam antibiotics. Central to the regulation of the ampC beta-lactamase in P. aeruginosa are the penicillin-binding proteins (PBPs), which serve as the primary targets of beta-lactam antibiotics. PBPs are essential enzymes involved in peptidoglycan biosynthesis and play a key role in maintaining cell wall integrity.^2,6^ During active growth, the bacterial cell wall undergoes constant remodelling, requiring controlled cleavage of peptidoglycan by dedicated hydrolases to permit insertion of new material and successful cell division. Although a portion of the resulting fragments is released into the extracellular milieu, the majority are transported back into the cytoplasm via the AmpG permease as part of the recycling pathway.^2,7^ Once internalized, the muropeptides [predominantly N-acetylglucosamine-1,6-anhydro-N-acetylmuramyl-peptides (NAG-anhNAM-peptides)] are sequentially processed: NagZ removes the terminal NAG, and AmpD hydrolyses the bond linking the 1,6-anhNAM to the peptide stem.^2,7–9^ This process liberates sugars and peptides that are reincorporated into peptidoglycan biosynthesis, ultimately contributing to the generation of UDP-N-acetylmuramyl-pentapeptide (UDP-NAM-P5), a critical precursor. Under basal conditions, UDP-NAM-P5 binds to the transcriptional regulator AmpR in a configuration that represses ampC expression, thus maintaining β-lactamase production at minimal levels in the absence of cell wall stress.^2^
However, exposure to inducer β-lactams, such as cefoxitin, disrupts the equilibrium of peptidoglycan turnover favouring autolysis by inhibiting LMW-PBPs, notably PBP4.^2,8^ The disruption of peptidoglycan recycling leads to the saturation of NagZ and AmpD leading to the transient accumulation of cell wall degradation products such as anhMurNAc-pentapeptides (anhNAM-P5). These molecules interact with the LysR-type transcriptional regulator AmpR, converting it from a repressor to an activator of ampC transcription.^2,6,10,11^ Mutations in ampD or to a lower extent related amidases (ampDh2, ampDh3) can lead to constitutive hyperexpression of ampC and β-lactam resistance.^8,12^
Furthermore, inactivation of the LMW-PBPs, PBP5/6 (dacC), PBP7 (pbpG), and particularly PBP4 (dacB), results in the constitutive accumulation of the cell wall degradation products which in turn lead to high-level β-lactam resistance.^2,6,10,11^ PBP4 inhibition and deletion have been shown to cause the activation of the CreBC two-component system in contrast with AmpD (AmpDh2 and AmpDh3) disruption, which has no effect on this system.^2,13,14^ The abnormal buildup of anhNAM-P5 following PBP4 inactivation acts as a key trigger activating CreBC through CreC sensor while also boosting ampC expression via AmpR. This over-accumulation of soluble anhNAM-P5 likely underlies the role of PBP4 as a cell wall stress sentinel, acting as the signal sensed by the membrane-bound CreC sensor. This, in turn, triggers CreBC activation and simultaneously promotes ampC expression through AmpR binding.^2,8,10,11,15,16^
In P. aeruginosa, ampC mRNA levels and β-lactamase activity are related but not interchangeable measures of resistance. Quantifying ampC transcripts gives an early readout of gene induction, often before any detectable enzymatic activity, but doesn’t always translate to functional resistance due to possible post-transcriptional or post-translational regulation.^6,11,17^ On the other hand, β-lactamase activity assays directly measure the hydrolysis of β-lactams, providing a functional snapshot, but they’re influenced by growth phase and metabolic state and can miss early low-level activity. Furthermore, post-transcriptional mechanisms, such as mRNA stability and translation efficiency, further modulate beta-lactamase expression and contribute to the overall antibiotic resistance phenotype.^15,18^ Relying on mRNA alone risks overlooking the actual resistance phenotype, while enzyme assays may not catch early shifts. Using both methods in parallel gives a more accurate picture of resistance progression in clinical isolates.^7,11,12,19–21^
It is essential to evaluate the actual inducibility potential of β-lactams, as their classification as either strong or weak ampC inducers significantly influences their likelihood of selecting for mutations that result in ampC constitutive derepression.^6,17,22^ Understanding the underlying determinants of ampC differential regulation is crucial for predicting antibiotic resistance selection and optimizing therapeutic interventions.
