A co-speciation dilemma and a lifestyle transition with genomic consequences in Wolbachia of Neotropical Drosophila
Konstantinos Papachristos, Wolfgang J. Miller, Lisa Klasson

TL;DR
This study explores the co-speciation and genome changes in Wolbachia bacteria infecting Neotropical fruit flies, revealing unexpected patterns in genome size and evolution.
Contribution
The study identifies a genome expansion in an obligate Wolbachia strain, contradicting genome reduction theories and highlighting unique evolutionary dynamics.
Findings
Wolbachia strains show topological congruence with host mitochondrial DNA but not nuclear DNA.
The obligate wPau strain has a larger genome due to IS4 element expansion, contrary to genome reduction expectations.
The Undecim gene cluster is uniquely duplicated in wPau, suggesting strong selection for these genes.
Abstract
Long-term persistent symbiotic associations may result in co-speciation and can be inferred if species trees of hosts and symbionts are congruent in topology and divergence times. Co-speciation has been seen to occur relatively frequently in obligate associations, but is less common in parasitic or facultative ones, mainly due to the difference in horizontal transmission rates. The long-term vertical inheritance and close host association of obligate endosymbionts also generally result in smaller genomes than in facultative endosymbionts. Here, we investigate co-speciation and genome reduction using highly similar strains of the endosymbiont Wolbachia infecting Drosophila species from the willistoni and saltans groups, where only one strain, wPau, infecting D. paulistorum, is obligate. We sequenced the Wolbachia genomes from five species of the willistoni and saltans groups and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
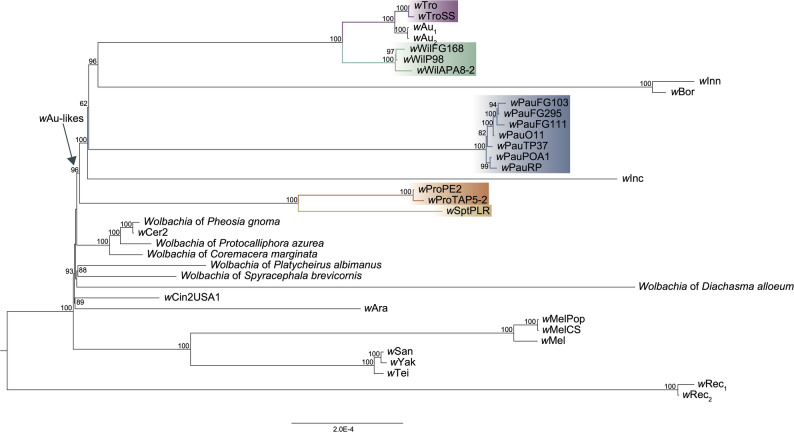 Figure 1
