Loss of LafB activity reverses daptomycin resistance in E. faecium
Suelen Scarpa de Mello, Bailey Schultz, Byoungsook Goh, Alexandra Grote, Terrance Shea, Jacob Muscato, Sungwhan F. Oh, Abigail L. Manson, Ashlee M. Earl, Suzanne Walker, Ilana L. B. C. Camargo, Michael S. Gilmore

TL;DR
Disabling the lafB gene in E. faecium makes daptomycin-resistant bacteria more sensitive to the antibiotic, offering a new way to combat resistance.
Contribution
Loss of lafB function prevents or reverses daptomycin resistance in E. faecium, suggesting LafB inhibition as a novel therapeutic strategy.
Findings
Daptomycin-resistant mutants do not emerge in vitro when lafB is nonfunctional.
Clinical daptomycin-resistant strains with lafB mutations become more susceptible to daptomycin.
LafB is critical for known daptomycin resistance mechanisms and has phenotypic dominance over other resistance mutations.
Abstract
Infections caused by multidrug-resistant enterococci, particularly vancomycin-resistant Enterococcus (VRE), present significant therapeutic challenges. Daptomycin, a last-line treatment for VRE, often loses efficacy due to the emergence of resistance. In this study, we revealed the critical role of the lafB gene as a key determinant of daptomycin susceptibility and resistance in E. faecium. We showed that in the absence of a functional lafB, daptomycin-resistant mutants did not emerge in vitro, and derivatives of clinical daptomycin-resistant strains engineered to lack functional lafB were rendered even more sensitive to daptomycin than wild-type daptomycin-susceptible strains. These findings indicated that functional lafB is critical for key known mechanisms of daptomycin resistance, and mutations in lafB have phenotypic dominance to those that otherwise confer resistance. Therefore,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
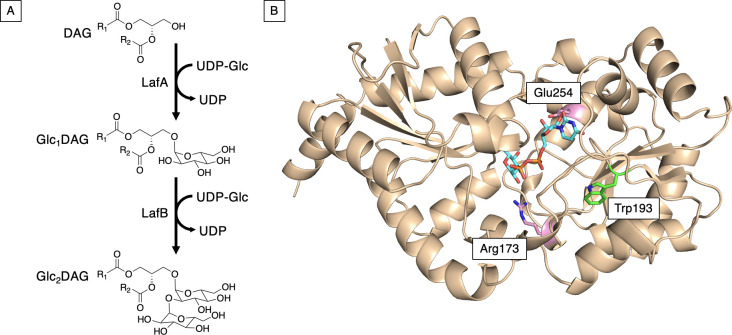 Fig 1
Fig 1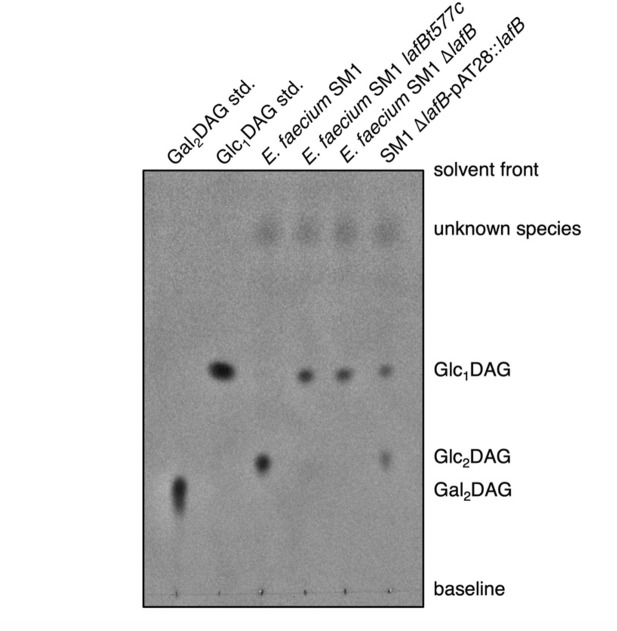 Fig 2
Fig 2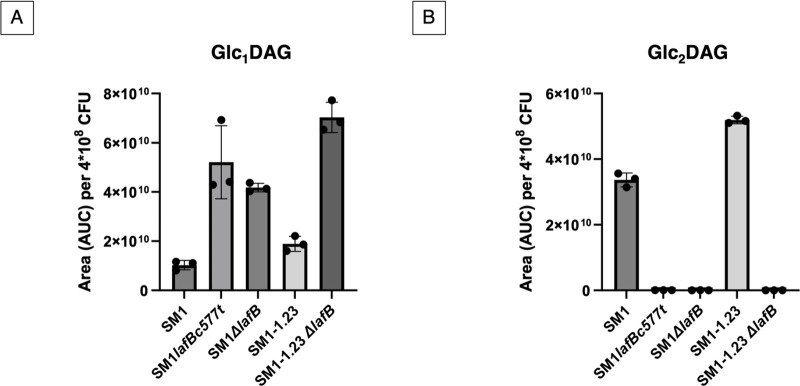 Fig 3
Fig 3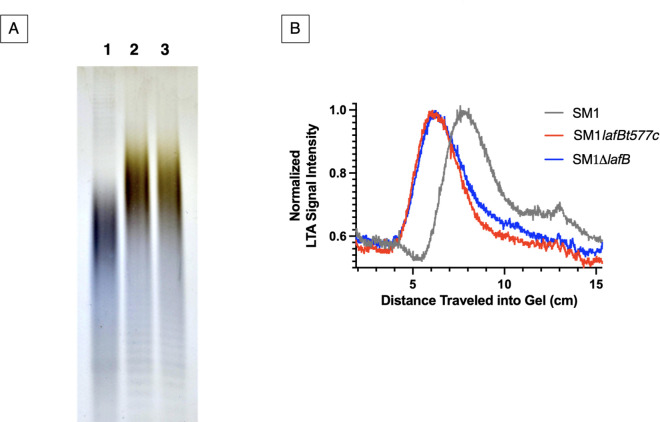 Fig 4
Fig 4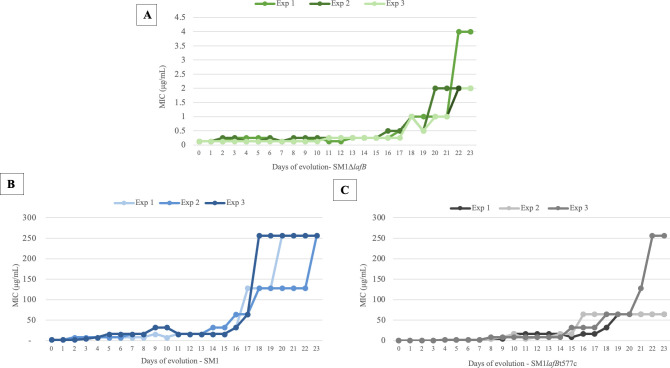 Fig 5
Fig 5| Strains | MIC (mg/mL) | |||||
|---|---|---|---|---|---|---|
| DAP | Colistin | Polymyxin B | Bacitracin | Gentamicin | Vancomycin | |
| 2 | 512 | 512 | 16 | 8 | 1 | |
|
|
|
|
|
|
| |
|
|
|
|
|
|
| |
| SM1 |
| NT | NT | NT | NT | NT |
| Parental strain | DAP | Annotation | Gene identification ( | Amino acid change |
|---|---|---|---|---|
| MIC range (mg/mL) | ||||
| SM1lafBt577c | 32–256 | cardiolipin synthase | HMPREF0351_11068 | Ala-20-Asp |
| SM1 | 256 | His-215-Arg | ||
| Asn-13-Ser | ||||
| SM1 | 128-256 | DAK2 protein | HMPREF0351_10092 | Ala-432-Asp |
| 256 | Gly-94-Glu | |||
| SM1lafBt577c | 64 | YycG two-component system sensor histidine kinase | HMPREF0351_12361 | Trp-609-STP |
| SM1 | 128–256 | Asp-272-Gly | ||
| 64 | Gly-564-Ser |
| Isolate | DAP MIC (mg/mL) | Annotation | Gene identification ( | Amino acid change |
|---|---|---|---|---|
| SM1ΔlafB 1-23 | 4 | alpha/beta hydrolase | HMPREF0351_11083 | Gly-123-Glu |
| HD protein | HMPREF0351_11978 | Leu-673-Leu | ||
| LTA synthase- ltaS | HMPREF0351_11744 | Tyr-427-Ser | ||
| SM1ΔlafB 2-23 | 2 | alpha/beta hydrolase | HMPREF0351_11083 | Asp-191-Tyr |
| UDP-N-acetylglucosamine 1-carboxyvinyltransferase 2 | HMPREF0351_12044 | Cys-263-Arg | ||
| LTA synthase- ltaS | HMPREF0351_11744 | Tyr-427-Ser | ||
| SM1ΔlafB 3-23 | 1 | alpha/beta hydrolase | HMPREF0351_11083 | Gly-32-Val |
| phage tail tape measure protein | HMPREF0351_10868 | Val-815-Glu |
| Strains | Mutations | DAP MIC (mg/mL) |
|---|---|---|
| Clinical isolates | ||
| HOU503 | 1–2 | |