In this manuscript, we aimed to unravel the triad of PBP-mediated signalling, mRNA dynamics, and β-lactamase activity underlying P. aeruginosa AmpC induction in wild-type PAO1 and its isogenic mutants of LMW-PBPs. By leveraging a multidisciplinary approach that integrates genetic, biochemical, and gene expression analyses, we seek to expand our understanding on the factors that influence AmpC expression dynamics.
Materials and methods
Bacterial strains and in vitro susceptibility testing
P. aeruginosa PAO1 and its isogenic LMW-PBPs knockouts, PAOΔdacB, PAOΔdacBdacC, and PAOΔdacBdacCpbpG (which our group previously documented as constitutively hyper-expressing AmpC), were utilized in this study.^11^ MICs for imipenem, doripenem, meropenem, ertapenem, cefepime, ceftazidime, cefoxitin, aztreonam, piperacillin, ticarcillin, carbenicillin, mecillinam, and the β-lactamase inhibitors avibactam, clavulanic acid, relebactam, tazobactam, and sulbactam (all from MedChem Express, Sollentuna, Sweden) were determined by Clinical and Laboratory Standards Institute (CLSI) broth microdilution.^23^
PBP binding assays
Binding to PBP2, PBP3, PBP4, and PBP5 was quantified in intact PAO1 at 30, 90, and 180 min after exposure to twelve β-lactams (four carbapenems, three cephalosporins, three antipseudomonal penicillins, one monobactam) plus four β-lactamase inhibitors. Overnight 100 mL LB cultures were harvested (4000 rpm, 20 min, RT), washed four times in PBS, and finally resuspended to 2 mL. Aliquots (150 µL) were dispensed into microtiter plates and exposed to 2-fold antibiotic dilutions. PBP labelling with Bocillin-FL and IC_50_ calculation followed the protocol previously described.^13,14^
Induction of AmpC expression
Cultures of PAO1 and its LMW-PBPs knockouts (PAOΔdacB, PAOΔdacBdacC, PAOΔdacBdacCpbpG) were grown in 30 mL Mueller Hinton broth (1:100 inoculum from overnight cultures) and incubated for 3 h at 37°C, 180 rpm with each drug at 0.5 × MIC (a dose that triggers AmpC signalling and avoids excessive killing during the 180-min activity assessment) and at the concentration that half maximally inhibited PBP4 after 30 min exposure (PBP4_30_IC_50_). Untreated controls ran in parallel. At 30, 90, and 180 min, 3 mL aliquots were harvested (4000 rpm, 15 min, 4°C), supernatants were discarded, and cell pellets were collected. Each pellet was then split: one half for total RNA isolation and qRT-PCR, the other for protein extraction and β-lactamase assays as detailed below.
