Scientific Opinion on the tolerable upper intake level for supplemental docosahexaenoic acid
Dominique Turck, Torsten Bohn, Montaña Cámara, Jacqueline Castenmiller, Stefaan de Henauw, Karen‐Ildico Hirsch‐Ernst, Angeles Jos, Alexandre Maciuk, Inge Mangelsdorf, Breige McNulty, Androniki Naska, Kristina Pentieva, Frank Thies, Alfonso Siani, Ionut Craciun

TL;DR
The paper concludes that a daily intake of up to 1 gram of supplemental docosahexaenoic acid (DHA) is safe for all population groups, including pregnant and lactating women.
Contribution
The study reaffirms the 2012 safe level of DHA intake without establishing a tolerable upper intake level due to insufficient data.
Findings
A safe level of 1 g/day of supplemental DHA is retained for all population groups.
No tolerable upper intake level (UL) could be established due to insufficient data on dose–response relationships.
DHA intake is safe in various chemical forms and sources with an EPA/DHA ratio < 0.3.
Abstract
Following a request from the European Commission, the Panel on Nutrition, Novel Foods and Food Allergens (NDA) was asked to deliver a scientific opinion on the revision of the safe level of intake for supplemental docosahexaenoic acid (DHA). Systematic reviews of the literature were conducted to identify human intervention studies administering supplemental DHA alone from a source with an eicosapentaenoic acid (EPA)/DHA ratio < 0.3 for at least 8 weeks, without restrictions on population or outcome. Hazard identification focused on bleeding complications (including bleeding time, platelet function and blood clotting parameters), glucose homeostasis, blood lipid profile, markers of lipid peroxidation, immune function, pregnancy endpoints and safety, tolerability and adverse events. The risk of spontaneous bleeding was selected as the critical effect on which to base the UL/safe level of…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
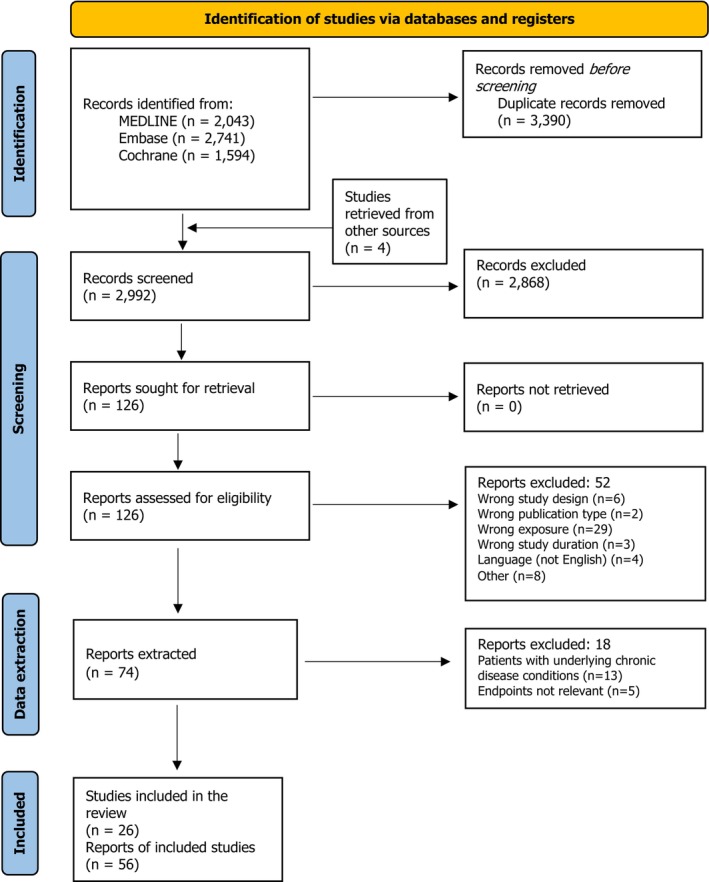 Figure 1
Figure 1| Sub‐questions | Method to address the sub‐questions |
|---|---|
| 1. What is the dose–response relationship between the intake of supplemental DHA ≥ 1 g/day and adverse health effects in humans? | Systematic review |
| 2. What are the potential mechanisms/mode(s) of action underlying the relationship between supplemental DHA intake ≥ 1 g/day and the identified adverse health effects? | Narrative review |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFatty Acid Research and Health · Antioxidant Activity and Oxidative Stress · Food Chemistry and Fat Analysis
INTRODUCTION
1
Background as provided by the European Commission
1.1
On 24 September 2024, EFSA adopted a scientific opinion on the “Safety of an extension of use of oil from Schizochytrium limacinum (strain FCC‐3204) as a novel food pursuant to Regulation (EU) 2015/2283”.1 In its opinion, EFSA concluded that the novel food oil from Schizochytrium limacinum (strain FCC‐3204) is safe under the new intended use.
The above‐mentioned opinion refers to EFSA's “Scientific Opinion related to the tolerable upper intake level of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid (DPA)” adopted by the EFSA NDA Panel (2012).
In its 2012 opinion, EFSA concluded that a Tolerable Upper Intake Level for DHA could not be established. However, the Panel noted that supplemental intakes of EPA and DHA combined at doses up to 5 g/day, and supplemental intakes of EPA alone up to 1.8 g/day, did not raise safety concerns for the adult population. The Panel also considered that supplemental intakes of DHA alone up to about 1 g/day did not raise safety concerns for the general population (safe level of intake). Limited data were available on the effects of long‐term supplementation with DHA alone at higher doses. The Panel noted that specific dietary recommendations for DHA for European adults and children were well below this amount.
In the recent 2024 opinion, EFSA provided anticipated intake estimates of DHA from the authorised novel food sources (excluding food supplements) showing that the anticipated intake of DHA from the authorised uses ranges from 0.1 to 2.4 g/day (mean intake) and from 0.3 to 4 g/day (95th percentile intake). According to these data, the intake of DHA from the authorised uses could be above 1 g DHA/day, the safe level of intake established by the NDA Panel in 2012 scientific opinion.
Terms of Reference as provided by the European Commission
1.2
In accordance with Article 29(1)(a) of Regulation (EC) 178/2002, the European Commission asks the European Food Safety Authority to provide a scientific opinion on the safety of supplemental docosahexaenoic acid (DHA).
In particular, EFSA is requested to:
- reassess the safe level of intake of 1 g/day for supplemental DHA alone established for the general population by the NDA Panel in 2012, in view of the scientific data that have become available since then. To that end, EFSA is requested to review new available data of long‐term supplementation with DHA alone at doses at or above the current safe level of intake of 1 g/day;
- establish a safe level of intake or, if data allow, a Tolerable Upper Intake Level (UL) for supplemental DHA alone for all population groups and, as appropriate, for specific vulnerable subgroups of the population.
Interpretation of the Terms of Reference and context of the assessment
1.3
The protocol developed for this assessment was endorsed by the NDA Panel on 24 March 2025 (Annex A). The context of the assessment, including the rationale for the definition of the exposure of interest, and the interpretation of the terms of reference for this mandate (problem formulation), can be found in that protocol (Sections 2 and 3, respectively).
The exposure of interest is DHA added to foods or consumed as food supplements in any chemical form (e.g. triacylglycerols, ethyl esters, phospholipids) from sources (e.g. fish oil concentrates, algal oils, krill oils) containing DHA alone or mostly DHA (i.e. EPA/DHA ratio < 0.3). For convenience, DHA alone will be used in this opinion to refer to the exposure of interest.
Briefly, the only assessment question for this mandate can be formulated as follows:
What is the maximum level of total chronic daily intake of supplemental DHA, which is not expected to pose a risk of adverse health effects to humans? (Hazard identification and characterisation).
In addressing this question and relying on previous assessments, the Panel considers that this maximum level of total chronic daily intake cannot be lower than 1 g/day.
The formulation of the assessment sub‐questions and the methods used to address them are shown in Table 1.