| HOU503Δ | Lacking | 0.06 |
| R496 | 16 | |
| R496Δ | Lacking | 0.125 |
| R496 | Knockout restored via allelic replacement | 16 |
| R497 | 32 | |
| R497Δ | Lacking | 0.125 |
| R497 | Knockout restored via allelic replacement | 32 |
| DAP evolved strains | ||
| SM1-1.23 | DAP-resistant mutant | 256 |
| SM1-1.23– Δ | DAP-resistant mutant lacking | 0.125 |
| SM1-1.23Δ | Empty plasmid | 0.125 |
| SM1-1.23-pAT28: | Complementation of | 32 |
| Controls | ||
| SM1 | WT | 2 |
| SM1lafBt577c | Clinical hypersusceptible strain | 0.125 |
| SM1 | Lacking | 0.125 |
| SM1 | Empty plasmid | 0.125 |
| SM1 | Complementation of | 2 |
| MIC (mg/mL) | ||
|---|---|---|
| Strains | DAP (mg/mL) | DAP + oleic acid (mg/mL) |
| SM1 | 2 | 4 |
| SM1lafBt577c | 0.125 | 0.125 |
| SM1 | 0.125 | 0.125 |
| SM1 | 2 | 4 |
| SM1 | 0.125 | 0.125 |
| SM1-1.23 | 256 | 512 |
| SM1-1.23- Δ | 0.125 | 0.125 |
| SM1-1.23- Δ | 32 | 64 |
| SM1-1.23- Δ | 0.125 | 0.125 |
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
- —National Institutes of Healthhttp://dx.doi.org/10.13039/100000002
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAntimicrobial Resistance in Staphylococcus · Probiotics and Fermented Foods · Bacterial Genetics and Biotechnology
INTRODUCTION
Enterococci are important causes of multidrug-resistant hospital infection (1, 2), and rates of vancomycin-resistant Enterococcus (VRE) can be as high as 70% for Enterococcus faecium in some hospitals (3). Daptomycin (DAP) is a cyclic lipopeptide antibiotic that is currently understood to exert its antibacterial effects through two mechanisms: it inhibits cell wall synthesis by binding to lipid II, causing lipid clustering; and it induces membrane depolarization independently of cell wall components (4). Its activity is enhanced by interacting with phosphatidylglycerol (PG), prevalent in gram-positive bacteria. In the presence of calcium ions, DAP forms a complex with lipid II and PG, disrupting cell wall synthesis and leading to membrane depolarization. This action can trigger autolysis in susceptible bacteria, with any delayed membrane permeability being a secondary effect related to autolysis rather than direct pore formation (4). DAP is a critical last-line drug for VRE, but its efficacy is compromised by the emergence of resistance during therapy (5–8). Mutations in genes encoding the LiaFSR cell membrane stress response system or cardiolipin synthase involved in phospholipid metabolism are common routes to DAP resistance in E. faecium (9–14).
Lipoteichoic acid (LTA) is an important component of the gram-positive cell membrane. In Enterococcus faecalis, the enterococcal species in which LTA is most well studied, defects in LTA biosynthesis lead to reduced biofilm formation, virulence, and pathogenesis (15–17). Because of its unique structure, enterococcal LTA has also been considered as a potential vaccine candidate (18, 19).
In a previous study, we identified and characterized an unusual DAP-hypersusceptible E. faecium isolate from a suspected urinary tract infection (20). This new and unexpected vulnerability to DAP was traced to a mutation in the gene lafB, which encodes a glycosyltransferase involved in LTA biosynthesis (20). When exposed to DAP in vitro, this DAP-hypersusceptible isolate consistently reverted the initial missense mutation back to the wild-type (WT) sequence, restoring typical WT levels of DAP susceptibility before acquiring any additional mutations that confer resistance. This raised the possibility that the mutation in lafB may be phenotypically dominant to the effects of other mutations known to confer DAP resistance (20).
