Stereodivergent Synthesis of Three Contiguous Stereogenic Centers by Cu/Ir-Catalyzed Borylallylation
Suman Das, Stanna K. Dorn, M. Kevin Brown

TL;DR
A new copper and iridium catalyzed method enables precise control over three stereogenic centers in complex molecule synthesis.
Contribution
A stereodivergent Cu/Ir-catalyzed borylallylation method for controlling three contiguous stereogenic centers is introduced.
Findings
The reaction uses a chiral Cu-complex and a chiral Ir-complex to achieve high stereocontrol.
The method enables stereodivergent synthesis of acyclic products with three stereogenic centers.
The functionalized products were used in the formal synthesis of the natural product (+)-(β)-Lycorane.
Abstract
Polyfunctional compounds bearing multiple stereogenic centers are synthetically valuable and important for complex molecule preparation. Herein, a Cu/Ir-catalyzed borylallylation of electron deficient alkenes is presented. The reaction operates by the cooperative function of a chiral Cu-complex and a chiral Ir-complex to generate products with high levels of stereocontrol. The method also allows for the stereodivergent synthesis of acyclic products with control of three stereogenic centers. Importantly, the products contain C–B, ester, and alkene functionalities, which allows for a diverse range of products to be generated. Finally, the highly functional nature of the products has been exploited in an efficient formal synthesis of the complex natural product (+)-(β)-Lycorane.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
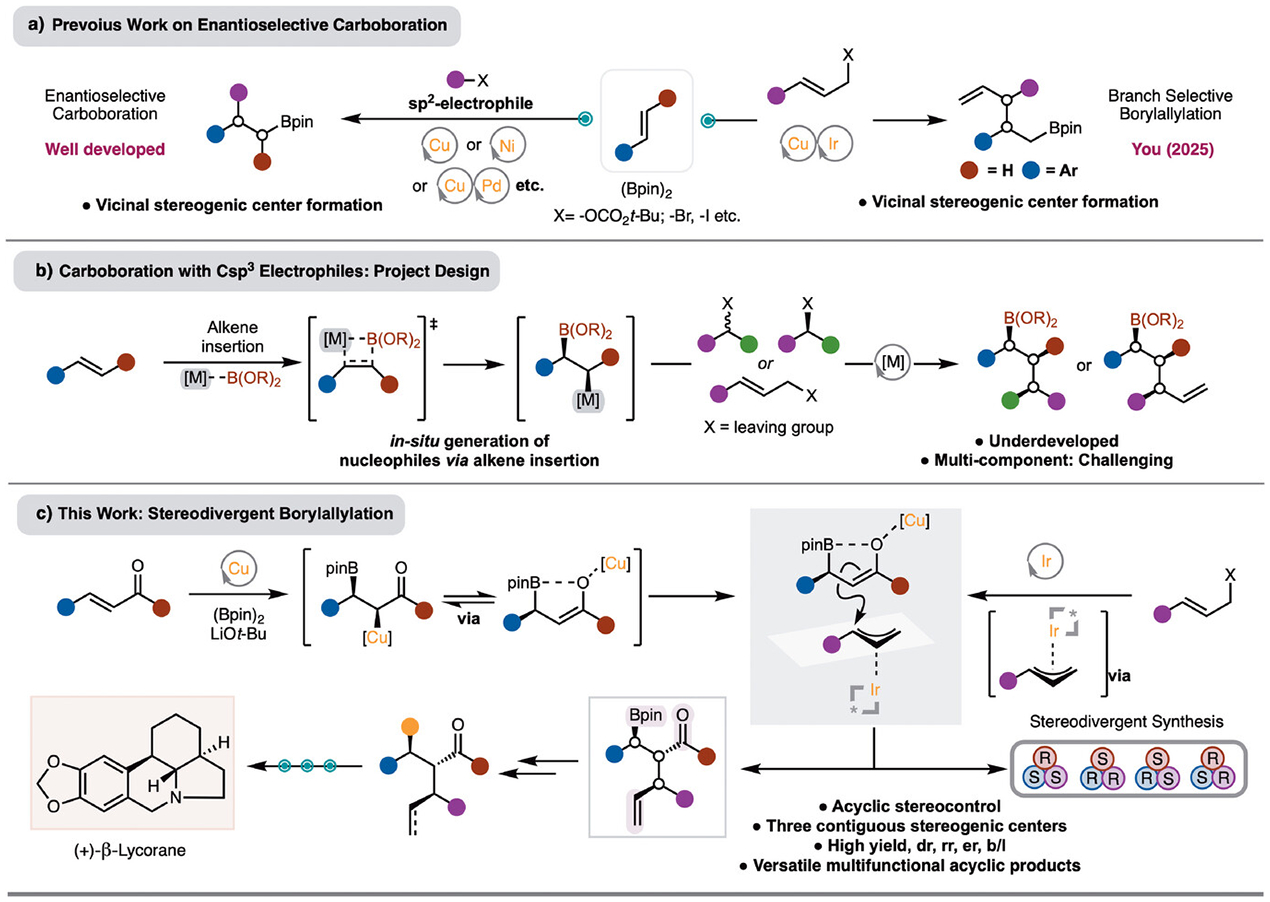 Figure 1
Figure 1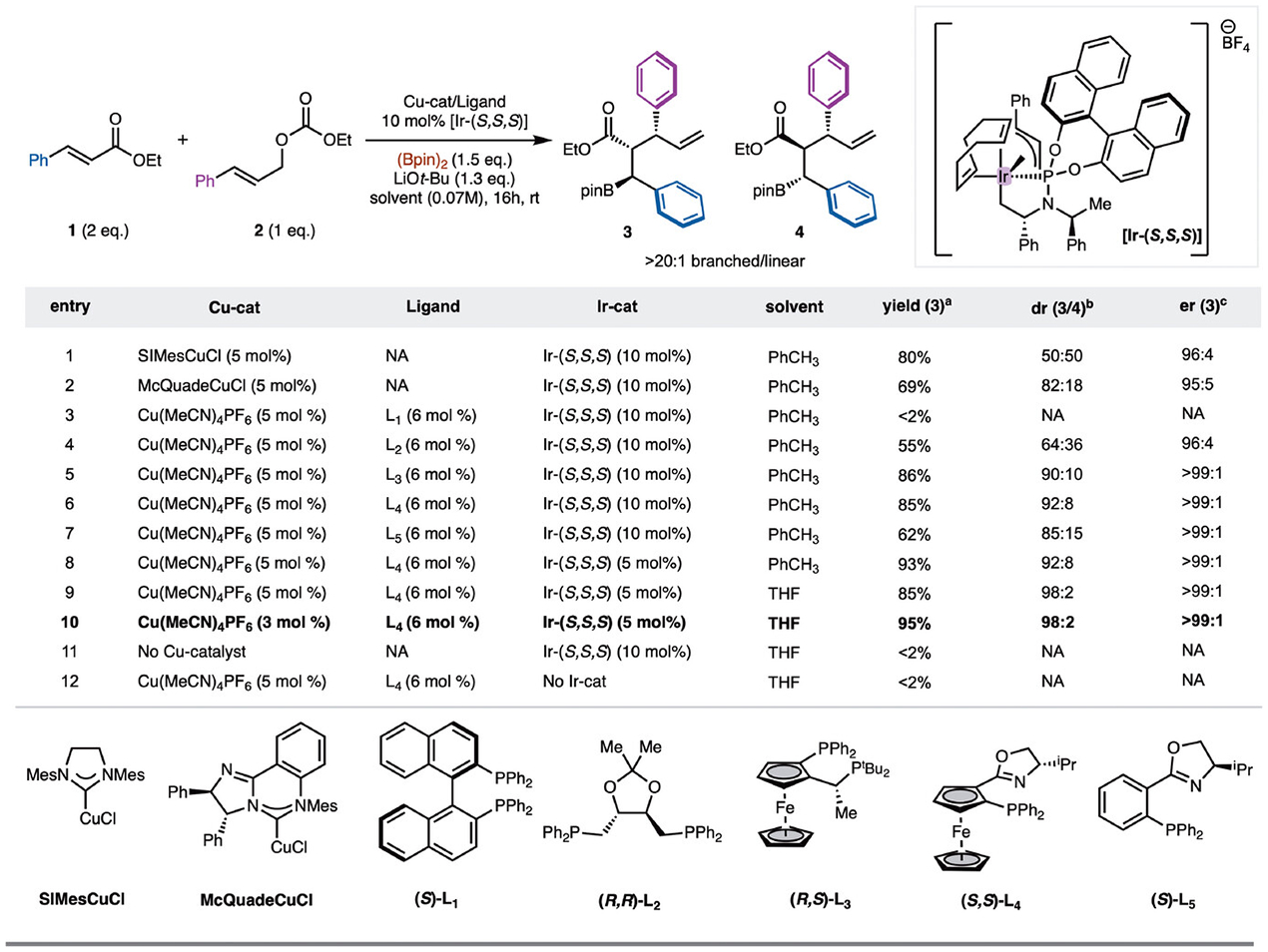 Figure 2
Figure 2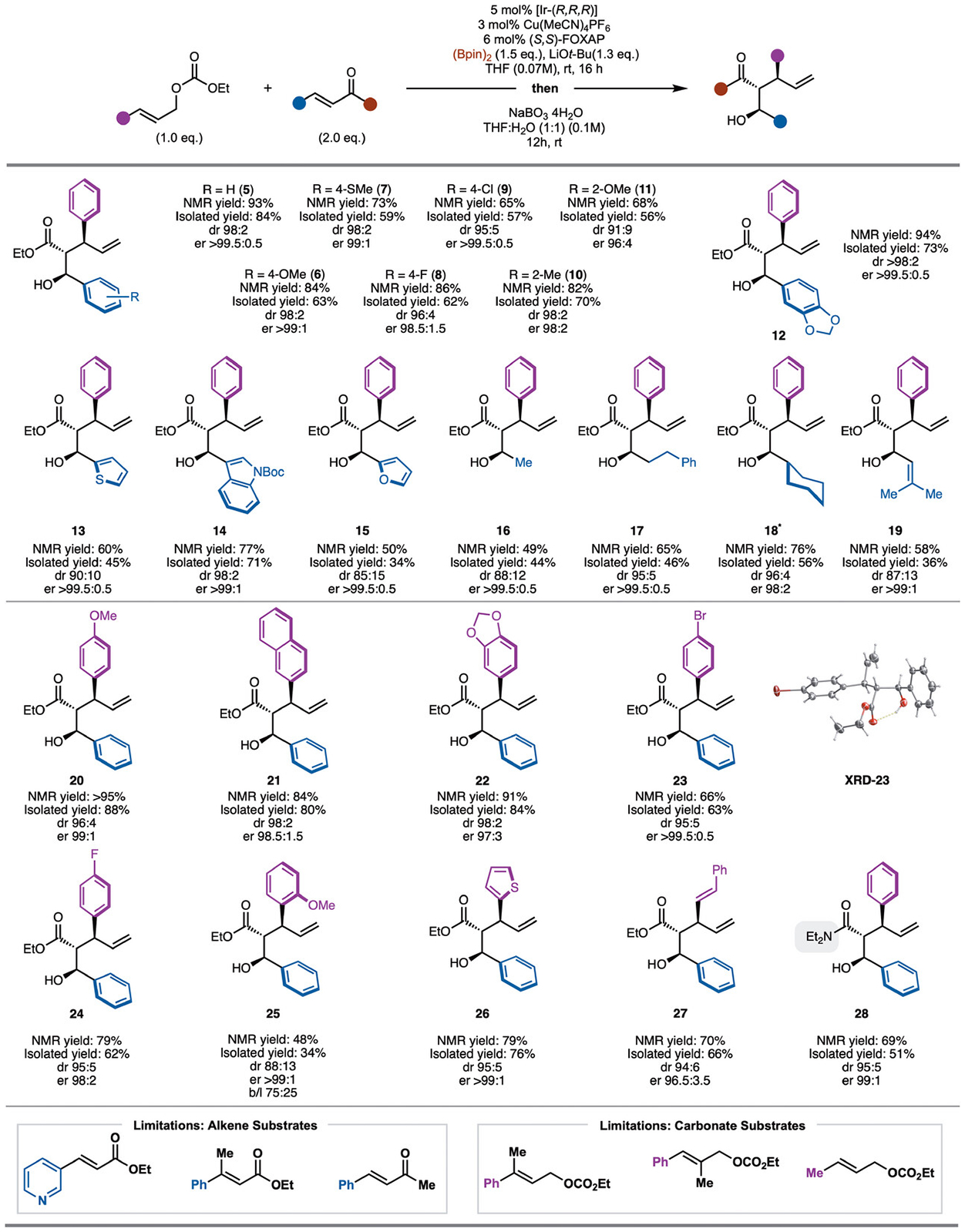 Figure 3
Figure 3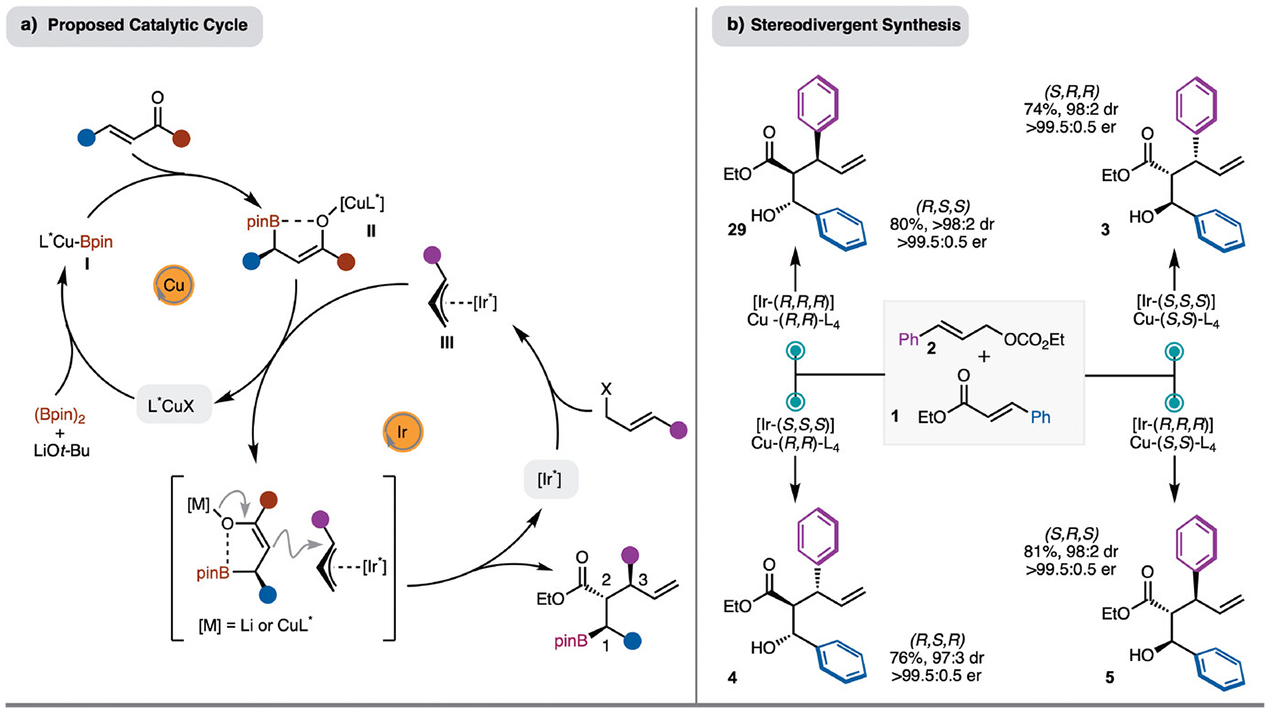 Figure 4
Figure 4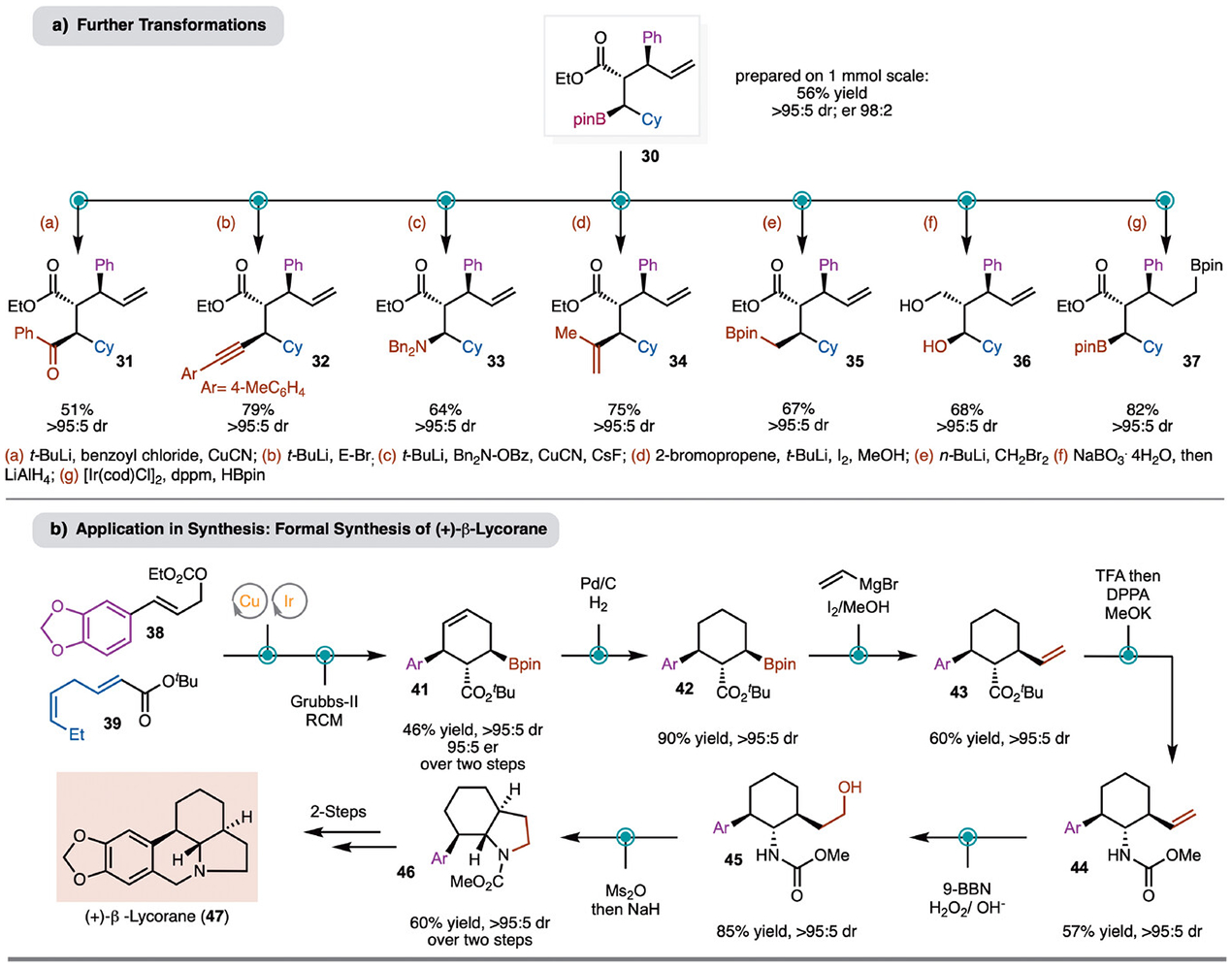 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChemical synthesis and alkaloids · Organoboron and organosilicon chemistry · Advanced Synthetic Organic Chemistry
Alkene difunctionalizations are a critical class of reaction for chemical synthesis as rapid molecular complexity can be generated from simple reagents.^[1–3]^ Of the many methods that have been reported, alkene carboboration reactions are of high significance.