DATA AND METHODOLOGIES
2
Data
2.1
For the sub‐question addressed through a systematic review, a brief description of the process used for evidence retrieval, study selection and data extraction is provided below.
Literature search
2.1.1
MEDLINE (Ovid), Embase (Ovid) and Cochrane Central Register of Controlled Trials were searched on 26 March 2025 to identify intervention studies and systematic reviews of intervention studies published in English from 1 January 2010 that had investigated the effects of supplemental DHA in humans. Specific search strings were developed by information specialists at EFSA for the mentioned databases to limit by type of study, publication type, publication year and language. Endpoints and population groups were not specified (no restriction). The search strategies and the number of records before and after de‐duplication can be found in Annex B. The reference lists of systematic reviews were screened to identify potentially eligible articles that might have not been captured by the literature search. None were identified. Backward citation searches were conducted on relevant publications to identify other articles reporting on the same study and addressing potentially relevant endpoints. Four additional publications were identified.
Study selection
2.1.2
RCTs and non‐randomised comparative studies of interventions investigating the effect of supplemental DHA at doses ≥ 1 g/day from a source with an EPA/DHA ratio < 0.3 given through the oral route for at least 8 weeks were eligible (Annex A). Title/abstract and full‐text screening was done in duplicate. Conflicts were solved by discussion.
The results of the different steps of the study selection process can be found in Appendix A.
Data extraction
2.1.3
Data from the studies eligible for the assessment as defined in the protocol (no restrictions regarding the study population or the endpoints investigated were established a priori) were extracted in a structured form which includes all references reporting on the same study, the study design, study population, dose and source of supplemental DHA, the EPA/DHA ratio, the number of individuals randomised to DHA and the number of individuals completing the DHA intervention, the duration of the intervention and the outcomes reported in the study.
Studies conducted in patients with underlying chronic disease conditions such as cystic fibrosis, HIV infection, rheumatoid arthritis or diabetes with unclear concomitant therapies or under standard pharmacological treatment for the disease were excluded from the assessment (n = 7, reported in 13 publications). This is because the disease and/or its treatment are expected to modify the effect of supplemental DHA on the outcomes investigated in these studies that are of interest for this assessment (i.e. glucose homeostasis, immune function and the blood lipid profile), which limits the extrapolation of the results to the general population for which the UL/safe level of intake for supplemental DHA is derived. Three additional studies reported in 5 publications were excluded at data extraction because the Panel considered that the endpoints investigated were not relevant for the safety assessment (Section 3.1).
The characteristics of the 26 studies included in the assessment and of the 10 studies excluded at data extraction, together with the rationale for their exclusion, can be found in Appendix B, Tables B.1 and B.2, respectively.
The results of the studies included in the assessment were extracted in tabular form for specific outcomes, where needed.
Methodologies
2.2
The methodologies used to address the assessment question and sub‐questions are depicted in the protocol (Annex A).
Public consultation
2.3
A draft opinion was endorsed by the NDA Panel on 30 September 2025 and was open for public consultation from 3 October to 14 November 2025 (PC‐1647). The draft opinion has been amended in view of the comments received, which have all been addressed and are published in a technical report (Annex C).
ASSESSMENT
3
Introduction
3.1
In 2012, the NDA Panel concluded that the available data were not sufficient to establish a UL2 for DHA. However, the Panel considered that supplemental intakes of DHA alone up to about 1 g/day do not raise safety concerns for the general population. This safe level of intake3 is based on long‐term human intervention studies that have generally not reported adverse effects of supplemental DHA at these levels of intake, and on the limited data available on the effects of long‐term supplementation with DHA alone at higher doses. Adverse effects which have been described in humans in association with high intakes of n‐3 long‐chain polyunsaturated fatty acids (LCPUFAs) include bleeding episodes, impaired immune function, increased lipid peroxidation and impaired lipid and glucose metabolism.
The human intervention studies eligible for this assessment were designed to investigate potential beneficial effects of DHA supplementation in healthy individuals or patients with well‐defined medical conditions at recruitment and were all controlled for supplemental fat intake. Vegetable oils and/or EPA were used for comparison in all the studies. Some of these studies report on several outcomes/endpoints that are not relevant for this safety assessment, including:
- The fatty acid profile in red blood cells (RBCs), plasma or other body fluids (e.g. cerebrospinal fluid), generally measured as markers of compliance with the intervention and/or to calculate the omega‐3 index;
- Endpoints related to cognitive function, MRI measures of brain volume and specific areas thereof (hippocampal volume), or fMRI measures of brain activation, in healthy individuals or in individuals with cognitive impairment, Alzheimer disease, dementia or bipolar disorder.
- Depression‐related endpoints in individuals with mild–moderate depression or post‐traumatic injury;
- Post‐stress or quality of life endpoints in patients with post‐traumatic injury; biomarkers of head trauma in American football players;
- Exercise performance, markers of muscle damage and muscle soreness post‐exercise in amateur endurance athletes;
- Gingival endpoints in individuals with periodontitis (except bleeding on probing);
- Endpoints related to vaginal microbiota composition and potential pathobionts in pregnant women with overweight or obesity;
- Sperm quality in males with history of infertility;
- Vision‐related endpoints in patients with X‐linked retinitis pigmentosa, Stargardt macular dystrophy or Best disease (macular degeneration);
- Anthropometry, body composition and blood pressure;
- Clinical endpoints in very pre‐term infants (< 29 weeks of gestation) of DHA‐supplemented mothers, as this population group is outside the target population for which ULs/safe levels of intake are derived.
Three of the initially eligible human intervention studies only investigated one or more of these endpoints and were excluded at data extraction (Section 2.1.3).
No adverse effects of DHA supplementation on any of these outcomes/endpoints have been reported in any of the eligible studies.
The Panel notes that results regarding gene expression/transcriptomics in peripheral white blood cells, monocytes or subcutaneous adipose tissue related to markers of inflammation, insulin resistance or lipid metabolism cannot be used to establish a UL/safe level of intake for DHA, but could be considered when discussing the mechanisms by which supplemental DHA could adversely affect other endpoints related to immune function, glucose homeostasis and/or blood lipids, if needed.
Of the eligible 26 human intervention studies, 13 have systematically investigated the effect of supplemental DHA on relevant outcomes for this safety assessment and 13 only report on safety/tolerability/adverse effects/adverse events in different ways (see Section 3.2.7). The characteristics of all the included studies by category of relevance for this assessment can be found in Appendix B, Table B.1.
Hazard identification
3.2
Bleeding complications, bleeding time, platelet function and blood clotting parameters
3.2.1
Safety concerns about a higher risk of bleeding complications associated with the consumption of high‐dose supplements of n‐3 LCPUFAs arise from the observation, mostly in short‐term intervention studies, that fish oils (EPA and DHA in combination), as well as either EPA or DHA alone (highly purified ethyl esters), increase bleeding time and decrease platelet aggregation in a dose–response manner at doses up to about 9 g/day (highest doses tested) (EFSA NDA Panel, 2012). Some episodes of epistaxis in 11 children and adolescents consuming 1.5 g/day EPA and DHA from fish oil for 6 months (single‐arm, uncontrolled study) have also been reported (Clarke et al., 1990).
In 2012 (EFSA NDA Panel, 2012), the Panel noted that the changes in bleeding time (for supplemental intakes of EPA and DHA combined up to about 6 g/day) and platelet aggregation (for supplemental intakes of EPA and DHA, either alone or in combination, up to about 4 g/day) observed in these short‐term (4–12 weeks) intervention studies could not be considered adverse, as they were not associated with an increased risk of clinical complications (e.g. spontaneous bleeding). However, conclusions on bleeding complications were reached based on a wealth of controlled human intervention studies on the effects of n‐3 LCPUFA conducted in population subgroups at high risk of bleeding, including patients on antiplatelet or antithrombotic medications (acetyl salicylic acid, clopidogrel, anticoagulants) undergoing invasive procedures, and pregnant women at delivery.