To determine whether the naturally occurring missense mutation in lafB (i) resulted in LafB loss of function, (ii) was phenotypically dominant to other known forms of DAP resistance, and (iii) could potentially lead to alternate mechanisms of DAP resistance that may be lafB-independent, we generated and examined the phenotypes of non-reverting deletion derivatives of lafB in both WT and DAP-resistant E. faecium backgrounds.
RESULTS
Loss of lafB function leads to DAP hypersusceptibility
To test whether the hypersusceptible phenotype we initially associated with a missense mutation in lafB (20) is caused by loss of LafB function, we deleted 116 codons out of a total 349 of the lafB reading frame by allelic replacement (21) to generate a non-revertible mutant of strain E. faecium SM1. Deletion of lafB recapitulated the DAP hypersusceptibility seen in the naturally occurring clinical isolate (20). Moreover, providing the lafB reading frame in trans restored the WT level of susceptibility, proving that the phenotype is ascribable solely to a loss of LafB function (Table 1).
We next examined whether the DAP-hypersusceptible lafB loss of function mutant possessed altered susceptibilities to other antimicrobials. To test this, we examined susceptibility to other antibiotics that target cell membrane components/cell envelope precursors (bacitracin and vancomycin), as well as cationic antibiotics including gentamicin, colistin, and polymyxin B, where changes to the highly polyanionic LTA polymers resulting from lafB loss of function may have an impact. In addition to conferring hypersusceptibility to DAP, mutation in lafB also rendered E. faecium about 10-fold more susceptible to polymyxin B and bacitracin, about fourfold more susceptible to colistin, and about twofold more susceptible to gentamicin and vancomycin (Table 1).
Modification of lafB leads to the production of an aberrant LTA
To assess the functional impact of lafB mutation in E. faecium, we examined a DAP-hypersusceptible clinical isolate harboring a W193R substitution in the glycosyltransferase LafB. We previously showed that this mutation decreases solubility and thermal stability of the protein, consistent with a loss-of-function phenotype (22). This enzyme, homologous to BgsA in E. faecalis, catalyzes the second step in LTA anchor biosynthesis by converting monoglucosyldiacylglycerol (Glc_₁_DAG) to diglucosyldiacylglycerol (Glc_₂_DAG) (15) (Fig. 1A). Structural modeling using AlphaFold predicted that the W193R substitution lies within the nucleotide-binding domain, near the UDP-glucose binding pocket, and likely disrupts the hydrophobic core necessary for proper folding and stability (Fig. 1B).
(A) Proposed pathway for LTA glycolipid biosynthesis in E. faecium. LafA transfers glucose onto diacylglycerol using UDP-glucose (UDP-Glc) as a substrate to make Glc1DAG. LafB then adds a second glucose to make an α-(1, 2)-linked Glc2DAG. (B) AlphaFold model of E. faecium LafB with UDP-glucose (cyan) modeled into the active site using the Chai Discovery-predicted LafB:UDP-Glc complex structure (23–25). Trp193 (green) is the WT residue replaced by an arginine as the result of a point mutation in the missense DAP-hypersusceptible strain. Chai and AlphaFold models of the protein were highly similar (RMSD: ~0.3 Å), and the predicted UDP-Glc binding site matched well with the UDP binding site for the Thermosynechococcus elongatus sucrose phosphate synthase crystal structure (PDBID: 6KIH). This sucrose phosphate synthase structure (26) was identified by the Dali server (27) as the experimental structure with the best structural match to the AlphaFold LafB structure (RMSD: ~2.4 Å). Glu254 and Arg173 (pink) are predicted to play roles in substrate binding based on homology to other similar glycosyltransferases.
To determine the effect of lafB loss of function on E. faecium LTA biosynthesis, total lipids were extracted from the cell membranes of both WT E. faecium SM1 and lafB missense and deletion mutants and analyzed by thin-layer chromatography (TLC) with N-(1-naphthyl) ethylenediamine staining to detect glycolipids (Fig. 2). This revealed an accumulation of the monoglucosylated Glc_1_DAG produced by the product of lafA but no formation of the diglucosylated Glc_2_DAG product in both lafB mutants (SM1lafBc577t and SM1 ΔlafB). The accumulation of Glc_1_DAG, compared to SM1 and SM1-1.23, and the absence of Glc_2_DAG in the lafB mutants (SM1lafBc577t, SM1ΔlafB, and SM1-1.23ΔlafB) were confirmed by mass spectrometry-based lipidomic analysis (Fig. 3).
TLC analysis of glycolipids in total membrane lipid extracts from E. faecium. Glycolipid profiles of E. faecium SM1, SM1lafBT577C, SM1ΔlafB, and SM1 ΔlafB/pAT28::lafB mutants were compared to WT E. faecium SM1. Purified lipid standards included Gal₂DAG [1,2-diacyl-3-O-(α-D-galactosyl(1→6)-β-D-galactosyl)-sn-glycerol] and Glc₁DAG [1,2-diacyl-3-O-(α-D-glucopyranosyl)-sn-glycerol] (Avanti Polar Lipids). Glycolipid species Glc₁DAG, Glc₂DAG, and Gal₂DAG were identified based on migration relative to standards. Additional bands representing unknown lipid species are also indicated. The absence of glycolipid bands in the ΔlafB mutant and their restoration in the complemented strain demonstrates the role of lafB in glycolipid biosynthesis. Data are representative of three independent experiments.
Quantitative comparison of glycolipids Glc1DAG (A) and Glc2DAG (B). The mean of the normalized area per 4 × 108 CFU is shown. Peaks were quantified using extracted ion counts (EICs), and the mass (m/z) values used to generate these plots are provided in the supplementary material (Table S2 and S3). E. faecium SM1- wild-type strain; E. faecium SM1lafBt577c: clinical DAP hypersusceptible strain; E. faecium SM1ΔlafB - wild-type strain lacking lafB; E. faecium SM1-1.23(dak Ala432Asp YycGAsp-272-Gly clsHis215Arg): DAP-R evolved in vitro from wild-type strain; E. faecium SM1-1.23 (dak Ala432Asp YycGAsp-272-Gly clsHis215Arg) ΔlafB– evolved strain lacking lafB.
Since an aberrant polymer of reduced mobility in polyacrylamide gel was observed previously in E. faecalis possessing a mutation in the homolog of lafB (15), we examined whether lafB inactivation in E. faecium led to the formation of a similar polymer. We compared the relative mobility of LTA purified from WT SM1 and the novel polymer produced by lafB mutants in polyacrylamide gels, following lipid tail removal by lipase treatment and visualization with Alcian blue/silver staining (Fig. 4A). As shown in Fig. 4B, loss of LafB functionality, whether due to a point mutation or deletion of a large portion of the reading frame, resulted in production of an aberrant polymer that migrated through polyacrylamide more slowly than the WT LTA polymer, similar to observations for E. faecalis (15). This indicates either a reduced charge-to-mass ratio or an increase in the size of the polymer produced when the typical Glc_2_DAG linker is unavailable.