Figure 1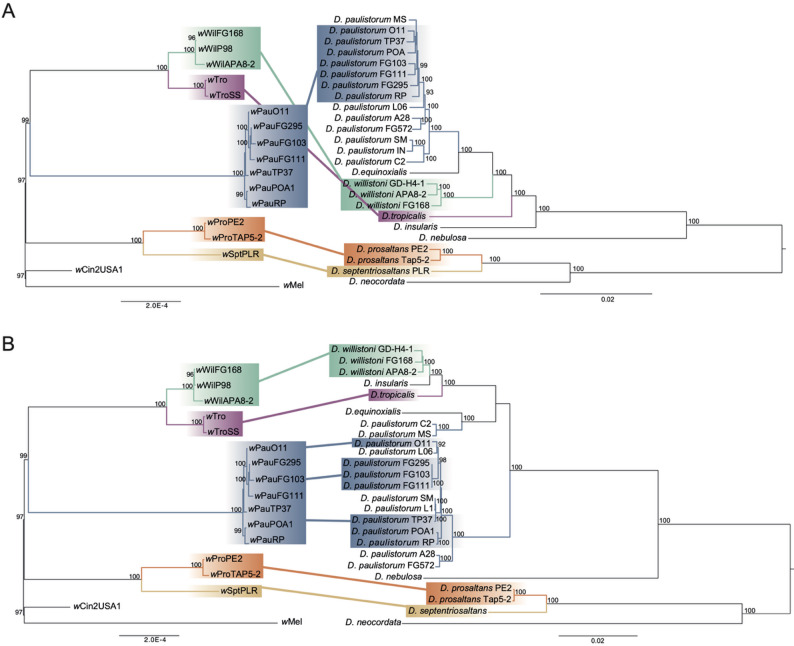 Figure 2
Figure 2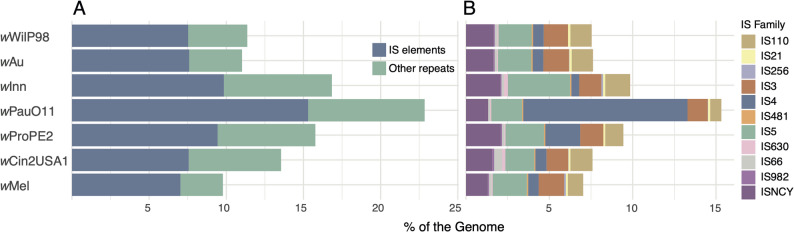 Figure 3
Figure 3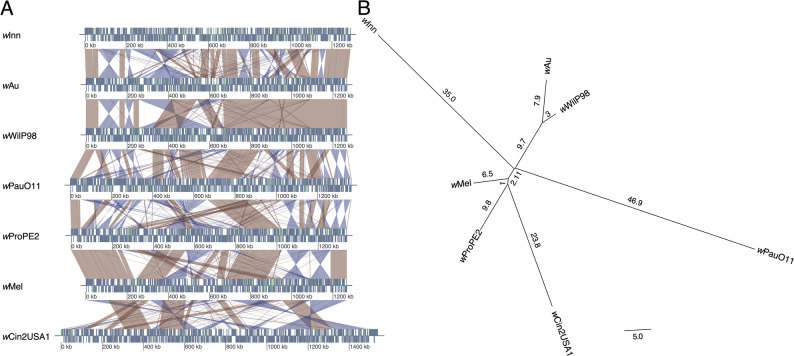 Figure 4
Figure 4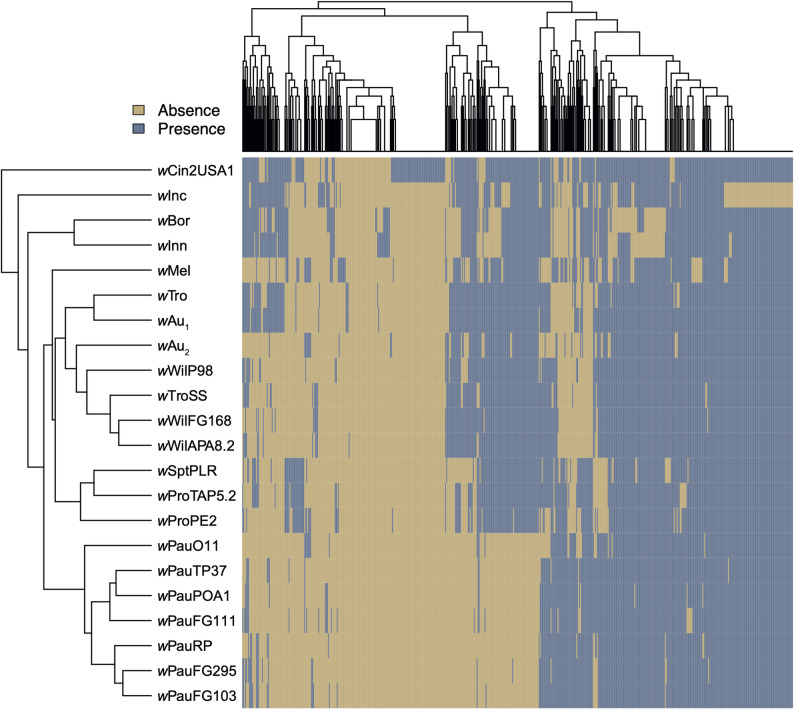 Figure 5
Figure 5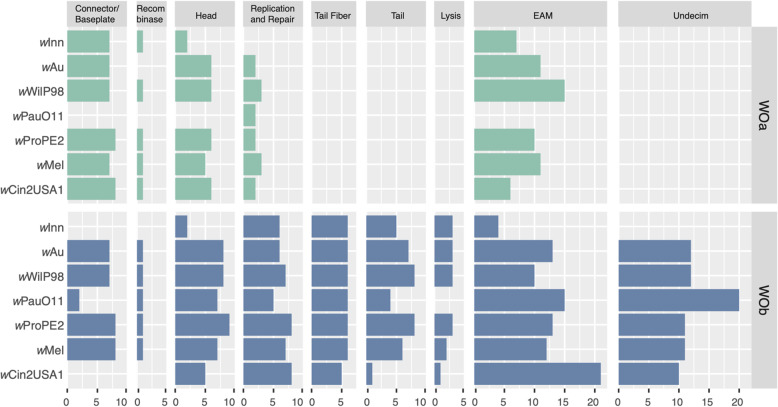 Figure 6
Figure 6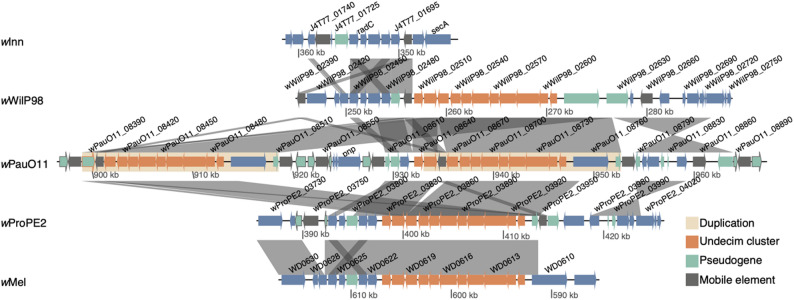 Figure 7
Figure 7- —Uppsala University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect symbiosis and bacterial influences · Trypanosoma species research and implications · Phytoplasmas and Hemiptera pathogens
Background
Bacterial symbionts are ubiquitous among animals but vary widely in phenotypic effects, level of dependency, and evolutionary persistence within host lineages. Mutualistic symbioses often involve tight associations with the host that may become obligate and thereby evolutionary persistent in a host lineage over time. In contrast, parasitic or pathogenic symbionts are usually facultative and evolutionarily short-lived unless they can manipulate their host to ensure their transmission across generations, by so-called reproductive parasitism [1]. Hence, the evolutionary persistence of a symbiont typically suggests that it is either an obligate mutualist or a reproductive parasite. The phenotypic effect, dependency, and persistence of a symbiotic association are thus interconnected and have been shown to influence how symbiont genomes evolve [2, 3]. Numerous studies [2, 4, 5] have demonstrated that obligate symbionts typically have smaller genomes, fewer pseudogenes, repeats, and mobile elements than facultative symbionts. The reduced genome size in obligate symbionts reflects their long-term reliance on the host, with the loss of non-essential gene functions and ongoing deletions facilitated by genetic drift. In contrast, facultative symbionts, which maintain a more transient association with their hosts, retain larger genomes that provide greater metabolic versatility and independence as well as more repeats and mobile elements.