In 2012, the Panel considered that supplemental intakes of EPA and DHA combined of up to about 5 g/day for up to 2 years and up to about 7 g/day for up to 6 months, do not increase the risk of spontaneous bleeding episodes or bleeding complications, even in subjects at high risk of bleeding (e.g. in antiplatelet or antithrombotic medications). The Panel noted that the data available were insufficient to conclude on whether the same doses administered mostly as EPA or mostly as DHA would have different effects on this outcome. The Panel also considered that intakes of EPA alone at doses up to 1.8 g/day for 2 years do not increase the risk of bleeding complications, based on the results of one open label human intervention study (Yokoyama et al., 2007) which investigated the effects of 1.8 g/day of EPA as ethyl esters consumed for 5 years in combination with statins (n = 9326) vs. statins alone (n = 9319) in hypercholesterolaemic, high fish consumers on the primary and secondary prevention of coronary heart disease which also assessed safety outcomes and the risk of stroke and its subclasses (Tanaka et al., 2008). No long‐term intervention studies with DHA alone at doses > 1 g/day reporting on bleeding complications were available at the time.
Bleeding complications
3.2.1.1
No long‐term intervention studies on supplemental DHA at doses ≥ 1 g/day reporting on spontaneous bleeding or bleeding complications were identified through the literature search conducted for this assessment. This is consistent with the studies retrieved in a recent systematic review and meta‐analysis of RCTs which investigated bleeding risk in individuals with (or at high risk of) cardiovascular events receiving supplemental EPA and DHA, either alone or in combination (Javaid et al., 2024). The systematic review was triggered by the results of the REDUCE‐IT (Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention) trial, in which 4 g/day of highly purified EPA consumed for a median of 4.9 years increased (although non‐significantly) the number of serious bleeding events as compared to a placebo mineral oil (2.7% vs. 2.1%, p = 0.06) among the 8179 patients enrolled (n = 4089 randomised to EPA), 70.7% of which were on secondary prevention of cardiovascular events (Bhatt et al., 2019). Trials with < 1000 patients were excluded to minimise the risk of type 1 statistical error. Trials of short duration were also excluded. A total of nine RCTs on EPA and DHA in combination and two RCTs on EPA alone were identified, all lasting at least 2 years. None used supplemental DHA alone.
None of the studies retrieved reported on spontaneous bleeding.
One double‐blind, parallel RCT investigated the effect of supplemental DHA on bleeding on probing in adults with mild periodontitis (Naqvi et al., 2014). Participants aged ≥ 40 years were randomised to consume 2 g/day supplemental DHA (n = 27, n = 24 completers) or a control oil (50% maize oil and 50% soybean oil; n = 28, n = 22 completers) for 3 months. All participants received low‐dose aspirin (81 mg/day). Clinical measurements included bleeding on probing scores (dichotomous, yes/no) at 6 sites per tooth. The authors report no significant differences in bleeding on probing scores between groups or changes in bleeding on probing scores throughout the study.
Another double‐blind, parallel RCT reports on postpartum haemorrhage as a predefined secondary outcome (Pellonperä et al., 2019). Pregnant women with overweight and obesity were randomised to supplemental DHA‐rich fish oil (1.9 g/day DHA, n = 109), ‘probiotics’ (n = 110), their combination (n = 109) or placebo (medium‐chain fatty acids; n = 110), from early pregnancy (mean 14 weeks' gestation) until 6 months postpartum. Among the women analysed for postpartum haemorrhage (n = 95, 96, 96 and 92 in the respective groups), median blood loss was 400 mL across all groups (p = 0.96). Severe haemorrhage (> 1000 mL) occurred in 5%–8% of women, with no significant differences among groups (p = 0.83).
The Panel considers that, in the absence of adequately powered, long‐term intervention studies investigating supplemental DHA alone at doses ≥ 1 g/day and reporting on spontaneous bleeding or bleeding complications, the available data remain insufficient to conclude on whether supplemental DHA alone would have different effects on the risk of bleeding than DHA in combination with EPA or EPA alone.
Bleeding time
3.2.1.2
None of the eligible studies has systematically investigated the effect of supplemental DHA on bleeding time. One RCT reports on the effect of supplemental DHA on the international normalised ratio (INR) of patients on warfarin because subtherapeutic INR was incidentally discovered in some patients after trial initiation.
In a double‐blind, parallel RCT, male and female adults with Alzheimer disease were randomised to consume 2 g/day DHA (n = 238, n = 171 completers) or a control oil (50% maize oil and 50% soybean oil; n = 164, n = 128 completers) for 1.5 years. A total of 32 patients were on warfarin at the time of randomisation. During the blinded phase of the trial, the data and safety monitoring board noted that 3 individuals taking warfarin had a subtherapeutic INR after starting the study product, and since then INR was tested monthly in all individuals taking warfarin. After unblinding, all three cases were found to receive DHA. One case of increased INR was detected in the control oil group. The number of patients on warfarin randomised to each intervention group was not reported (Quinn et al., 2010).
The Panel notes that no eligible studies have systematically investigated the effect of supplemental DHA alone on bleeding time.
Platelet function and blood clotting parameters
3.2.1.3
In a double‐blind, parallel RCT, males aged 7–31 years with diagnosis of the X‐linked form of retinitis pigmentosa (XLRP) were randomised to consume 30 mg/kg bw/day (mean = 1.74 g/day, range = 0.6–3.6 g/day) of DHA (n = 41, n = 29 completers) or a control oil (50% maize oil and 50% soybean oil; n = 37, n = 22 completers) for 4 years. As part of the safety assessment of the intervention, whole blood platelet aggregation was monitored every 6 months to address concerns of increased bleeding due to inactivation of platelet activation responses (Hughbanks‐Wheaton et al., 2014). Platelet aggregation was measured in citrate‐anticoagulated whole blood using type I collagen as agonist. No group differences over the 4‐year trial were found. The Panel notes the highly variable doses of supplemental DHA across participants in absolute amounts.
In another double‐blind, parallel RCT, male and female adults 18–65 years of age that were overweight or obese (BMI 25–39.9 kg/m^2^) were randomised to consume 2 g/day DHA (n = 19) or a control oil (50% maize oil and 50% soybean oil; n = 17) for 4.5 months. The outpatient intervention period was preceded and followed by inpatient periods of 21 days each. Prothrombin time and the activated partial thromboplastin time were measured on fasting plasma drawn on day 14 of each inpatient period with standard clot‐based assays using reagents. Fibrinogen was measured on fasting plasma with the Clauss method. The authors report that, compared with the control oil, DHA had no significant effect on clotting parameters, including prothrombin time, activated partial thromboplastin time and fibrinogen. Data are not shown in the publication (Neff et al., 2011).
In the RCT in adults with Alzheimer disease described above (Quinn et al., 2010), the data and safety monitoring board also noted during the blinded phase of the trial that thrombotic events were occurring at a rate higher than expected overall, and thereafter they were closely monitored through the trial. After unblinding, there were no significant differences between the DHA and control groups in the incidence of thrombotic events (3.4% and 1.2% in the DHA and control groups, respectively).
The Panel notes that the studies identified investigating supplemental DHA alone at doses of about 2 g/day do not report adverse effects on platelet function or blood clotting parameters. These findings are consistent with the Panel's previous conclusion that changes in platelet function observed at supplemental intakes of DHA alone up to about 4 g/day in mostly small and relatively short‐term intervention studies are not considered adverse, as they are not associated with an increased risk of clinical complications (e.g. spontaneous bleeding).
Conclusions on bleeding complications, bleeding time, platelet function and clotting parameters
3.2.1.4
The Panel considers that no new data have become available to revise its previous conclusions on supplemental DHA alone in relation to the risk of bleeding, as no long‐term intervention studies conducted in a sufficiently large number of individuals with supplemental DHA alone at doses ≥ 1 g/day reporting on spontaneous bleeding or bleeding complications are available.