Determination of ampC expression through quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted with the RNeasy Mini Kit (Qiagen, Hilden, Germany), treated with TURBO DNase (Ambion, Austin, TX, USA), and quantified on a NanoDrop One (Thermo Fisher Scientific, Waltham, MA, USA). Normalized (50 ng/mL) RNA aliquots were subjected to one-step reverse transcription and qPCR with the QuantiTect SYBR Green kit (Qiagen) on a CFX Connect thermocycler (Bio-Rad, Hercules, CA, USA). Primer pairs ACrnaF/ACrnaR (ampC) and rpsL-1/rpsL-2 (housekeeping ribosomal protein S12; rpsL) were used as previously reported.^16^ Relative expression was calculated by the 2^−ΔΔCt^ method from at least two biological and two technical replicates, normalized to each strain’s t = 0 min baseline.^24^
Assessment of the specific enzymatic activity (SEA) of the β-lactamase AmpC
The specific enzymatic activity (SEA) of AmpC was quantified as nmol nitrocefin hydrolysed min^−1^ mg^−1^ protein by monitoring A_482_ in the presence of 10^−5^ M nitrocefin, exactly as described previously.^25^ Proteins were harvested from three compartments: crude extract, periplasm, and culture supernatant, and activities were expressed relative to the untreated PAO1 (or mutant) sample at t = 0 min.^16^ To confirm that hydrolysis was not substrate-dependent, parallel assays were run with cefalotin (10 mM, ΔA_260_). AmpC specificity was verified by pre-incubating crude extracts with cloxacillin at concentrations of 50 and 100 μM (equivalent to approximately 23.8 and 47.6 mg/L, respectively).^16,25,26^ Periplasmic fractions were prepared by lysozyme/MgCl_2_ spheroplasting as in our earlier work;^27^ extracellular activity was measured directly in cell-free supernatants collected at the same time points.^28^
Statistical data analysis
The GraphPad Prism software (version v9.01) was used for graphical representation and statistics. One-way ANOVA with post hoc Tukey’s multiple comparison tests was run to determine the statistically significant differences over time. Unpaired Student’s t-tests were applied to determine statistically significant differences between mRNA, qRT-PCR, and SEA determinations (Table S1, available as Supplementary data at JAC Online).
Results
Differential induction patterns among carbapenems, cephalosporins, and penicillins
At 0.5 × MIC (Table 1), carbapenems were the most potent ampC inducers. Imipenem drove a 700-fold rise in transcripts within 30 min and achieved higher expression thereafter (1900-fold at 180 min); meropenem and doripenem produced similar profiles peaking at 875- and 377-fold, respectively, whereas ertapenem triggered only minimal transcription. Relative mRNA and enzymatic outputs tracked closely for all four drugs. Cefoxitin, another strong inducer, sustained parallel increases in ampC transcripts (385–500-fold) and β-lactamase activity (77–296-fold), while cefepime caused virtually no response. Ceftazidime and aztreonam each produced a late, ∼80-fold jump in mRNA at 180 min, yet neither yielded detectable enzyme activity. Piperacillin and mecillinam were poor inducers, but ticarcillin and carbenicillin boosted transcripts 347- and 404-fold, respectively, again without measurable activity (Figure 1).
*The relative expression (baseline measurements at t = 0 min) of ampC (mRNA) in P. aeruginosa strain PAO1, as measured by qRT-PCR, and the corresponding relative specific enzymatic activity (SEA) were assessed following exposure to 0.5 × MIC of various β-lactams and β-lactamase inhibitors at time points of 0, 30, and 180 min. Results are averages of at least two sets (biological replicates) of two technical replicates. One-way ANOVA with post hoc Tukey’s multiple comparison test was run to determine the statistically significant differences over time (*P < 0.05; **P < 0.01; **P < 0.005). Unpaired Student’s t-tests were applied to determine statistically significant differences between mRNA and SEA determinations (#P < 0.05; ##P < 0.01; ###P < 0.001). Drug abbreviations are detailed in Table 1, while the concentrations employed are defined in Table 2.
Among β-lactamase inhibitors, only clavulanic acid and avibactam raised ampC mRNA, and this transcriptional signal did not translate into increased enzyme output.