Altered migration of LTA in lafB mutant strains. (A) Alcian blue/silver-stained LTA isolated from WT E. faecium and lafB mutants, separated by electrophoresis through 20% polyacrylamide. Lane 1: LTA isolated from WT E. faecium SM1; Lane 2: LTA isolated from E. faecium SM1lafBt577c; Lane 3: LTA isolated from E. faecium SM1ΔlafB. The bulk of polymers extracted from lafB mutant lines migrate more slowly than those from the WT. Results shown here are representative of three independent experiments. (B) Densitometric analysis of Fig. 4A quantifies the leftward shift in polymer migration, reflecting altered size, structure, and/or composition compared to WT LTA.
Defects in lafB block mutational pathways to DAP resistance
We next sought to determine whether pathways exist for the emergence of DAP resistance that are independent of a functional lafB. To do this, we exposed strains to stepwise increases in DAP concentration over the course of 23 days (Fig. S2; Table S1). As controls, the original clinical isolate possessing the missense point mutation in lafB, SM1lafBt577c, and its WT revertant SM1 were compared to the non-revertible lafB deletion strain SM1ΔlafB. Following extended stepwise exposure to DAP over 23 days, the lafB-deleted strain SM1ΔlafB never evolved resistance. In contrast, both control strains rapidly evolved mutants ultimately exhibiting over 100-fold increases in resistance to DAP (Fig. 5; Fig. S2; Table S1). Consistent with our prior work (20), the missense SM1lafBt577c loss of function control invariably reverted lafB to the WT sequence as the first step in the evolution of DAP resistance. This was followed by the appearance of a mutation in cardiolipin synthase (cls), a key enzyme in phospholipid biosynthesis (10, 11, 28–30) in one DAP-resistant derivative of SM1lafBt577c. In two out of the three replicates, DAP-resistant mutants derived from WT SM1 developed mutations in cls and in dihydroxyacetone kinase-like gene (dak), which encodes an enzyme that phosphorylates exogenous fatty acids for incorporation into membrane phospholipids (20, 31).
In vitro evolution of DAP resistance in E. faecium strains over 23 days. Graphs show changes in MIC values for E. faecium SM1ΔlafB (A), E. faecium SM1 (B), and E. faecium SM1lafBT577c (C) at each time point. Data represent individual wells in three biological replicates.
Three out of six replicates derived from both SM1 and SM1lafBt577c had mutations in YycG (EF1194, also known as WalK), which is predicted to function as a sensor histidine kinase within the YycF/YycG two-component system. YycG plays a crucial role in cell envelope biosynthesis in Enterococcus and is often mutated in DAP-resistant strains of E. faecium (13, 32–34). These three genes have all been implicated in enterococcal DAP resistance (Table 2). However, none of the mutations leading to high-level DAP resistance observed in WT or revertible lafB strains appeared in SM1ΔlafB over at least 115 generations.
To identify the genetic changes that could be associated with the small decreases in DAP susceptibility observed in SM1ΔlafB, we performed whole genome sequencing of those exhibiting the highest DAP minimum inhibitory concentration (MIC) levels. Derivatives of SM1ΔlafB were obtained exhibiting DAP MICs of 1–4 µg/mL, below levels of DAP resistance which is defined as an MIC greater than or equal to 8 µg/mL (35). Mutations in an alpha/beta hydrolase were identified in all three independently evolved SM1ΔlafB derived lineages (Table 3). A mutation in the LTA synthase (ltaS) gene, a transmembrane protein required to produce the polyglycerol-phosphate chain of LTA using PG as a substrate (36), occurred in two out of the three DAP mutant strains derived from SM1ΔlafB (Table 3). In Staphylococcus aureus, ugtP encodes the glycolipid synthase that catalyzes both glycosylation steps in Glc_2_DAG biosynthesis. In ugtP loss of function mutants, second site mutations in ltaS arise that generate shorter, less abundant LTA polymers presumably compensating for the effect of functional ugtP loss (37).
Deletion of lafB in DAP-resistant mutants restores susceptibility
To prove that lafB loss of function mutation was phenotypically dominant to mutations observed in clinical isolates that render them DAP-resistant, allelic replacement was used to delete lafB in an evolved DAP-resistant strain containing mutations in dak, yycG, and cls (strain SM1-1.23; MIC = 256 µg/mL), as well as in well-characterized DAP-resistant clinical isolates. In evolved DAP-resistant mutant SM1-1.23, deletion of lafB reduced the DAP MIC from 256 µg/mL to hypersusceptible levels of 0.125 µg/mL, comparable to the MIC of the lafB deletion made in the SM1 WT background. Thus, the additional mutations occurring in dak, yycG, and cls had no demonstrable effect in the absence of functional lafB. Moreover, DAP resistance could be partially restored by complementing the lafB mutant in trans with the WT lafB allele (Table 4).
Mutations in the liaFSR operon*,* especially the liaR^W73C^ variant, contribute to DAP resistance in clinical strains by constitutive induction of the cell wall stress response regulon (38). Given the importance of mutations in the liaFSR operon in clinical isolates, we investigated whether lafB is essential for this phenotype as well. Deletion of lafB decreased DAP MIC from 32 to 0.125 µg/mL in strain R496 (liaF ^I142T^ cls ^N13I)^, and from 16 to 0.125 µg/mL in strain R497 (liaS^T120A^ liaR ^W73C^ cls ^insertion110^ ^of MPL^) (Table 4). For the clinical isolate HOU503, which is mutated in both liaR and liaS (LiaR^W73C^ and LiaS^T120A^), deletion of lafB decreased the DAP MIC from 1 to 0.06 µg/mL. These results show that depriving the cell of a functional LafB was phenotypically dominant to mutations in the liaFSR operon and to yvcG, dak, and cls genes, reverting DAP resistance driven by the most known resistance mechanisms (Table 4).