Wolbachia is an intracellular symbiont that infects many different arthropod species as well as some nematodes [6] and forms both facultative and obligate host associations. It is mainly vertically transmitted via the maternal line to the next generation [7] but is also frequently horizontally transmitted between host species [8]. Wolbachia can affect many aspects of its host’s biology, including fecundity, immunity, and behaviour [6]. However, it is best known in arthropods for its ability to alter host reproduction. One such reproductive phenotype is cytoplasmic incompatibility (CI), where infected males are unable to produce offspring with uninfected females. The reproductive advantage thereby gained by infected females over uninfected females can result in the spread of Wolbachia into a host population [9] as well as persistence in a population once the infection has reached a high frequency [10]. Even so, only a handful of examples of evolutionary persistent Wolbachia are currently known. Phylogenetic congruence, which is a hallmark of evolutionary persistence and co-speciation, has, for example, been observed between obligate mutualistic Wolbachia in filarial nematodes [11] and bed bugs [12], but also in non-obligate associations between Wolbachia and Nasonia wasps where Wolbachia induces CI [13] and Nomada solitary bees [14] where the phenotypic effect of Wolbachia is unknown. However, phylogenetic congruence between host mitochondrial DNA and Wolbachia is regularly observed for closely related host species, suggesting that the cytoplasmic co-inheritance of mitochondria and Wolbachia is frequently maintained at least over shorter times [15–19], although exceptions also exist [20].