Correlating PBP4 inhibition with ampC induction: mRNA levels and enzymatic activity in classic inducers
Binding to PBP4 is a recognized trigger for ampC induction. Canonical inducers, cefoxitin, imipenem, and the BLIs clavulanic acid and avibactam, all promote ampC transcription via this pathway. To test whether a partial PBP4 occupancy (50% binding at 30 min, PBP4_30_IC_50_; Table 1) elicited comparable responses, we repeated the induction assays at these lower concentrations (Figure S1 and Table S2) following prior determination of the IC_50_ values for PBP2, PBP3, PBP4, and PBP5/6 (Table 2). Carbapenem IC_50_ values were 12–50-fold below 0.5 × MIC; penicillins were 1.5–5-fold lower (mecillinam required 35-fold more); cephalosporins were 1.5–2-fold lower, except cefoxitin (25-fold lower); β-lactamase inhibitor (BLI) values varied ±2-fold from the standard 4 mg/L (Table 1).
Under PBP4 IC_50_ conditions, ampC mRNA induction remained below the 10-fold threshold for most drugs; only cefoxitin, aztreonam, ticarcillin, carbenicillin, avibactam, and clavulanic acid exceeded this threshold (Figure 2). However, enzyme output exhibited higher significant changes (Table S1): canonical inducers imipenem and cefoxitin boosted AmpC peak activity ∼550-fold and ∼50-fold, 30 and 90 min post-exposure, respectively, while meropenem, doripenem, and mecillinam produced 20–45-fold increases at 90 min and ∼25-fold at 180 min.
*Relative ampC qRT-PCR (mRNA) expression and relative specific enzymatic activity (SEA) in P. aeruginosa strain PAO1 after 0, 30, and 180 min exposure to the concentration that half maximally inhibits PBP4 after 30 min (PBP430IC50) of drug exposure of different β-lactams and β-lactamase inhibitors relative to basal (ctrl; time = 0 min). Results are averages of at least two sets (biological replicates) of two technical replicates. One-way ANOVA with post hoc Tukey’s multiple comparison test was run to determine the statistically significant differences over time (*P< 0.05; **P< 0.01; **P< 0.005). Unpaired Student’s t-tests were applied to determine statistically significant differences between mRNA and SEA determinations (#P< 0.05; ##P< 0.01; ###P< 0.001). Drug abbreviations are detailed in Table 1, while the concentrations employed are defined in Table 2.
Limited ampC inducibility in LMW-PBP-deficient mutants
Our investigation into the binding of antibiotics to PBP4 and its role in ampC induction in PAO1 prompted us to examine the contribution of all the LMW-PBPs when fully inhibited. To this end, we performed parallel experiments using isogenic knockout mutants PAOΔdacB, PAOΔdacBdacC, and PAOΔdacBdacCpbpG.^11^
Sequential inactivation of the LMW-PBP set (PAOΔdacB, PAOΔdacBdacC, and PAOΔdacBdacCpbpG) drove a stepwise rise in constitutive ampC expression of 24-, 1500-, and 7000-fold above PAO1, respectively, yet relative AmpC activity climbed more modestly (24-, 902-, and 1154-fold) (Figure S2). SEA peaked at 1521 pmol nitrocefin min^−1^ mg^−1^ of protein in the triple mutant after 3 h. However, further inducibility proved limited: only the single mutant showed modest mRNA boosts with cefoxitin, imipenem, or ceftazidime. Although ceftazidime (PBP-3 binding; Table 2 and Table S2) further increased transcripts in the double and triple mutants, the enzyme activity remained unaltered (Figure 3).
*The relative expression (baseline measurements of each strain at t = 0 min) of ampC (mRNA) in P. aeruginosa strains PAO1 and isogenic mutants PAOΔdacB, PAOΔdacBdacC, and PAOΔdacBdacCpbpG as measured by qRT-PCR, and the corresponding relative specific enzymatic activity (SEA) were measured after exposing the strains to 0.5 × MIC of various β-lactams and β-lactamase inhibitors at intervals of 0, 30, and 180 min. Results are averages of at least two sets (biological replicates) of two technical replicates. One-way ANOVA with post hoc Tukey’s multiple comparison test was run to determine the statistically significant differences over time (*P < 0.05; **P < 0.01; **P < 0.005). Unpaired Student’s t-tests were applied to determine statistically significant differences between mRNA and SEA determinations (#P < 0.05; ##P < 0.01; ###P < 0.001). Drug abbreviations are detailed in Table 1, while the concentrations employed are defined in Table 2.