Partial and complete complementation in E. faecium ΔlafB mutants
To validate that the observed phenotypes were specifically due to LafB loss and not other unknown mutations, we first complemented the SM1-23 ΔlafB null mutant with lafB expressed from the pAT28 plasmid as noted above. This construct partially restored DAP susceptibility, increasing the MIC from 0.125 to 32 µg/mL, but did not fully restore to the original SM1-23 MIC level of 256 µg/mL. The partial restoration is consistent with TLC results, which showed partial recovery of Glc_₂_DAG glycolipid production in the plasmid-complemented strain (Fig. 2), presumably stemming from either low-level expression despite the multicopy nature of the plasmid, or lack of potentially coordinate expression with other LTA biosynthetic functions. To achieve full complementation, we used allelic replacement to specifically restore only the lafB gene in strain R496 and R497 null mutant backgrounds. In both cases, this fully restored DAP resistance to MIC levels observed prior to lafB deletion. These results confirm that lafB is essential for Glc_₂_DAG biosynthesis and plays a critical role in modulating DAP susceptibility in both laboratory and clinical E. faecium backgrounds (Table 4).
Exogenous oleic acid supplementation enhances DAP tolerance in WT but not lafB mutant
Previous reports found that E. faecalis can increase tolerance to DAP by incorporating exogenous fatty acids such as oleic acid which is abundant in human serum, into cell membrane lipids (39–41). To determine whether exogenous fatty acids could rescue the DAP hypersusceptible phenotype of lafB strains, we cultured WT and lafB mutant strains in the presence or absence of 20 mg/mL oleic acid. Supplementation of oleic acid to the growth medium (added exclusively in the MIC plate) resulted in a twofold increase in DAP tolerance for both WT and DAP-resistant strains, but no change in the DAP susceptibility was observed for lafB mutants (Table 5).
DISCUSSION
Our previous work (20) raised the possibility that mutation of the lafB glucosyltransferase, and therefore loss of the α-(1→2)-linked second glucose on the diacylglycerol precursor of LTA, may be phenotypically dominant over key known mechanisms of DAP resistance in E. faecium. Here, we unambiguously prove this through several lines of work, showing that (i) non-revertible mutation of lafB prevents the emergence of DAP-resistant mutants under conditions where WT cells readily develop DAP resistance, (ii) deletion of lafB from either clinical or laboratory-derived DAP-resistant E. faecium strains carrying known resistance determinants reverses the resistance phenotype and renders the strains hypersusceptible, and (iii) in all cases tested, providing LafB functionality by trans complementation or allelic replacement partially or completely restores DAP resistance. In addition, the loss of LafB activity and associated alteration in LTA structure are accompanied by increased susceptibility to other cell wall-active and cationic antibiotics.
Loss of LafB activity led to a predicted absence of Glc_₂_DAG pools and accumulation of Glc_₁_DAG, consistent with disruption of the LTA glycolipid biosynthesis pathway, as previously observed in E. faecalis (15, 42). As in E. faecalis, mutants instead produced an altered form of LTA with reduced electrophoretic mobility. In S. aureus lacking a functional copy of ugtP, which catalyzes addition of both glucose units to DAG, analogously aberrant polymers are built on PG anchors and contain more glycerol-phosphate repeat units than WT LTA polymers (43). Whether the aberrant molecules observed in E. faecium lafB mutants or E. faecalis bgsA mutants (15) arise from a similar mechanism, potentially involving polymer assembly on the accumulating monoglycosylated DAG stem which is not available in ugt mutants of S. aureus remains unknown. Regardless, production of this altered polymer does not compensate for functional membrane changes that lead to DAP hypersusceptibility, even in strains that are otherwise highly DAP-resistant.
Loss of Glc_2_DAG production in E. faecalis has been linked to altered virulence in diseases such as endocarditis and peritonitis (17, 42, 44). By fundamentally changing the organization of the cell membrane, these disruptions likely cause pleiotropic effects, and the precise mechanisms by which defects in LTA biosynthesis contribute to loss of virulence remain unclear.
In sum, we demonstrate that lafB deletion prevents the emergence of DAP resistance, fully suppresses established DAP resistance phenotypes, and limits the potential effect of incorporation of exogenous fatty acids into membrane lipids on DAP susceptibility. These results highlight LafB as a promising therapeutic target for both blocking the emergence of DAP resistance during treatment and for restoring DAP susceptibility in resistant E. faecium infections. Additionally, this work demonstrates the pivotal role of LTA biosynthesis in modulating DAP susceptibility, offering new insights into the mechanisms underlying antibiotic resistance in this important pathogen.
MATERIALS AND METHODS
Genetic manipulation of E. faecium
A deletion mutant of E. faecium SM1 was created by allelic replacement of glycosyltransferase lafB gene. DNA segments upstream (741 bp) and downstream (738 bp) of the gene to be deleted were amplified by PCR with the following primers: F1 up-forward, R1 up-reverse; F2 down-forward and R2 down-reverse (Table S2). The PCR products from both regions were fused to generate a 1.479 bp PCR product lacking nucleotides 655–1004 of the lafB gene, which was then further digested using BamHI and PstI and ligated with BamHI/PstI-digested pLT06 (21) by using T4 DNA ligase (NEB). The ligation product was transformed into Escherichia coli EC1000 for propagation and sequence verification. The confirmed deletion construct was then introduced to E. faecium SM1 strain by electroporation (45). The SM1ΔlafB strain was obtained through temperature shifts and p-chlorophenylalanine counterselection, as previously described (21). Colonies were screened for lafB deletion by PCR using primers LafB_Int_F and LafB_Int_R (Table S2). The desired mutant was confirmed by Sanger sequencing. The strains used in this study are listed in Table S5.
To selectively repair the lafB deletion in the process of excluding other factors from contributing to the phenotypes observed, the full-length lafB gene along with its native promoter region was reintroduced into the lafB deleted strains of clinical isolates R496 and R497 by allelic replacement using the same methodology described above. The lafB gene was amplified by PCR using primers A and B (Table S2), generating a product that was cloned into the pLT06 vector digested with BamHI and PstI, followed by ligation and transformation into E. coli EC1000 for propagation and sequence verification. The verified construct was then electroporated into strains R496ΔlafB and R497ΔlafB. Single and double crossover recombinants were selected by the same temperature shifts and p-chlorophenylalanine counterselection procedure as above, and the presence of the restored lafB gene was confirmed by PCR and Sanger sequencing.
Antimicrobial susceptibility testing
E. faecium strains were grown at 37°C in brain heart infusion (BHI) for all experimental conditions. Broth microdilution was employed to determine the MIC of DAP, polymyxin B, colistin, vancomycin, gentamycin, and bacitracin according to the Clinical and Laboratory Standards Institute guidelines, essentially as described (35). For assessing DAP susceptibility, cation-adjusted Mueller-Hinton broth was used and supplemented with 50 mg/L Ca^2+^ as recommended (35). In some assays, oleic acid (obtained from Sigma-Aldrich) was added to the broth to a final concentration of 20 µg/mL. All experiments were conducted in biological triplicates.