The Panel concludes that the long‐term safety of supplemental DHA alone at doses > 1 g/day in relation to the risk of bleeding cannot be established for any population group.
Glucose homeostasis
3.2.2
In 2005, the IoM advised that subjects with ‘impaired glucose tolerance or diabetic conditions requiring increased doses of hypoglycaemic agents’ should take EPA and DHA supplements with caution (IOM, 2005) owing to a number of human intervention studies, mostly uncontrolled, that have described adverse effects of supplemental n‐3 LCPUFA (≥ 10 g/day) on glucose homeostasis, such as increased insulin requirements, an increase in glycated haemoglobin (HbA1c) and an increase in fasting and postprandial glycaemia, in patients with type 1 and type 2 diabetes (see (De Caterina et al., 2007) for review).
In 2012, the NDA Panel noted that human intervention studies that were controlled for fat intake generally do not show a differential effect of vegetable oils and supplemental fish oil at doses up to 5 g/day of EPA and DHA consumed for 12 weeks on blood glucose control in diabetic subjects, or on insulin sensitivity in healthy or diabetic subjects, but data were insufficient to conclude on whether the same doses administered mostly as EPA or mostly as DHA would have different effects on this outcome (EFSA NDA Panel, 2012).
Among the human intervention studies eligible for the current safety assessment, seven RCTs report on the effect of supplemental DHA in one or more measures of glucose homeostasis. The characteristics of the studies and the endpoints measured in relation to glucose homeostasis are shown in Appendix C.
One additional study measured blood glucose levels as part of the routine clinical biochemistry, but the results for this endpoint were not reported (McNamara et al., 2010). This study will not be considered further in this section. Measures of glucose homeostasis which follow the natural history of type 2 diabetes, i.e. from those that are expected to be impaired first to those expected to be impaired later in time, can be grouped in lines of evidence (LoE) as follows (EFSA NDA Panel, 2022):
- Measures of insulin sensitivity obtained either in steady‐state conditions (during an euglycaemic hyperinsulinaemic clamp) or in non‐steady state conditions (e.g. during an intravenous glucose administration with frequent sampling/minimal model assessment (IVGTT)).
- Indices of insulin sensitivity/resistance and indices of insulin secretion/beta cell function, either derived from the fasting state (e.g. HOMA‐IR, HOMA‐beta) or from an oral glucose tolerance test (OGTT) (e.g. Matsuda index of insulin sensitivity).
- Measures of glucose tolerance, either derived from the fasting state (fasting glucose and insulin) or from an OGTT, including glucose and insulin at 120 min and areas under the curve (AUC) for glucose and insulin.
- Measures of blood glucose control, including fructosamine, glycated albumin and glycated haemoglobin (HbA1c). These are not expected to change significantly in non‐diabetic individuals.
None of the available studies have measured insulin sensitivity directly (LoE a). As for LoE b, three studies report on indices of insulin sensitivity/resistance derived from the fasted state (HOMA‐IR, QUICKI; (Félix‐Soriano et al., 2021; Kelley et al., 2012; Neff et al., 2011)), of which one (Kelley et al., 2012) also calculated the Matsuda index in the post‐prandial state. In relation to LoE c, three studies assessed fasting glucose as part of the routine clinical biochemistry (Chang et al., 2021; Hughbanks‐Wheaton et al., 2014; Klingel et al., 2019) and four other studies measured fasting glucose and insulin (Félix‐Soriano et al., 2021; Kelley et al., 2012; Neff et al., 2011; Singhal et al., 2013), two of which have also reported on the 120‐min AUC for glucose and insulin after an OGTT (Neff et al., 2011) or after a standardised breakfast (Kelley et al., 2012). Finally, one study (Neff et al., 2011) measured HbA1c (LoE d). The Panel notes that, although this study lasted 16 weeks, which is sufficient to detect differences in HbA1c in persons with diabetes, HbA1c is not expected to change significantly over such short period of time in non‐diabetic individuals.
None of the eligible human intervention studies, which were all controlled for supplemental fat intake, showed a differential effect of supplemental DHA as compared to vegetable oils or EPA‐rich oils on fasting glucose or insulin, indices of insulin sensitivity/resistance thereof, indices of insulin sensitivity/resistance in the post‐prandial state, or HbA1c. The doses of supplemental DHA tested ranged from 1.6 to 3 g/day, and the duration of the intervention from 10 to 16 weeks (up to 4 years) in most studies. All the studies were conducted in non‐diabetic adults and only one, which measured fasting glucose only as part of the routine clinical biochemistry, included also children (Hughbanks‐Wheaton et al., 2014). The Panel notes that the studies' design, including sample size and trial duration, were generally adequate to detect any biologically relevant adverse effect of the intervention on the endpoints measured within each study.
Conclusions on glucose homeostasis
3.2.2.1
The Panel considers that supplemental intakes of DHA alone up to 3 g/day consumed for up to 10–16 weeks do not adversely affect glucose homeostasis in adult healthy individuals. Data do not allow concluding on children or patients with diabetes.
Blood lipid profile
3.2.3
In 2012, the Panel noted that supplemental intakes of EPA and DHA combined of 2–6 g/day, and supplemental intakes of mostly DHA of 2–4 g/day, increase blood concentrations of low‐density lipoprotein (LDL)‐cholesterol by about 3%, and that such increase is accompanied by a decrease in triglycerides (TG) with no changes in total (or non‐high‐density lipoprotein (non‐HDL)) cholesterol concentrations.
A total of nine RCTs eligible for the present assessment investigated the effects of supplemental DHA on the blood lipid profile as compared to EPA or a control vegetable oil. Among the blood lipid parameters investigated in these studies, the Panel decided to consider only those that are recommended to estimate the risk of atherosclerotic cardiovascular disease (CVD) and/or that are treatment targets for dyslipidaemias (Mach et al., 2019), namely plasma concentrations of total cholesterol (TC), LDL‐cholesterol (LDL‐C), HDL‐cholesterol (HDL‐C), TG, apolipoprotein B (ApoB) and non‐HDL‐C. Where plasma concentrations of non‐HDL‐C are not reported, the TC/HDL‐C ratio will be considered instead. The Panel acknowledges that the AUC for post‐prandial lipaemia and lipoprotein particle size are being investigated in relation to CVD risk, and that very high lipoprotein(a) levels represent a genetically determined risk factor for atherosclerosis in certain individuals. However, the Panel notes that the independent role of any of these variables in determining the risk of CVD in the general population is to be established.
The characteristics of the nine eligible RCTs and the results for the blood lipid variables that are relevant for this safety assessment can be found in Appendix D.
Changes in blood lipids during the intervention did not differ between the supplemental DHA and the control groups in 3 studies conducted in adults with hypertriglyceridaemia (1.6 g/day for 10 weeks; (Dittrich et al., 2015)), low‐grade chronic inflammation (3 g/day for 10 weeks; (So et al., 2022)) or adults and children with XLRP (30 mg/kg bw/day, mean 1.740 g/day for 4 years; (Hughbanks‐Wheaton et al., 2014)). The number of participants in the supplemental DHA arm ranged from 19 to 24 individuals in the studies with cross‐over designs and up to 41 individuals in the RCT with a parallel design (Hughbanks‐Wheaton et al., 2014). Vegetable oils and EPA (3 g/day; (So et al., 2022)) served as controls.
In three additional studies, only a significant decrease in blood TG between 22 and 33% with supplemental DHA as compared to control oils was reported in healthy adults of both sexes (Klingel et al., 2019; MacIntyre et al., 2023; Singhal et al., 2013) and in post‐menopausal women with overweight or obesity (Félix‐Soriano et al., 2021). All these studies had a parallel design, used olive oil as comparator, lasted between 12 and 16 weeks and tested doses of supplemental DHA between 1.6 and 3 g/day. The number of participants in the supplemental DHA arm ranged from 21 to 162 individuals (15 and 135 completers). In the RCT using the highest dose of supplemental DHA (3 g/day), also EPA was tested at the same daily dose (Klingel et al., 2019; MacIntyre et al., 2023). No significant differences between the effect of these n‐3 LCPUFA on the blood lipid profile were reported.