^[4,5]^ The utility of these reactions largely stems from the generation of a new C–C bond and a synthetically useful C–B bond in one step.^[6]^ Of the methods that have been developed, Cu-catalyzed,^[7–10]^ Cu/Pd-catalyzed carboboration,^[11–19]^ and Ni-catalyzed^[20–24]^ processes have been the most widely investigated (Scheme 1a). Analysis of these methods reveal that effective protocols have been developed for generation of two stereogenic centers from bond formation at the positions of the alkene (Scheme 1a). Recently, You reported a method for generation of two stereogenic centers by reaction of styrenes with Ir-π-allyl complexes (Scheme 1a).^[25]^ Methods that allow for control of three stereogenic centers and use secondary Csp3 electrophiles that allow for stereogenic center formation are not known. Synthesis of molecules with three or more contiguous stereogenic centers is challenging and often requires multistep approaches. Development of methods that can precisely control stereochemistry with three or more contiguous stereogenic centers would be of high value to streamline synthesis (Scheme 1b).
To address this challenge, we elected to combine a Cu-catalyzed alkene borylation with an Ir-catalyzed allylation reaction (Scheme 1c). In recent years, Ir-catalyzed allylation has been extensively developed for the enantioselective combination of either linear or branched allylic alcohols with a variety of nucleophiles to deliver the branched products.^[26–31]^ In particular, (cooperative) catalytic methods with prochiral and chiral nucleophiles have been advanced.^[32–37]^ There is substantial literature precedent for Ir-catalyzed allylation with enolate nucleophiles to generated two stereogenic centers.^[38–45]^ However, forming three contiguous stereogenic centers with control of enantioselectivity and diastereoselectivity are rare and limited to cyclic systems, and thus heavily favored to achieve anti-selectivity, or acyclic systems with sterically biased substituents, which limits scope.^[46,47]^
We endeavored to develop the Cu/Ir-borylallyation of α,β-unsaturated esters (Scheme 1c). This is important because the products would contain three different functional