Lipid extraction and analysis of glycolipid content by TLC
A modified Bligh-Dyer method (46, 47) was used to extract total lipids from the bacteria. Strains were streaked out from glycerol stocks onto BHI (from Hardy Diagnostics)-agar plates and grown at 30°C overnight. For each strain, 3 mL of BHI was inoculated with a single colony, and cultures were grown overnight at 30°C with aeration via shaking. Subcultures were then started with a 1:100 dilution of overnight culture into 50 mL fresh BHI media, and these sub-cultures were grown at 37°C, with aeration via shaking, until reaching an OD_600_ value of ~1.0–1.2. Sub-cultures were normalized to an OD_600_ value of 1.00, and then cells from 50 mL of the normalized sub-culture were pelleted by centrifugation at 10,000 × g for 10 min. Supernatant was removed, and the pellets were resuspended in 50 mL of 150 mM NaCl. Cells were then pelleted again by centrifugation at 10,000 × g for 10 min. Supernatant was discarded, and cell pellets were snap-frozen in liquid nitrogen and stored at −80°C for future use.
For lipid extraction, cell pellets were thawed on ice and then resuspended in 1 mL of 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) (pH 6.5) with 150 mM NaCl. More of the same buffer was added to each sample to reach a total volume of 1.5 mL. This resuspension was then added to a glass tube containing 3.75 mL methanol and 1.875 mL chloroform. Samples were vortexed vigorously ~5 times each over the course of 10 min. Cell debris was pelleted by centrifugation at 2,000 × g for 10 min in a swinging bucket rotor. Supernatant was transferred to a new glass tube, to which 1.875 mL chloroform and 1.875 mL Milli-Q water was then added, giving a 1:1:0.9 final ratio of methanol:chloroform:water. Samples were again vortexed vigorously ~5 times each over the course of 10 min before centrifugation at 2,000 × g for 10 min in a swinging bucket rotor to separate the phases. The lower (organic) phase was carefully removed and transferred to a scintillation vial, and solvent was removed using a nitrogen dryer. Samples were then resuspended in 1 mL of chloroform and transferred to smaller glass vials for storage at −20°C.
For TLC analysis, solvent was fully removed from samples using a nitrogen dryer, and then samples were resuspended in 50 µL chloroform. Approximately 5 µL of each of the two standards (Gal_2_DAG: 1,2-diacyl-3-O-(α-d-galactosyl1-6)-β-d-galactosyl-sn-glycerol Avanti Polar Lipids 840524P, Glc_1_DAG: 1,2-diacyl-3-O-(α-D-glucopyranosyl)-sn-glycerol Avanti Polar Lipids 840522P) and ~15 µL of each sample was spotted on the baseline of a Silica gel 60 TLC plate. Once spots were fully dry, the plate was exposed to the running solvent (9:2 chloroform: methanol) in a pre-equilibrated beaker. When the solvent front neared the top of the plate, the plate was removed from the chamber and allowed to dry completely. The plate was then quickly dipped into the stain: 6.5 mM N-(1-naphthyl) ethylenediamine dihydrochloride (Sigma-Aldrich) in 97% methanol/3% concentrated sulfuric acid. Finally, the plate was placed onto a 100°C hot plate until spots appeared and then imaged using both a Nikon D3400 DSLR camera with an AF Micro-Nikkor 60 mm f/2.8D lens and an Azure Biosystems c150 imaging system. All experiments were conducted in biological triplicates.
Isolation of LTA from E. faecium
This protocol was adapted from previous studies (37, 48–51). Strains were streaked out from glycerol stocks onto BHI agar plates and grown at 30°C overnight. For each strain, 10 mL of BHI media was inoculated with a single colony, and cultures were grown for 8 hours at 37°C with aeration via shaking. Large cultures were then started with a 1:200 dilution of the 8-hour culture into 1.0 L fresh BHI media, and these large cultures were grown at 30°C, with aeration via shaking, overnight (~16 hours). Cells were harvested by centrifugation at 5,000 × g for 20 min at 4°C. Supernatant was removed, and pellets were resuspended in phosphate-buffered saline (pH 7.4) to a total volume of ~48 mL. To the resuspension was added benzonase nuclease, MgCl_2_, lysozyme, and mutanolysin (Sigma-Aldrich M9901) to final concentrations of 10 U/mL, 5 mM, 1 mg/mL, and 10 U/mL in 50 mL total. Samples were tumbled end-over-end at 37°C for 1 hour, and then cells were lysed on an Avestin EmulsiFlex-C3 cell disruptor by passaging through ~8 times at 15,000 psi. Samples were then subjected to ultracentrifugation (140,000 × g for 45 min. at 4°C), and the supernatant was discarded. A Dounce tissue grinder (Wheaton) was then used to resuspend the membrane/cell debris material in 6 mL 50 mM sodium citrate (pH 4.7). More of the same buffer was then added to reach a final volume of 15 mL. At this point, tubes were snap-frozen in liquid nitrogen and stored at −80°C for future use.
For extraction of the LTAs, tubes were thawed, and then 15 mL of n-butanol was added. Samples were vortexed to mix and then tumbled at room temperature for 30 min, vortexing every ~5 min. Insoluble material was pelleted via centrifugation at 13,000 × g for 20 min at 4°C, and both layers of supernatant were moved together to a separate tube. Eight milliliters of the 50 mM sodium citrate (pH 4.7) buffer and 8 mL of n-butanol were added to the insoluble material for further extraction—vortexing followed by 30 min of tumbling at room temperature with more vortexing every ~5 min. These samples, plus the original supernatants in their separate tubes, were subjected to centrifugation at 13,000 × g for 20 min at 4°C. The upper organic layers were removed and discarded, and the lower aqueous layers were removed by pipetting through the insoluble interface and combined. An equal volume of n-butanol was added to the aqueous samples, and samples were vortexed to mix. Samples were then subjected to centrifugation at 13,000 × g for 15 min at 4°C. Upper organic layers were removed and discarded, and the lower aqueous layer was moved to a new tube. Once again, an equal volume of n-butanol was added to the aqueous layers, and the vortexing, centrifugation, and layer removal steps were repeated. 2.5 mL of methanol was added to the final aqueous layer samples (which now are ~12.5 mL), and samples were dried down by rotary evaporation to near-but-not-total dryness. Samples were stored in the glass flasks at −20°C overnight.