In the remaining three studies, conducted in adults of both sexes with overweight/obesity (Neff et al., 2011), abdominal obesity and low‐grade chronic inflammation (Allaire et al., 2016; Allaire et al., 2018) or males with hypertriglyceridaemia (Kelley et al., 2007; Kelley et al., 2008), supplemental DHA at doses between 2 and 3 g/day consumed for 10–16 weeks also significantly reduced blood TG (by between 13% and about 26%) as compared to olive oil or a 1:1 mixture of maize and soy oil. However, these studies also report a significant increase in TC, LDL‐c, ApoB or a combination of these in the supplemental DHA group as compared to the control which, on their own, could be considered adverse. The Panel notes the small number of participants in the supplemental DHA arm in the two studies with a parallel design, which was 20 (17 completers) (Kelley et al., 2007; Kelley et al., 2008) and 19 individuals (Neff et al., 2011).
In males with hypertriglyceridaemia (Kelley et al., 2007; Kelley et al., 2008), LDL‐c concentrations significantly increased (by 15.8%) with 3 g/day supplemental DHA given for 90 days as compared to olive oil. However, blood concentrations of TC, apolipoprotein B and the total:HDL cholesterol ratio were not affected. In adults with overweight (Neff et al., 2011), TC significantly increased (by 4.8%) with 2 g/day supplemental DHA given for 16 weeks as compared to maize/soy oil. LDL‐c and HDL‐c were not significantly affected. ApoB was not measured, and non‐HDL‐c or the total: HDL‐c ratio were not calculated. Finally, the largest RCT (154 individuals, 123 completers) had a three‐way cross‐over design and tested the effect of supplemental DHA, EPA and maize oil at doses of 2.7 g/day for 10 weeks, with washout periods of 9 weeks in between (Allaire et al., 2016; Allaire et al., 2018). Supplemental DHA significantly increased TC (by 3.8%), LDL‐c (by 6.9%) and ApoB (by 4.5%), but also HLD‐c (by 7.6%), leading to a significant 2.5% decrease in the total: HDL‐c ratio as compared to maize oil. A significant increase in TC (by 3.6%), LDL‐c (by 3.2%) and HDL‐c (% difference cannot be calculated), and a significant decrease in the total: HDL‐c (% difference cannot be calculated) were also reported for DHA as compared to EPA, whereas no significant differences were observed for ApoB. DHA also showed a significant reduction in TG (by 7.1%) as compared to EPA.
Overall, the Panel notes that the effect of supplemental DHA on the blood lipid profile depends on the dose of supplemental DHA, but also on the oil used as control. When compared to vegetable oils (and to a lower extent with EPA), doses of supplemental DHA between 2 and 3 g/day generally show a hypotriglyceridaemic effect, which may be accompanied by a significant increase in TC and/or LDL‐c but also by an increase in HDL‐c, whereas the total: HDL‐c ratio is either not affected or modestly reduced.
Conclusions on the blood lipid profile
3.2.3.1
The Panel considers that the new data that have become available are consistent with its previous conclusions that supplemental DHA alone at doses of 2–4 g/day for up to 16 weeks does not induce changes in the blood lipid profile that may be considered adverse in relation to CVD risk.
Markers of lipid peroxidation
3.2.4
Early observations linking DHA intake with increased lipid peroxidation and oxidative damage to cells and molecules in laboratory animals may have been confounded by the presence of primary and secondary oxidation products in supplements lacking antioxidants, as the effect was reversed when DHA was administered with supplemental vitamin E (EFSA NDA Panel, 2012).
In 2012, the Panel concluded that supplemental intakes of EPA and DHA consumed either alone or in combination at doses up to about 5 g/day for up to 16 weeks do not induce changes in lipid peroxidation which might raise concern in relation to CVD risk as long as the oxidative stability of these n‐3 LCPUFAs is guaranteed. This conclusion was based on the results of controlled human intervention studies which assessed the effects of EPA and DHA, alone or in combination, on plasma or urine F2‐isoprostanes and/or in vivo LDL‐oxidation. Most studies used fish oil in combination with antioxidants and vegetable oils as control, although some studies did not report on whether sources of EPA, DHA, or both, contained antioxidants or not, and few had measured secondary oxidation products in the supplements administered. The addition of antioxidants to food supplements containing n‐3 LCPUFA to ensure product stability appeared to be optional (GOED, 2012).
The Panel notes that, despite best practice guidelines have been issued to control oxidation products in EPA and DHA supplements (GOED, 2012), the compliance of EPA/DHA supplements on the market appears to be variable (Bannenberg et al., 2017).
F2‐isoprostanes
3.2.4.1
Two studies assessed the effect of supplemental DHA on plasma F2‐isoprostanes, one of which used an ELISA kit for 8‐F2c‐isoprostane measurements (Dong et al., 2021). The Panel notes that immunological techniques, owing to their lack of specificity due to possible cross reactions with other prostanoids, are not appropriate for measuring F2‐isoprostanes (EFSA NDA Panel, 2018), and so this study will not be considered further in this section.
In the second study (Shichiri et al., 2014), 34 men with hypertriglyceridaemia were randomised to consume 3 g/day of supplemental DHA or olive oil (control) for 90 days (n = 17 per group). Ascorbyl palmitate and mixed tocopherol (250 ppm each) were added as antioxidants to both oils (Kelley et al., 2007). Plasma and RBC concentrations of 8‐isoprostaglandin‐F_2α_ (8‐iso‐PG F2α) and other markers of lipid peroxidation were measured on days 1, 45 and 90 of the study using liquid chromatography–tandem mass spectrometry (LC–MS/MS). No significant differences between the DHA and control groups in any of these markers were observed in plasma or RBC for the duration of the study.
Oxidation of LDL
3.2.4.2
Two studies assessed the effects of supplemental DHA on in vivo plasma oxidised LDL (oxLDL).
The first study (Hughbanks‐Wheaton et al., 2014) is the double‐blind, parallel RCT in males aged 7–31 years with diagnosis of XLRP described in section 3.2.1.3 in relation to platelet function. Patients were randomised to consume DHA supplements (30 mg/kg bw per day; mean = 1.74 g/day, range = 0.6–3.6 g/day; n = 41, n = 29 completers) or a control oil (50% maize oil and 50% soybean oil; n = 37, n = 22 completers) for 4 years. All study capsules contained small amounts of vitamins E and C as antioxidants (amounts not reported). Plasma oxLDL were measured every 6 months as part of the safety assessment of the intervention. No between‐group differences over the 4‐year trial were found.
The second study (Dittrich et al., 2015) is a RCT in which 59 male and female adults with hypertriglyceridaemia were randomised to DHA oil (1.6 g/day, n = 19, 16 completed), echium oil or linseed oil (n = 18, 15 completed in both groups) for 10 weeks. All groups also received sunflower oil (control) for 10 weeks in a crossover design, with a 10‐week washout period in between. The DHA oil contained 10.67 mg/100 g α‐tocopherol. Plasma oxLDL were measured at the beginning and end of each intervention. No differences were observed between the intervention oils and the control, or among the intervention oils, on plasma oxLDL.
Other markers of lipid peroxidation
3.2.4.3
Several studies have investigated the effect of supplemental DHA on non‐specific changes in the overall antioxidant capacity of plasma using methods such as TRAP, FRAP, TEAC, ORAC or FOX assays, and/or other markers of lipid peroxidation (e.g. thiobarbituric acid reactive substances (TBARS), malondialdehyde (MDA), conjugated dienes, lipid hydroperoxides). The Panel notes that changes in any of these markers do not necessarily imply changes in oxidative damage to molecules (EFSA NDA Panel, 2018) and therefore will not be considered further.