Wolbachia genomes generally conform to the genome reduction theory. Facultative Wolbachia strains infecting various arthropods have genomes ranging between 1.1 and 1.8 Mb in size [8, 21] with a substantial fraction of mobile elements and other repeats and relatively many pseudogenes. Obligate mutualistic Wolbachia of nematodes have smaller genomes, ranging from 0.5 to 1.0 Mb, with few pseudogenes and a low fraction of mobile elements or repeats [22, 23]. Additionally, prophage WO regions are present in nearly all Wolbachia genomes except in Wolbachia of nematodes, where they are absent or highly degraded [24, 25]. Prophage WO is important in Wolbachia biology as it may contain genes responsible for reproductive parasitism, such as cifA and cifB responsible for CI [26, 27] and wmk involved in male-killing [28], as well as the Octomom region affecting Wolbachia titer and virulence [29, 30]. Furthermore, the Eukaryotic Association Module (EAM) of prophage WO encodes several proteins suggested to play a role in Wolbachia-host interactions [31].
In the Neotropical Drosophila of the willistoni and saltans groups, most tested species carry Wolbachia with high similarity to each other and the well-known wAu strain from Drosophila simulans (hereafter called wAu*-like) [32]. The high frequency of infections and similarity between the Wolbachia strains found in the two Drosophila groups indicate that their ancestor may have been infected with a wAu-*like Wolbachia and that the two partners may have been co-speciating. However, this observation is puzzling, as co-speciation suggests evolutionary persistence and almost all the Wolbachia strains in willistoni and saltans group flies are believed to be facultative [33] and, in some cases, unable to induce CI and/or lack the cif genes [34, 35]. Only the Wolbachia strain wPau infecting D. paulistorum from the willistoni group, a superspecies consisting of multiple reproductively isolated semispecies, is considered obligate mutualistic, as previous results have shown that all collected D. paulistorum flies are infected. Additionally, complete removal of wPau by antibiotic treatment causes lethality and reducing the wPau titer results in distorted ovary development [36], thus affecting fecundity. Moreover, wPau is likely involved in the ongoing host speciation of D. paulistorum as it contributes to assortative mating between different semispecies of D. paulistorum [36], affects pheromone profiles [37] and contributes to the distinct gene expression pattern in heads and abdomens of three semispecies [38]. D. paulistorum is also known to carry two distinct mitotypes, α and β, and multiple mitochondrial introgressions with related Neotropical Drosophila species likely occurred during divergence of its semispecies, as indicated by discordant mitochondrial and nuclear phylogenies [39]. Thus, if a potential mitochondrial donor species carried Wolbachia, wPau could have entered D. paulistorum via introgression.
The Neotropical Drosophila system thus offers the opportunity to investigate evolutionary persistence and co-speciation in non-obligate Wolbachia associations and study genomic differences between very closely related facultative and obligate Wolbachia strains. To do so, we sequenced and assembled Wolbachia genomes from five Drosophila species: D. paulistorum,* D. willistoni* and D. tropicalis from the willistoni group, and D. prosaltans and D. septentriosaltans from the saltans group and inferred whole genome phylogenies. We found congruence between four of five Wolbachia strains and host nuclear genes and full congruence between Wolbachia and host mitochondrial genomes. Additionally, we discovered that the genome of the obligate Wolbachia strain wPau is larger than closely related facultative strains due to the expansion of IS4 elements. However, the wPau genome also has features commonly associated with ongoing genome reduction, like a lower coding percentage and more pseudogenes. Finally, despite an overall reduced prophage WO content, wPau carries intact prophage WO-associated cif genes and a unique duplication of the Undecim cluster, suggesting these genes are under selection and thus potentially important for the symbiosis between wPau and its host.
Materials and methods
DNA extraction and sequencing.
Drosophila lines used in this study are summarized in Table S1. For D. paulistorum O11, total DNA for Oxford Nanopore (ONT) sequencing was extracted from many adult flies of both sexes using the MagAttract HMW DNA Kit (Qiagen) to obtain high molecular weight DNA [39]. For Wolbachia strains wWilP98, wProPE2, wPauFG111, wPauPOA1 and wPauTP37, DNA samples from 20 to 30 embryos used for PacBio sequencing were enriched for Wolbachia using a differential centrifugation and filtration procedure to obtain a Wolbachia cell pellet, which was then subjected to whole genome amplification using the Repli-g midi kit (Qiagen) as described in [40]. For Illumina sequencing, total DNA was extracted from 20 ovaries of 3-day-old adult females as described in [39], except for wWilP98 and wProPE2, for which Illumina sequences enriched for Wolbachia were obtained using embryos and the same protocol as described for PacBio sequencing above.