To isolate the LTAs, samples were dissolved in 5 mL of 5% isopropanol/95% water and then transferred to conical tubes. Insoluble material was pelleted by centrifugation at 3,200 × g for 10 min at 4°C. Samples were added to a pre-equilibrated (750 µL methanol, then 2 × 750 µL 20 mM sodium citrate, pH 4.7, with 5% isopropanol; all pushed through with air pressure) C18 Bakerbond SPE columns (silica packing material, 100 mg bed weight, 1 mL capacity, 40 µm particle size, 60 pore size) and allowed to flow through by gravity (note: this will take a few hours). For washing, air pressure was used to push liquid through columns that were washed twice with 900 µL of 20 mM sodium citrate, pH 4.7, with 5% isopropanol: then, sequentially, with 20 mM sodium citrate, pH 4.7, 15%/25%/35%/45%/65% isopropanol. Each wash was collected separately. All samples were dried down using a Vacufuge plus (Eppendorf) vacuum concentrator and stored at −20°C overnight.
The washes were resuspended in 100 µL of water each, tested for the presence of LTA by dot immunoblotting (next section), and then the 15%, 25%, 35%, and 45% isopropanol washes were combined based on the immunoblotting results. The combined washes were then snap-frozen in liquid nitrogen and then lyophilized for 2 days. Samples were resuspended in Milli-Q water to a final concentration of 50 mg dried sample per mL of water.
Detection of LTA by dot immunoblotting
For each sample, 2.00 µL was spotted onto polyvinylidene difluoride (PVDF) membrane (pre-treated with methanol, then water, then Trans-Blot Turbo transfer buffer) that had been placed on top of a transfer buffer-soaked piece of extra thick blot/filter paper, which was itself on top of a dry piece of extra thick blot/filter paper and dry paper towels. The PVDF membrane was allowed to dry for ~5 min before sample spotting. After spotting, samples were allowed to soak into the membrane for 45 min, and then, the membrane was placed into a solution of 5% wt/vol milk powder in Tris-buffered saline with Tween 20 (TBST) to be rocked for 1 hour at room temperature. The membrane was then washed thrice with TBST for 5 min each time and then incubated in TBST with a 1:500 dilution of -LTA antibody (Hycult Biotech, mAb clone 55) for 1.5 hours at room temperature. The membrane was again washed thrice with TBST for 5 min each time and then incubated with a 1:1,000 dilution of mouse IgG, horseradish peroxidase (HRP)-linked antibody (Cell Signaling Technology 7076) for 1 hour at room temperature. Finally, the membrane was washed thrice with TBST for 5 min each before imaging by enhanced chemiluminescence with SuperSignal West Pico PLUS substrate (Thermo Scientific) on an Azure c600 gel/blot imaging system.
Polyacrylamide gel electrophoresis analysis of E. faecium LTAs
This protocol was adapted from previous studies (37, 49, 50, 52, 53). Samples were prepared by adding 5.0 L 1.0 M Tris buffer (pH 7.5) to 20.0 L of 50 mg/mL LTA sample (note: the mass in the LTA sample is not purely from LTAs). The pH was then adjusted to ~8.0 via addition of 2.0 L 1.0 M NaOH and 4.0 L Milli-Q water. 1.0 L Resinase-HT (Strem Chemicals) was then added, and the samples were incubated overnight (~16 hours) at 50°C to allow for removal of the lipid tail acyl chains. Next, 1.0 L proteinase K (New England Biolabs) was added to remove protein contaminants, and samples were incubated for a further 5 hours at 50°C. Finally, samples were brought to room temperature, and 4.0 L of 100 mM AEBSF-HCl was added to quench the Proteinase K over a period of ~30 min.
A 20 cm × 20 cm (overall) polyacrylamide gel was cast with a 20% polyacrylamide resolving gel (with 6% of the total acrylamide added being bisacrylamide) and a ~2 cm 3% polyacrylamide stacking gel (with 3.3% of the total acrylamide being bisacrylamide) on top. Both portions of the gel were prepared with Tris pH 8.5 buffer (1.0 M final concentration for the resolving gel; 900 mM final concentration for the stacking gel), 0.1% wt/vol ammonium persulfate and 0.01% vol/vol tetramethylethylenediamine. The gel cassette was placed into the Protean II xi Cell electrophoresis system (Bio-Rad) along with 1.5 L total running buffer (100 mM Tris-tricine, pH 8.2). Samples were mixed with an appropriate amount of 3 loading buffer (50% vol/vol glycerol, 100 mM Tris-tricine pH 8.2, 0.02% wt/vol bromophenol blue), and 15 L per sample was loaded into the gel wells. The gel was run at a constant 35 mA current for ~20–24 hours.
To visualize the LTAs, the gel was first rinsed in Milli-Q water (three 5 min) and then stained in 1.0 mg/mL Alcian blue stain for at least 2 hours. Next, the gel was rinsed repeatedly (five 5 min) in Milli-Q water and then rinsed again for 2 hours and again overnight. Finally, the gel was stained according to the Silver Stain Plus kit instructions (Bio-Rad): 20 min incubation in 40% ethanol (vol/vol)/5% acetic acid (vol/vol) in water, two 10 min water incubations, and then incubation with the freshly mixed silver stain solution until LTAs are observable at the desired intensity. The silver stain was then removed, and the gel was treated with 5% acetic acid (vol/vol) in water for 15 min. Gel images were captured with a Nikon D3400 252 DSLR camera fitted with an AF Micro-Nikkor 60 mm f/2.8D lens. The line densitometry analysis was done in Fiji, and large spikes in intensity from visible and identifiable staining aberrations (dark spots/particles) were removed from the plots for clarity.
DAP evolution assay
Single isolated colony of the E. faecium SM1 ΔlafB was used for each technical triplicate of an in vitro evolution experiment to generate increasingly resistant variants; the protocol was adapted from our previously published work (10). The evolution experiment started with overnight culture of each colony grown in BHI supplemented with 50 mg/L Ca^2+^ in 25 mL at 37 ˚C. After 22- to 24-hour incubation in 50 mL falcon tubes, cultures of each line were examined for visible bacterial growth. From these, 1 mL (10^9^ CFU/mL) of overnight culture was inoculated into tubes containing 25 mL of BHI supplemented with 50 mg/L Ca^2+^ and containing DAP at 1/2× MIC, 1× MIC, 2× MIC, and 4× MIC. All tubes were incubated at 37°C overnight with shaking (100 rpm). The following day, the falcon tube with visible growth at the highest DAP concentration was used as inoculum for the next series of tubes with increasing drug concentration. An aliquot of this growth was stored at −80°C for the remaining assays. This procedure was repeated for 23 days. The same strategy was applied for the hypersusceptible E. faecium SM1lafBt577c and E. faecium SM1 WT strains. The schematic representation is shown in Fig. S3.