Conclusions on markers of lipid peroxidation
3.2.4.4
The Panel considers that the new data that have become available are consistent with its previous conclusion that supplemental DHA alone at doses up to about 5 g/day for up to 16 weeks does not induce changes in lipid peroxidation which might raise concern in relation to CVD risk, as long as the oxidative stability of DHA supplements is guaranteed.
Immune function
3.2.5
In its previous opinion (EFSA NDA Panel, 2012), the NDA Panel noted that, whereas EPA and DHA had been shown to decrease the expression of cytokines and the proliferation of peripheral white blood cells at doses as low as 0.9 g/day EPA and 0.6 g/day DHA consumed as fish oil for 6–8 weeks in ex vivo and in vitro studies, the clinical relevance of these changes in vivo was unknown. The Panel also noted that, whereas immunosuppression, if sustained, may increase the risk of infections, there were no human intervention studies available which had investigated the effects of n‐3 LCPUFA supplementation on the risk of infections in vivo.
None of the eligible studies have investigated the effect of supplemental DHA on the risk of infections.
In 2012, the NDA Panel noted that chronic and/or inappropriate activation of inflammatory responses (innate immunity) can also lead to disease, and that some markers of the so‐called low‐grade systemic (e.g. high‐sensitivity C‐reactive protein and some cytokines) and vascular (e.g. sICAM‐1, VCAM‐1 and E‐selectin) inflammation have been associated with an increased risk of cardiovascular events in healthy and high‐risk subjects. However, there was no information available on the effect of high intakes of n‐3 LCPUFA on the risk of chronic diseases of inflammatory origin, and there was no evidence that changes induced by diet or drugs in any of these inflammatory markers modify the risk of disease per se. The Panel also noted that most studies conducted with EPA and DHA combined at doses up to 5 g/day had either no effect or showed a decrease in markers of vascular and/or systemic inflammation, but the data available were insufficient to conclude on whether the same doses administered mostly as EPA or mostly as DHA would have different effects on this outcome (EFSA NDA Panel, 2012).
A total of 10 eligible RCTs have investigated the effect of supplemental DHA on markers of systemic or vascular inflammation in plasma or serum. Details on the study characteristics and the specific endpoints measured can be found in Appendix E. Changes in markers of inflammation (i.e. hs‐CRP, IL‐6 and IL‐1ß) in crevicular gingival fluid among individuals with periodontitis that were investigated in one of these studies (Naqvi et al., 2014) will not be considered further, as their clinical relevance for the general population is unclear.
One additional study assessed inflammatory markers in breastmilk (Fougère et al., 2023); however, the clinical relevance of this endpoint for infant health‐related outcomes remains to be elucidated. Moreover, there is a lack of well‐established reference ranges or defined threshold values for inflammatory markers in breastmilk that would indicate a potential risk to the infant. Therefore, this study was not considered in the assessment for this outcome.
None of the available studies reported an adverse effect (i.e. a significant increase in ‘pro‐inflammatory markers’ and/or a significant decrease in ‘anti‐inflammatory’ markers, such as IL‐10 or adiponectin) of supplemental DHA as compared to the control (vegetable) oils. Daily doses of supplemental DHA ranged from 1 to 3 g/day, and the duration of the intervention from 8 to 21 weeks. All the studies were conducted in adults, including two in pregnant women, which provided supplemental DHA at doses of 1 and 1.9 g/day.
Conclusions on immune function
3.2.5.1
The Panel considers that supplemental intakes of DHA alone up to about 3 g/day for up to 16 weeks are unlikely to induce changes in immune functions which might raise concern in relation to the risk of infections or inappropriate activation of inflammatory responses.
Pregnancy endpoints
3.2.6
One double‐blind, RCT investigated the effect of two doses of supplemental DHA (1 g/day, n = 576; and 0.2 g/day, n = 524) given to adult (> 18 years) pregnant women from 12 to 20 weeks of pregnancy until delivery on the incidence of early preterm birth (EPB), defined as delivery < 34 weeks of gestation (Carlson et al., 2021). Participants were randomised to one of the two DHA groups with a maximum number of pregnant women of 1100 enrolments and 5% expected dropout rate following a planned Bayesian adaptive design. The primary outcome was EPB by dose and by DHA status at baseline (low/high: RBC phospholipid DHA < 6%/≥ 6%). A total of 540 women in the 1 g/day DHA group and 492 women in the 0.2 g/day DHA group entered data analysis. Bayesian posterior probabilities (pp) were determined for planned efficacy and safety outcomes using intention‐to‐treat analyses. Women in the higher supplemental DHA dose (1 g/day) had a lower EPB rate [1.7% (9/540) vs. 2.4% (12/492), pp. = 0.81], especially those with a lower DHA status at enrolment [2.0% (5/249) vs. 4.1%, (9/219), pp. = 0.93], whereas no difference was seen in women with a high DHA status [1.4% (4/289) vs. 1.1% (3/271), pp. = 0.57]. The higher supplemental DHA dose was also associated with fewer serious adverse events (maternal: chorioamnionitis, premature rupture of membranes and pyelonephritis; neonatal: feeding, genitourinary and neurological problems, all pp. > 0.90).
Another double‐blind, placebo‐controlled, parallel RCT already described in Section 3.2.1.1 (Pellonperä et al., 2019) investigated the effect of supplemental DHA‐rich fish oil (1.9 g/day DHA, n = 109), ‘probiotics’ (n = 110) and their combination (n = 109) versus placebo (medium‐chain fatty acids; n = 110) from early pregnancy (mean 14 weeks' gestation) until 6 months postpartum in overweight and obese pregnant women. The primary outcome was incidence of gestational diabetes mellitus (GDM). No significant differences among groups were reported in relation to the incidence of GDM, fasting glucose or insulin concentrations, or the HOMA‐IR. Likewise, there were no significant differences in the incidence of gestational hypertensive disorders (pregnancy‐induced hypertension and pre‐eclampsia) among the groups. Neonatal outcomes, including birth weight, macrosomia (> 90th percentile), small for gestational age, gestational age at delivery, Apgar score, umbilical artery pH, hypoglycaemia, admission to the intensive care unit and congenital malformations, showed no significant differences among groups.
The Panel notes that only one study in pregnant women was identified using doses > 1 g/day. The Panel considers that the two studies identified support the safety of supplemental DHA alone at doses up to 1 g/day on pregnancy related endpoints.
Safety, tolerability and adverse events
3.2.7
A total of 21 out of the 26 RCTs eligible for the assessment reported on (or mentioned) safety, tolerability and/or adverse effects/events. These studies were designed to investigate potential beneficial effects of DHA supplementation in healthy individuals (children, adults and pregnant women) or patients with well‐defined medical conditions, rather than to establish the safety of supplemental DHA. The characteristics of these 21 studies, including the safety endpoints assessed, the timing and methods of assessment (when reported), and the results, are presented in Appendix F.
Across these studies, the methods for capturing adverse events varied considerably and were often poorly described. One study combined physical examinations (including vital signs) and clinical biochemistry at baseline and week 8 with a structured side‐effect interview administered at baseline, week 4 and week 8 (McNamara et al., 2010). In another trial conducted in children, parents were contacted by phone or email at weeks 4, 10 and 15 to report any adverse events (Milte et al., 2015), while in another study, serious adverse events were recorded during weekly telephone calls, and data on adverse effects and tolerance over the preceding 7 days were collected during monthly calls (Singhal et al., 2013). Two US FDA Investigational New Drug (IND) studies monitored treatment‐emergent adverse events (Carlson et al., 2021; Hughbanks‐Wheaton et al., 2014). In one study, participants self‐reported adverse events via quarterly diaries and at annual visits, and blood chemistry abnormalities were assessed using the DAIDS4 criteria (Hughbanks‐Wheaton et al., 2014). In the other study, adverse events were recorded by investigators and assessed yearly by a data safety monitoring committee (Carlson et al., 2017). In both studies, serious adverse events (e.g. death, hospitalisation, life‐threatening events, disability/incapacity, congenital anomalies) were reported immediately to investigators. In a perinatal trial, maternal events were recorded up to 40 weeks post‐menstrual age, and neonatal safety documented in case report forms (MOBIDIck trial). In another study involving pregnant women with overweight and obesity, participants were instructed to maintain a weekly diary to document any potential adverse effects (Pellonperä et al., 2019). Most other studies, however, simply noted that adverse effects or side effects were ‘assessed’ at various intervals (e.g. every 1–2 weeks, at 6 and 12 weeks, monthly or quarterly), without providing details on the methods used for the assessment. Overall, the heterogeneity in assessment methods and frequency, and the lack of methodological details, limit comparisons across studies.