To exclude sample-handling losses as the cause of the mismatch between ampC transcripts and β-lactamase output under ceftazidime treatment (PBP3-binding, filamentation), we determined activity in supernatant, periplasm, and crude extract after ceftazidime incubation, using imipenem (PBP2-binding, spheroplast formation) as a comparator. In PAO1, imipenem drove extracellular, crude extract, and periplasmic activities up 20–60-, 416-, and 637-fold, respectively, yet no fraction of the double or triple mutants registered any differential value after imipenem or ceftazidime exposure (Figure 4).
*The relative specific enzymatic activity of different cellular compartments [crude extract (SEA), periplasmic (PER), and extracellular medium (EM)] of AmpC in P. aeruginosa strains PAO1 and isogenic mutants, PAOΔdacB, PAOΔdacBdacC, and PAΔdacBdacCpbpG, was assessed after treatment with 0.5 × MIC (per each strain) of different β-lactams and β-lactamase inhibitors. Measurements were taken at the time intervals of 0, 30, and 180 min. Results are averages of at least two sets (biological replicates) of two technical replicates. One-way ANOVA with post hoc Tukey’s multiple comparison test was run to determine the statistically significant differences over time (*P < 0.05; **P < 0.01; **P < 0.005). Unpaired Student’s t-tests were applied to determine statistically significant differences between mRNA and SEA determinations (#P < 0.05; ##P < 0.01; ###P < 0.001). Drug abbreviations are detailed in Table 1, while the concentrations employed are defined in Table 2.
To confirm that our enzyme readings were not conditioned by the use of nitrocefin as substrate, we repeated the inductions in PAO1 with imipenem, cefoxitin, and ticarcillin (0.5 × MIC and PBP4_30_IC_50_) using cefalotin as an alternative substrate (Table S3). Specific enzymatic activities (SEA) with cefalotin closely tracked those obtained with nitrocefin throughout the 180 min time course, showing only minor point-to-point variation. Thus, AmpC activity proved substrate-independent under all conditions tested. As before, ceftazidime failed to elicit activity in any LMW-PBP knockout, regardless of substrate. Finally, adding 500 mg/L cloxacillin to crude extracts of the triple mutant (PAOΔdacBdacCpbpG), which constitutively overproduces AmpC, nearly abolished activity with both substrates (>95% reduction in the hydrolysis rates), confirming that the results obtained were AmpC-specific (Table S4).
Discussion
Understanding the regulatory dynamics of ampC expression in P. aeruginosa is essential to anticipate emerging β-lactam resistance. In this study, we investigate the relationship between ampC transcriptional activity and β-lactamase production across a representative panel of β-lactam and β-lactamase inhibitors, with a particular focus on the differential roles of PBPs.