groups – C–B bond, alkene, and ester – that are all easily amenable to functionalization. This feature has been exploited in the efficient formal synthesis of the complex natural product Lycorane. Finally, a key goal is to achieve the synthesis of multiple stereoisomers by changing the enantiomer of chiral ligands of either Ir- and/or Cu-catalysts.
Initial studies focused on reaction of ethyl cinnamate (1) with linear allylic alcohol derivative 2 promoted by Cu- and Ir-catalysts (Scheme 2). Reactivity was probed with NHC-Cu-complexes due to their demonstrated high reactivity in alkene borylation reactions.^[48–50]^ Pleasingly, good reactivity was observed with SIMesCuCl and [Ir-(S,S,S)]^[51]^ (Scheme 2, entry 1). Branched allylic carbonates were also evaluated, however, low yields were observed (see Supporting Information for details). Due to the achiral nature of the Cu-complexes a 1:1 mixture of diastereomers 3 and 4 was observed; however, the chiral Ir-complex allowed for high facial selectivity (96:4 er for formation of 3). To achieve a diastereoselective reaction, chiral NHC–Cu complexes were explored. While moderate success was found with McQuadeCuCl (Scheme 2, entry 2),^[52]^ challenges with modification of chiral NHC complexes prompted the exploration of commercially available chiral bisphosphine ligands with Cu-complexes. While several chiral bisphosphines were screened, an initial hit was observed with (R,R)-DIOP (Scheme 2, entry 4). Notably, the use of ferrocene derived JosiPhos type ligand L_3_ allowed for synthesis of 3/4 90:10 dr and >99:1 er (Scheme 1c, entry 5).^[53,54]^ Continued evaluation led to the finding that ferrocene derived (S,S)-Foxap L_4_ led to formation of 3 in 92:8 dr and >99:1 er (Scheme 2, entry 6).^[55–57]^ It is worth nothing that the diasteromeric (S,R)-Foxap ligand led to lower levels of yield and diastereoselectivity (See Supporting Information for details). Optimization of this result with evaluation of solvent and catalyst loading led to the formation of 3 in 98:2 dr and >99:1 er (Scheme 2, entry 10). Further control experiments were carried out to demonstrate that Cu- and Ir-catalysts are both necessary for the reaction to proceed. (Scheme 2, entries 11 and 12).