Across the included RCTs, DHA supplementation was generally well tolerated, with most studies reporting either no adverse events or only mild, self‐limited effects such as gastrointestinal (GI) symptoms, burping, unpleasant breath, or headache. In several trials, investigators explicitly stated that no adverse events were reported or that the intervention was well tolerated, though details on what was assessed or how were often lacking.
Only three studies have reported the occurrence of bleeding events during DHA supplementation.
In a double‐blind, 12‐month RCT with a 3‐way cross over design, 90 children 7–12 years of age with ADHD (of which 54 completed the study) consumed 1.032 g/day of DHA (DHA‐rich fish oil), EPA (EPA‐rich fish oil) and linoleic acid (LA) (safflower oil, control) for 4 months each. One case of nose bleeding was reported during the DHA treatment (Milte et al., 2015). This case was already noted in a previous publication from the same study, where the results of the first 4 months of the intervention (3 parallel arms with EPA, DHA or LA) are reported (Milte et al., 2012). No further information was provided.
In another double‐blind, RCT with an ABAB cross‐over design (MacDonald & Sieving, 2018), 11 adult males and females with autosomal recessive Stargardt macular dystrophy received 2 g/day of DHA and of a mixture of maize and soy oil (control) for two periods of 3 months each with no washout in between. A case of serious vaginal bleeding requiring hysterectomy was reported in the study arm receiving the control oil first. The exact timing of the event is not reported. All eight adverse events recorded, including the bleeding episode, were considered not to be related to the intervention.
In the MOBIDIck double‐blind, placebo‐controlled RCT (Marc et al., 2020), 461 lactating women who delivered before 29 weeks of gestation were randomised to receive 1.2 g/day of DHA or placebo until 36 weeks' postmenstrual age. Maternal bleeding requiring treatment or hospitalisation was a prespecified secondary outcome and recorded as a maternal serious adverse event. It occurred in 2 of 232 mothers (0.9%) in the DHA group and 4 of 229 (1.8%) in the placebo group. In addition, one case of haemorrhage was reported in the DHA group as a maternal adverse event. All adverse events and serious adverse events were documented throughout the study period, irrespective of severity or suspected relationship to the intervention.
The Panel notes the lack of methodological detail regarding the assessment of adverse events in most of these studies, the heterogeneity in assessment methods and frequency across studies, and that the reported adverse effects/events were mostly mild and self‐limiting.
Hazard characterisation
3.3
Selection of the critical effect
3.3.1
The critical effect on which the safe level of intake of 1 g/day for supplemental DHA alone was established in 2012 (EFSA NDA Panel, 2012) for all population groups was the risk of spontaneous bleeding.
Whereas long‐term, adequately powered intervention studies were used to establish the safety of supplemental EPA and DHA in combination (up to 5 g/day) and for EPA alone (up to 1.8 g/day), such studies were lacking for DHA alone.
The new studies that have become available are consistent with the previous conclusions of the Panel that changes in platelet function observed at supplemental intakes of DHA alone up to about 4 g/day in mostly small and relatively short‐term intervention studies are not considered adverse, as they are not associated with an increased risk of clinical complications (e.g. spontaneous bleeding). However, the Panel notes that the long‐term effects of supplemental DHA alone on the risk of spontaneous bleeding/bleeding complications, which are rare and complex events, have not been adequately investigated in humans at doses > 1 g/day.
As for other potential adverse effects previously identified in relation to the intake of supplemental EPA and DHA either alone or in combination, the Panel considers that supplemental DHA alone at doses up to 3 g/day do not adversely affect glucose homeostasis, blood lipids, markers of lipid peroxidation or immune function. Data are only available for adults, excluding pregnant and lactating women and individuals with diabetes.
Based on the available evidence, the Panel decided to select risk of spontaneous bleeding as the critical effect on which to base the UL/safe level of intake for supplemental DHA alone.
Derivation of health‐based guidance values
3.3.2
In the absence of adequate data to characterise a dose–response relationship and identify a reference point, no UL for supplemental DHA alone can be established for any population group. Therefore, a safe level of intake will be derived by the Panel (see Section 1.2).
Different from a UL, a safe level of intake is based on data which characterise levels of intake up to which no adverse effects have been observed. A safe level of intake may be based on the highest dose at which no adverse effects are observed in human studies, among others (EFSA NDA Panel, 2024).
Based on the available evidence, the Panel retains the safe level of intake of 1 g/day for supplemental DHA alone established in 2012 (EFSA NDA Panel, 2012) for all population groups (i.e. infants, children, adolescents and adults, including pregnant and lactating women).
This safe level of intake applies to DHA added to foods or consumed as food supplements in any chemical form (e.g. triacylglycerols, ethyl esters, phospholipids) from sources (e.g. fish oil concentrates, algal oils, krill oils) containing DHA alone or mostly DHA (i.e. EPA/DHA ratio < 0.3).
This safe level of intake does not apply to:
- DHA from the background diet (i.e. naturally occurring in food),
- Supplemental DHA from sources (e.g. fish oil, krill oil, algal oil) containing DHA in combination with EPA (EPA/DHA ratio ≥ 0.3)
Risk characterisation
3.4
The Panel notes that the application of safe levels of intake for risk assessment and risk management is more limited than an UL because (EFSA NDA Panel, 2024):
- intakes above the safe level of intake do not necessarily mean that there is an increased risk of adverse effects, and.
- the proportion of people at risk of adverse effects in a population or subgroups thereof cannot be estimated, as the intake level at which the risk of adverse effects starts to increase is not defined.
Therefore, in the absence of a UL, the risk associated with the consumption of supplemental DHA alone cannot be characterised.
CONCLUSIONS
4
The Panel considers that the available data are not sufficient to establish a UL for supplemental DHA alone for any population group.
Based on the data available, the Panel concludes that supplemental intakes of DHA alone up to 1 g/day do not raise safety concerns for the general population and retains the previously established safe level of intake of 1 g/day for all population groups (i.e., infants, children, adolescents and adults, including pregnant and lactating women), as set in 2012.
The safe level of intake applies to DHA added to foods or consumed as food supplements in any chemical form (e.g. triacylglycerols, ethyl esters, phospholipids) from sources (e.g. fish oil concentrates, algal oils, krill oils) containing DHA alone or mostly DHA (i.e. EPA/DHA ratio < 0.3).