The differential induction patterns observed among carbapenems, cephalosporins, and penicillins underscore the complexity of ampC regulation in response to structurally distinct β-lactams. While carbapenems such as imipenem and meropenem consistently triggered robust ampC transcription accompanied by measurable β-lactamase activity, the lack of enzymatic response—despite pronounced mRNA upregulation—observed with late-phase inducers like ceftazidime and aztreonam suggests the involvement of distinct regulatory pathways that remain to be elucidated.^16,29^ A similar dissociation between transcriptional activation and enzymatic output was noted for ticarcillin and carbenicillin.^30–32^ These compounds predominantly target PBP3, a non-essential high-molecular-weight DD-transpeptidase involved in septal peptidoglycan synthesis, whose inhibition does not generate the muropeptide signals typically associated with LMW-PBPs such as PBP4.^10,11,20^ Consequently, PBP3 inhibition fails to activate the CreBC two-component system, which has been implicated in the full induction of ampC and its post-transcriptional regulation.^2,10,11,20,33^ This mechanistic divergence accounts for the increased propensity of third-generation cephalosporins and aztreonam to select for constitutively derepressed AmpC mutants, as opposed to agents that primarily target LMW-PBPs.^29,31,34,35^
In contrast, cefepime combines minimal inducibility with certain structural resilience to P. aeruginosa AmpC-mediated hydrolysis, owing to the formation of a stable acyl-enzyme complex that significantly reduces catalytic turnover. Indeed, only approximately 5%–10% of AmpC-producing strains—typically those exhibiting both marked derepression and efflux upregulation—display clinical resistance to this compound.^12,22,36,37^
Among β-lactamase inhibitors, only clavulanic acid and avibactam raised ampC mRNA, and this transcriptional signal did not translate into increased enzyme output.^17,38,39^ Avibactam, a diazabicyclooctane inhibitor, binds reversibly to AmpC with a Kd of ∼0.5 μM and a deacylation half-life of ∼6 min, enabling dynamic equilibrium and frequent rebinding.^40,41^ This reversible acylation masks hydrolytic activity in vitro. Furthermore, clavulanic acid did not induce ampC activity in our assays using concentrations of ≤4 mg/L (clinically and mechanistically relevant range), mirroring early reports that showed significant β-lactamase activity induction only at above-physiological peak concentrations of 8–50 mg/L.^38,39,42^
These findings underscore the pivotal role of PBP4 in ampC induction, while highlighting that its inhibition alone may be insufficient for full derepression. Although ampC transcriptional responses at PBP4_30_IC_50_ concentrations remained low for most of the compounds, strong enzymatic activity was still observed with imipenem and cefoxitin. This suggests that additional factors beyond PBP4 binding, such as PBP cooperativity or post-transcriptional regulation, may shape AmpC activity.^22,43^ Notably, while ampC mRNA levels and β-lactamase activity are positively correlated, the relationship is not linear: even modest increases in transcription can result in disproportionately high enzymatic activity and MIC values. This non-linear dynamic suggests that β-lactamase regulation is finely tuned and highly sensitive to subtle shifts in transcriptional input, possibly due to through enzyme accumulation or enhanced catalytic turnover.^31,44,45^
To confirm and further elucidate the central role of LMW-PBPs in ampC regulation in P. aeruginosa, we quantified ampC mRNA expression and β-lactamase activity in isogenic mutants with sequential deletions of LMW-PBPs. Stepwise inactivation of the LMW-PBP-encoding genes dacB, dacC, and pbpG resulted in a progressive increase in ampC mRNA levels, consistent with previous findings linking the loss of LMW-PBPs to muropeptide accumulation and constitutive ampC derepression via AmpR.^2,10–12^ Notably, despite transcript levels rising by up to ∼7000-fold, the corresponding increase in β-lactamase activity was comparatively modest (up to ∼1100-fold, or 1521 pmol nitrocefin min^−1^ mg^−1^ of protein), suggesting the existence of a post-transcriptional or post-translational bottleneck.^12,46^ The graded increase in ampC transcription observed under ceftazidime treatment across the LMW-PBP mutants can be attributed, at least in part, to the use of 0.5 × MIC relative to each strain, resulting in higher concentration and effective induction in the triple mutant. The absence of detectable β-lactamase activity following ceftazidime exposure, together with unaltered periplasmic and extracellular enzyme levels, further supports the notion of differential regulation or delayed enzyme maturation in this context.^10,12,35,46^