With an optimized set of conditions in hand, the scope of the process was explored (Scheme 3). It should be noted that the products were oxidized to facilitate purification and enantiomeric ratio determination by HPLC with a chiral column. Initially, investigation of the α,β-unsaturated ester unit was conducted. Modification of the cinnamate ester revealed that substitution with electron-releasing (products 6, 7, and 11), electron-withdrawing (products 8 and 9), and sterically demanding groups (products 10 and 11) were tolerated. However, for the 2-OMe aryl-based substrate, slightly lower levels of diastereoselectivity were observed likely due to steric bulk of the –OMe group. In addition, various heteroaryl groups did not impede the reaction such as thiophene (product 13), indole (product 14), and furan (product 15). In the case of alkyl groups, reaction with ethyl crotonate worked well, but in slightly reduced dr (product 16). With larger alkyl groups such as n-alkyl (product 17), or cyclohexyl (product 18) high levels of dr were observed. Finally, α,β,γ, and δ-unsaturated esters can be used to provide access to 1,6-dienes (product 19).
With respect to the allylic alcohol derivatives, aryl groups with electron-donating (products 20 and 22) and withdrawing (products 23 and 24) function smoothly with high diastereoselectivity and enantioselectivity. For the 4-Br aryl substrate a crystal structure was obtained to confirm the absolute stereochemistry of the products using (S,S)-Foxap and [Ir-(R,R,R)]. Sterically demanding substituents worked, but in reduced yield (product 25). In this case, the linear product was observed likely due to steric pressure incurred during addition to the internal position of the Ir-π-allyl complex. Finally, tolerance to thiophene (product 26) and use of dienyl allylic carbonate (product 27) was shown. In one example, it was demonstrated that the ester is not required as an α,β-unsaturated amide worked well (product 28).
A proposed catalytic cycle for the process is presented in Scheme 4a. Based on literature precedence, generation of L_n_CuBpin (I) and borylcupration of the α,β-unsaturated ester likely leads to formation of II. It is probable that the enolate coordinates with the Bpin unit to rigidify the structure (II).^[49–50]^ At the same time, Ir-π-allyl complex III is formed by reaction of [Ir*] and the allylic carbonate. Addition of enolate (II) occurs from the face opposite the β-substituent at the internal position of the Ir-π-allyl complex III to generate the observed product. Based on the proposed catalytic cycle (Scheme 4a), control of stereochemistry at C1 is dictated by the enantiomer of chiral Cu-complex, the stereochemistry at C2 is based on substrate control, and finally, the stereochemistry at C3 is established by the enantiomer of chiral Ir-catalyst. Therefore, of the possible eight stereoisomers that could be formed, four should be accessible through various combination of the chiral catalysts. This is demonstrated in Scheme 4b. With either [Ir-(R,R,R)] or [Ir-(S,S,S)] and either [Cu-(R,R)-L_4_] or [Cu-(S,S)-L_4_] the four stereoisomers 3–5, 29 can be prepared in high yield, diastereoselectivity, and enantioselectivity. It does not appear that a significant matched or mismatched effect between chiral catalysts was observed (Scheme 4b).