ABBREVIATIONSApoBapolipoprotein BADHDattention deficit hyperactivity disorderAIDSacquired immunodeficiency syndromeASAacetylsalicylic acidAUCarea under the curveBMIbody mass indexbwbody weightCRPC‐reactive proteinCVDcardiovascular diseaseDAIDSDivision of AIDS (NIH grading scale)DBdouble blindDHAdocosahexaenoic acidDHAX trialdocosahexaenoic acid in X‐linked retinitis pigmentosa trialDPAdocosapentaenoic acidELISAenzyme‐linked immunosorbent assayEPAeicosapentaenoic acidEPBearly preterm birthFDAFood and Drug AdministrationfMRIfunctional magnetic resonance imagingFOXferrous oxidation‐xylenol orangeFRAPferric reducing antioxidant powerGAgestational ageG‐CSFgranulocyte colony‐stimulating factorGIgastrointestinalGM‐CSFgranulocyte monocyte‐colony stimulating factorGOEDGlobal Organization for EPA and DHA Omega‐3sHbA1cglycated haemoglobinHDL‐chigh‐density lipoprotein cholesterolHIVhuman immunodeficiency virusHOMA‐IRHomeostatic Model of Assessment of Insulin Resistancehs‐CRPhigh‐sensitivity C‐reactive proteinICAM‐1intercellular adhesion molecule‐1INDinvestigational new drugiAUCincremental area under the curveINFγinterferon gammaIL‐1βinterleukin 1 betaIL‐6interleukin 6IL‐10interleukin 10INRInternational Normalised RatioIOMInstitute of MedicineIVGTTintravenous glucose tolerance testLAlinoleic acidLC–MS/MSliquid chromatography–tandem mass spectrometryLCPUFAlong‐chain polyunsaturated fatty acidsLDL‐cLow‐density lipoprotein cholesterolLoEline of evidenceLPBlipopolysaccharide binding proteinMCP‐1monocyte chemoattractant protein‐1MMP‐2matrix metalloproteinase‐2MDAmalondialdehydeMOBIDIckmaternal omega‐3 supplementation to reduce bronchopulmonary dysplasia in very preterm infants trialMRImagnetic resonance imagingNDA PanelEFSA Panel on Nutrition, Novel Foods and Food Allergensn‐3omega‐3OGTToral glucose tolerance testOLopen labelORACoxygen radical absorbance capacityoxLDLoxidised low‐density lipoproteinPGprostaglandinppposterior probabilitiesQUICKIQuantitative Insulin Sensitivity Check IndexRBCred blood cellsRCTrandomised controlled trialREDUCE‐ITReduction of Cardiovascular Events with Icosapent‐Ethyl interventionSAAserum amyloid AsICAM‐1soluble intracellular adhesion molecule‐1SDStandard deviationsRAGEsoluble receptor for advanced glycation end productsTBARSthiobarbituric acid reactive substancesTCtotal cholesterolTEACTrolox Equivalent Antioxidant CapacityTGtriglyceridesTNF‐αtumour necrosis factor‐alphaTNFRtumour necrosis factor receptorTRAPtotal radical‐trapping antioxidant parameterULtolerable upper intake levelVCAM‐1vascular cell adhesion molecule‐1VEGFvascular endothelial growth factorWBCwhite blood cellsXLRPX‐linked retinitis pigmentosa
REQUESTOR
European Commission
QUESTION NUMBER
EFSA‐Q‐2025‐00054
COPYRIGHT FOR NON‐EFSA CONTENT
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
PANEL MEMBERS
Dominique Turck, Torsten Bohn, Montaña Cámara, Jacqueline Castenmiller, Stefaan De Henauw, Karen Ildico Hirsch‐Ernst, Angeles Jos, Alexandre Maciuk, Inge Mangelsdorf, Breige McNulty, Androniki Naska, Kristina Pentieva, Alfonso Siani and Frank Thies.
Supporting information
Annex A – Protocol for the Scientific Opinion on the Tolerable Upper Intake Level for supplemental docosahexaenoic acid
Annex B – Literature searches for the Scientific Opinion on the Tolerable Upper Intake Level for supplemental docosahexaenoic acid
Annex C: Public consultation on the draft scientific opinion on the Tolerable Upper Intake Level for supplemental docosahexaenoic acid
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alicandro, G. , Faelli, N. , Gagliardini, R. , Santini, B. , Magazzù, G. , Biffi, A. , Risé, P. , Galli, C. , Tirelli, A. S. , Loi, S. , Valmarana, L. , Cirilli, N. , Palmas, T. , Vieni, G. , Bianchi, M. L. , Agostoni, C. , & Colombo, C. (2013). A randomized placebo‐controlled study on high‐dose oral algal docosahexaenoic acid supplementation in children with cystic fibrosis. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 88, 163–169. 10.1016/j.plefa.2012.10.002 232662 · doi ↗ · pubmed ↗
- 2Allaire, J. , Couture, P. , Leclerc, M. , Charest, A. , Marin, J. , Lépine, M. C. , Talbot, D. , Tchernof, A. , & Lamarche, B. (2016). A randomized, crossover, head‐to‐head comparison of eicosapentaenoic acid and docosahexaenoic acid supplementation to reduce inflammation markers in men and women: The comparing EPA to DHA (Compar ED) study. American Journal of Clinical Nutrition, 104, 280–287. 10.3945/ajcn.116.131896 27281302 · doi ↗ · pubmed ↗
- 3Allaire, J. , Harris, W. S. , Vors, C. , Charest, A. , Marin, J. , Jackson, K. H. , Tchernof, A. , Couture, P. , & Lamarche, B. (2017). Supplementation with high‐dose docosahexaenoic acid increases the Omega‐3 index more than high‐dose eicosapentaenoic acid. Prostaglandins, Leukotrienes, and Essential Fatty Acids, 120, 8–14. 10.1016/j.plefa.2017.03.008 28515020 · doi ↗ · pubmed ↗
- 4Allaire, J. , Vors, C. , Harris, W. S. , Jackson, K. H. , Tchernof, A. , Couture, P. , & Lamarche, B. (2019). Comparing the serum TAG response to high‐dose supplementation of either DHA or EPA among individuals with increased cardiovascular risk: The Compar ED study. British Journal of Nutrition, 121, 1223–1234. 10.1017/S 0007114519000552 30854986 · doi ↗ · pubmed ↗
- 5Allaire, J. , Vors, C. , Tremblay, A. J. , Marin, J. , Charest, A. , Tchernof, A. , Couture, P. , & Lamarche, B. (2018). High‐dose DHA has more profound effects on LDL‐related features than high‐dose EPA: The Compar ED study. Journal of Clinical Endocrinology and Metabolism, 103, 2909–2917. 10.1210/jc.2017-02745 29846653 · doi ↗ · pubmed ↗
- 6Angoa, G. , Pronovost, E. , Ndiaye, A. B. K. T. , Lavoie, P. M. , Lemyre, B. , Mohamed, I. , Simonyan, D. , Qureshi, M. , Afifi, J. , Yusuf, K. , Sériès, T. , Guillot, M. , Piedboeuf, B. , Fraser, W. D. , Nuyt, A. M. , Mâsse, B. , Lacaze‐Masmonteil, T. , & Marc, I. (2022). Effect of maternal docosahexaenoic acid supplementation on very preterm infant growth: Secondary outcome of a randomized clinical trial. Neonatology, 119, 377–385. 10.1159/000524147 35413719 · doi ↗ · pubmed ↗
- 7Arellanes, I. C. , Choe, N. , Solomon, V. , He, X. , Kavin, B. , Martinez, A. E. , Kono, N. , Buennagel, D. P. , Hazra, N. , Kim, G. , D'Orazio, L. M. , Mc Cleary, C. , Sagare, A. , Zlokovic, B. V. , Hodis, H. N. , Mack, W. J. , Chui, H. C. , Harrington, M. G. , Braskie, M. N. , … Yassine, H. N. (2020). Brain delivery of supplemental docosahexaenoic acid (DHA): A randomized placebo‐controlled clinical trial. e Bio Medicine, 59, 102883. 10.1016/j.ebiom.2020.102883 32690472 PMC 750 · doi ↗ · pubmed ↗
- 8Ayats‐Vidal, R. , Bosque‐García, M. , Cordobilla, B. , Asensio‐De la Cruz, O. , García‐González, M. , Castro‐Marrero, J. , López‐Rico, I. , & Domingo, J. C. (2023). Changes of erythrocyte fatty acids after supplementation with highly concentrated docosahexaenoic acid (DHA) in pediatric cystic fibrosis: A randomized double‐blind controlled trial. Journal of Clinical Medicine, 12, 3704. 10.3390/jcm 12113704 37297899 PMC 10253750 · doi ↗ · pubmed ↗