Furthermore, canonical inducers such as imipenem and cefoxitin elicited modest or even negligible transcriptional responses in LMW-PBP (derepressed ampC) mutants. Notably, in the complete absence of LMW-PBPs (PAOΔdacBdacCpbpG), ceftazidime was the only compound capable of triggering a transcriptional response, further reinforcing the concept of differential ampC induction via PBP3 inhibition.^29,37^ Nevertheless, consistent with earlier data in PAO1, ceftazidime exposure failed to elicit any appreciable increase in β-lactamase activity, highlighting a persistent dissociation between ampC mRNA upregulation and enzymatic output for this compound. This may reflect a delayed or reduced production of active AmpC enzyme, as illustrated by the moderate ∼6-fold increase in PAO1 after 6 h of ceftazidime and aztreonam treatment and the absence of a significant rise in the triple mutant PAOΔdacBdacCpbpG (data not shown).^29,46^ These findings underscore that ampC transcriptional activation does not inherently predict functional enzyme production, with post-transcriptional and translational (fitness-preserving) constraints likely playing a key regulatory role.^8,15^
A precise characterization of ampC induction—integrating PBP-binding affinities (IC_50_), transcriptional responses, and enzymatic activity—is essential for informing therapeutic strategies against P. aeruginosa. These findings have direct clinical implications, particularly in the context of rational antibiotic selection and the design of novel combination therapies involving β-lactamase inhibitors, where the interplay between induction and inhibition must be carefully assessed. The present findings establish a framework for future investigations into the regulatory and structural determinants of alternative ampC control and their role in shaping β-lactam resistance phenotypes.
Supplementary Material
dkaf408_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Moyá B, Beceiro A, Cabot G et al Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob Agents Chemother 2012; 56: 4771–8. 10.1128/AAC.00680-1222733064 PMC 3421878 · doi ↗ · pubmed ↗
- 2Torrens G, Hernández SB, Ayala JA et al Regulation of Amp C-driven β-lactam resistance in Pseudomonas aeruginosa: different pathways, different signaling. m Systems 2019; 4: e 00524-19. 10.1128/m Systems.00524-19.31796566 PMC 6890930 · doi ↗ · pubmed ↗
- 3Freed S Jr, Hanson ND. Ampc induction by imipenem in Pseudomonas aeruginosa occurs in the absence of Opr D and impacts imipenem/relebactam susceptibility. Microbiol Spectr 2024; 12: e 0014224. 10.1128/spectrum.00142-2439315808 PMC 11537110 · doi ↗ · pubmed ↗
- 4Zincke D, Balasubramanian D, Silver LL et al Characterization of a carbapenem-hydrolyzing enzyme, Pox B, in Pseudomonas aeruginosa PAO 1. Antimicrob Agents Chemother 2016; 60: 936–45. 10.1128/AAC.01807-1526621621 PMC 4750667 · doi ↗ · pubmed ↗
- 5Gomis-Font MA, Cabot G, López-Argüello S et al Comparative analysis of in vitro dynamics and mechanisms of ceftolozane/tazobactam and imipenem/relebactam resistance development in Pseudomonas aeruginosa XDR high-risk clones. J Antimicrob Chemother 2022; 77: 957–68. 10.1093/jac/dkab 49635084040 · doi ↗ · pubmed ↗
- 6Sanders CC, Bradford PA, Ehrhardt AF et al Penicillin-binding proteins and induction of Amp C beta-lactamase. Antimicrob Agents Chemother 1997; 41: 2013–5. 10.1128/AAC.41.9.20139303404 PMC 164055 · doi ↗ · pubmed ↗
- 7Barceló IM, Torrens G, Escobar-Salom M et al Impact of peptidoglycan recycling blockade and expression of horizontally acquired β-lactamases on Pseudomonas aeruginosa virulence. Microbiol Spectr 2022; 10: e 0201921. 10.1128/spectrum.02019-2135171032 PMC 8849096 · doi ↗ · pubmed ↗
- 8Pérez-Gallego M, Torrens G, Castillo-Vera J et al Impact of Amp C derepression on fitness and virulence: the mechanism or the pathway? m Bio 2016; 7: e 01783-16. 10.1128/m Bio.01783-1627795406 PMC 5080387 · doi ↗ · pubmed ↗