A scale up synthesis was conducted on a 1.0 mmol scale between cyclohexyl substituted α,β-unsaturated ester and allylic carbonate 2 to achieve the allyl-borylated product 30 in 56% yield, >95:5 dr, and >98:2 er (Scheme 5a). Later, further transformation of the products was carried out (Scheme 5a). Based on recent developments from the Morken group, stereospecific Cu-catalyzed benzoylation and alkynylation can be carried out (products 31 and 32) with high yields and no erosion of diastereoselectivity.^[58]^ In addition to oxidation of the C–B bond (see Scheme 2), Cu-catalyzed amination is also effective (product 33). Additional stereospecific transformation of the C–B bond by Evans–Zweifel alkenylation (product 34)^[59–60]^ and Matteson homologation with dibromomethane worked smoothly (product 35).^[61]^ Finally, reduction of the ester leads to chiral 1,3-diol 36 and Ir-catalyzed hydroboration of the alkene allows for synthesis of 1,5-diborylated compound 37.^[62]^
It is important to note that due to the presence of three distinct functional groups within the direct products of borylallylation, independent and facile modification can be carried out to arrive at a diverse range of structures. In this regard, a formal synthesis of (+)-(β)-Lycorane is presented here utilizing this methodology as a key step (Scheme 5b). The synthesis commenced with Cu/Ir-catalyzed allylboration of allyl carbonate 38 and α,β-unsaturated ester 39 to obtain the corresponding product in 65%–69% NMR yield and >95:5 dr (er 95:5 after oxidation of C–Bpin; details in Supporting Information). This product was directly subjected to a ring-closing metathesis (RCM) to achieve cyclohexene derived product (41) in 46% overall yield and >95:5 dr. Subsequent, hydrogenation allowed for synthesis of cyclohexane 42 (>95% yield; >95:5 dr). Zweifel alkynylation of the secondary boronate ester led to installation of the vinyl group in product 43. Treatment with TFA resulted in formation of the corresponding carboxylic acid, which was subjected to DPPA to induce a Curtius rearrangement to provide carbamate 44. Hydroboration–oxidation of 44 resulted in formation of the corresponding alcohol 45, which upon treatment with Ms_2_O then NaH led to formation of carbamate 46. This intermediate was previously reported by Yang et al. in their total synthesis of (+)-(β)-Lycorane (47).^[63]^
In conclusion, we have developed a stereodivergent process for the Cu/Ir-catalyzed borylallylation of unsaturated esters. The method allows for the stereodivergent synthesis of three contiguous stereogenic centers. In addition, the products are of high utility due to the presence of three distinct functional groups, which has allowed for the concise enantioselective synthesis of the complex natural product Lycorane. Further advancement of the Cu-catalyzed borylation reactions in the area of cooperative catalysis are underway.
Supplementary Material
Supplemental Material
Data
Additional supporting information can be found online in the Supporting Information section
The authors have cited additional references within the Supporting Information.^[30,31]^
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Li Y, Wu D, Cheng H, Yin G, Angew. Chem. Int. Ed 2020, 59, 7990–8003.
- 2Wickham LM, Giri R, Acc. Chem. Res 2021, 54, 3415–3437, 10.1021/acs.accounts.1c 00329.34383469 PMC 8860250 · doi ↗ · pubmed ↗
- 3Zhi S, Ma X, Zhang W, Molecules 2024, 29, 2559, 10.3390/molecules 29112559.38893437 PMC 11173560 · doi ↗ · pubmed ↗
- 4Liu Z, Gao Y, Zeng T, Engle KM, Isr. J. Chem 2020, 60, 219–229, 10.1002/ijch.201900087.33785969 PMC 8006804 · doi ↗ · pubmed ↗
- 5Dorn SK, Brown MK, ACS Catal. 2022, 12, 2058–2063, 10.1021/acscatal.1c 05696.36212545 PMC 9540610 · doi ↗ · pubmed ↗
- 6Sandford C, Aggarwal VK, Chem. Commun 2017, 53 5481–5494, 10.1039/C 7CC 01254 C. · doi ↗
- 7Yoshida H, Kageyuki I, Takaki K, Org. Lett 2013, 15, 4, 952–955, 10.1021/ol 4001526. · doi ↗
- 8Smith KB, Huang Y, Brown MK, Angew. Chem. Int. Ed 2018, 57, 6146–6149, 10.1002/anie.201801139. · doi ↗