Whole genome sequencing and variant analysis
Total genomic DNA was extracted from cultures of each strain using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA).
Illumina sequencing libraries were prepared using the tagmentation-based and PCR-based Illumina DNA Prep kit and custom Integrated DNA Technologies (IDT) 10 bp unique dual indices with a target insert size of 320 bp. No additional DNA fragmentation or size selection steps were performed. Illumina sequencing was performed on an Illumina NovaSeq 6000 sequencer in multiplexed shared-flow-cell runs, producing 2 × 151 bp paired-end reads. Demultiplexing, quality control, and adapter trimming were performed with bcl-convert1 (v4.1.5).
The sample library for Oxford Nanopore sequencing of E. faecium SM1 was prepared using Oxford Nanopore Technologies Native Barcoding Kit 24 V14 (SQK-NBD114.24) to manufacturer’s specifications. The sample was run on a Nanopore R10.4.1 flow cell on a GridION. Samples were demultiplexed using Guppy (v6.3.8), with the “Super Accurate” base calling model (54).
A high-quality reference assembly for the genome of E. faecium HBSJRP18-2.7 was constructed using a combination of Illumina and Oxford Nanopore sequencing technologies. The hybrid assembly was performed using Unicycler v0.4.4 (55). The genome coverage was 317-fold (94-fold of Oxford Nanopore reads and 223-fold of Illumina reads).
Variants for the DAP evolved strains were called by first using BWA mem (version 0.7.12) (56) to align whole genome sequencing reads to the hybrid E. faecium SM1 reference genome and then using Pilon (version 1.12) (57) to call variants arising between timepoints. VCF files were filtered for variants containing the filter status PASS, and variants labeled as imprecise were removed. FastQ files were deposited under BioProject PRJNA1216963 NCBI’s SRA.
LafB complementation
A construct in the shuttle vector pAT28 harboring a cloned lafB gene previously shown to be expressed, pAT28lafB (20), was transformed into E. faecium SM1ΔlafB to show that deletion of lafB in that recipient was solely responsible for its DAP hypersusceptible phenotype.
Lipidomic analyses
Lipids were extracted from bacterial pellets using a mixture of methyl tert-butyl ether, methanol, and water (58). Perdeuterated (d35) beta-galactosylceramide (Matreya LLC) was added to each sample prior to extraction as an internal standard. Extracted lipids were analyzed by an ultra-high-performance liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) system, consisting of a Vanquish UHPLC and Q-Exactive Orbitrap (Thermo Scientific). Agilent Zorbax C18 column (4.6 mm × 75 mm × 1.8 µm, 600 µL min^−1^) was used for the lipid separation. The following LC gradient was used: 40% 2-propanol/40% acetonitrile/0.125% formic acid/10 mM ammonium formate isocratic for 2 min, then a linear change to 85% 2-propanol/10% acetonitrile/0.125% formic acid/10 mM ammonium formate over 3 min, and hold for 10 min, then return to the initial conditions over 0.1 min, and hold for 4.9 min. The column temperature was set at 40°C. The following heated electrospray ionization parameters were set: spray voltage, 3 kV; sheath gas, 60 AU; auxiliary gas, 15 AU; capillary temperature, 320°C; Aux gas heater temperature, 400°C; S-lens RF level, 65.0 AU. MS full scan was acquired from 300 to 1,600 m/z in positive ion mode (R = 70,000 at m/z 200). A top 3 data-dependent MS/MS was employed to obtain MS/MS spectra (R = 17,500 at m/z 200, normalized collision energy: 20 and 40 (AU), and isolation window: 1 m/z). Data processing, including peak alignment, lipid annotation, and peak quantitation, was performed with the MS-DIAL platform (59). The internal standard peak was quantified from each sample and used to calculate recovery values based on an external calibration curve. Peak areas of individual Glc_1_DAG and Glc_2_DAG were normalized by these recovery values to correct for variations in extraction and MS performance. Total areas for Glc_1_DAG and Glc_2_DAG were calculated by summing the normalized peak areas of the individual Glc_1_DAG and Glc_2_DAG species.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Zhou X, Willems RJL, Friedrich AW, Rossen JWA, Bathoorn E. 2020. Enterococcus faecium: from microbiological insights to practical recommendations for infection control and diagnostics. Antimicrob Resist Infect Control 9:130. doi:10.1186/s 13756-020-00770-132778149 PMC 7418317 · doi ↗ · pubmed ↗
- 2Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM. 2016. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the national healthcare safety network at the centers for disease control and prevention, 2011-2014. Infect Control Hosp Epidemiol 37:1288–1301. doi:10.1017/ice.2016.17427573805 PMC 6857725 · doi ↗ · pubmed ↗
- 3Arias CA, Murray BE. 2012. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol 10:266–278. doi:10.1038/nrmicro 276122421879 PMC 3621121 · doi ↗ · pubmed ↗
- 4Buttress JA, Schäfer A-B, Koh A, Wheatley J, Mickiewicz K, Wenzel M, Strahl H. 2024. The last resort antibiotic daptomycin exhibits two independent antibacterial mechanisms of action. bio Rxiv. doi:10.1101/2024.12.20.629673 PMC 1264503841285733 · doi ↗ · pubmed ↗
- 5Kohinke RM, Pakyz AL. 2017. Treatment of vancomycin-resistant enterococci: focus on daptomycin. Curr Infect Dis Rep 19:33. doi:10.1007/s 11908-017-0589-228779348 · doi ↗ · pubmed ↗
- 6Munoz-Price LS, Lolans K, Quinn JP. 2005. Emergence of resistance to daptomycin during treatment of vancomycin-resistant Enterococcus faecalis infection. Clin Infect Dis 41:565–566. doi:10.1086/43212116028170 · doi ↗ · pubmed ↗
- 7Lewis JS, Owens A, Cadena J, Sabol K, Patterson JE, Jorgensen JH. 2005. Emergence of daptomycin resistance in Enterococcus faecium during daptomycin therapy. Antimicrob Agents Chemother 49:1664–1665. doi:10.1128/AAC.49.4.1664-1665.200515793168 PMC 1068653 · doi ↗ · pubmed ↗
- 8Miller WR, Arias CA. 2024. ESKAPE pathogens: antimicrobial resistance, epidemiology, clinical impact and therapeutics. Nat Rev Microbiol 22:598–616. doi:10.1038/s 41579-024-01054-w 38831030 PMC 13147291 · doi ↗ · pubmed ↗