The ONT library was produced and sequenced using an R9.4 PromethION flow cell at the Uppsala Genome Center, Uppsala, Sweden. Basecalling was performed with Guppy 4.3.4 and the HAC model. Illumina TruSeq libraries were produced and sequenced at the Uppsala SNP and Seq platform on an Illumina 2500 HiSeq machine, generating 2 × 125 bp reads (for total DNA, it is the same sequencing runs as in [39]). The Illumina reads were quality filtered and trimmed using Trimmomatic-0.30 with parameters –phred33 ILLUMINACLIP: illumina_adapter.fasta:2:40:15 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:95 [41] and error-corrected using SPAdes-3.5.0 --sc --only-errorcorrection [42]. For PacBio, 5 kb SMRTbell libraries were created. Each library was run with P6-C4 chemistry in one SMRT cell on an RSII PacBio instrument at Uppsala Genome Center.
Genome assemblies
Wolbachia genomes were assembled using several types of data and pipelines. For wPauO11, reads from total DNA sequencing (i.e. including the Drosophila host) were filtered using Filt-long v0.2.1 with quality priority and the parameters “--target_bases 12500000000 --min_length 1000 --mean_q_weight 10” and assembled with Nextdenovo v2.5.0 using correction options: read_cutoff = 1k, genome_size = 250 m, sort_options = -m 7 g -t 2, minimap2_options_raw = -t 3, pa_correction = 17, correction_options = -p 2 and assemble options: minimap2_options_cns = -t 3, nextgraph_options = -a 1. The produced assembly contained only one Wolbachia contig that was subsequently polished with Illumina reads using Pilon v1.24 [43] with the --fix indels parameter and circularized. The genomes of the two strains wWilP98 and wProPE2 were assembled using Illumina and PacBio reads from amplified DNA enriched for Wolbachia. For these, the Illumina reads were assembled using Spades v3.15.5 [42] with kmer sizes 97,103,107,113 and the PacBio reads were used for scaffolding with the --pacbio parameter. Illumina and PacBio reads were mapped against the assemblies using BWA-mem [44] with default parameters for Illumina reads and using PacBio settings for PacBio reads to check the correctness of the genome assembly. Overlapping contigs were merged in Consed [45]. For all other assemblies, BWA-mem v0.7.17-r1198-dirty with default parameters was used to map Illumina reads on the wPauO11, wProPE and wWilP98 genomes and a set of concatenated Wolbachia genomes representing the Wolbachia diversity (81 genomes from supergroups A, B, C, D, E, F, L and M, Table S2). Mapped reads were filtered using samtools v.1.13 [46] view with the -f 1 -F 12 parameters to include only pairs where both were mapped. Filtered reads were then subsampled to approximately 100X with seqtk v1.3-r117-dirty sample -s 100 before assembly. The reads were then assembled using Spades v3.15.5 with kmer sizes 97,103,107,113, and the wPauFG111, wPauPOA1 and wPauTP37 assemblies were scaffolded with PacBio reads using the flag -pacbio.
The Illumina assemblies were then filtered by removing contigs that deviated from the mean sample distribution in kmer coverage and GC content. The kmer coverage of the contigs was determined from the fasta headers in the output from Spades. In cases where kmer coverage had a bimodal distribution, the minimum between the two peaks was identified using the quantmod package in R [47] and used as threshold. Contigs with kmer coverage below this threshold were blasted against the assembled contigs and were removed if the sequence existed. This procedure ensured that redundant low coverage contigs were removed. Remaining low coverage contigs were blasted against additional Wolbachia genomes and proteins. Contigs with a blast alignment length longer than 100 aa and percent identity over 60%, or blast alignment length longer than 200 nucleotides and percent identity over 70%, were kept. Any contig with an extreme GC value (outside the 99% interval of a normal distribution of GC) was removed. Finally, all remaining contigs were searched with Kraken against a custom database (containing a selection of archaea, bacteria, plasmids, viruses, human, D. melanogaster and D. willistoni sequences) and the contigs with a match to Drosophila were removed. All assemblies were polished once using Pilon with the Illumina reads used for assembly.
Intrastrain variation between wAu-like genomes
To estimate the similarity between the wAu-like strains infecting the same host species, we calculated the Average Nucleotide Identity (ANI) between the genomes using FastANI [48].
Genome annotation
For the complete genomes, wPauO11, wProPE2 and wWilP98, Prokka v1.14.6 [49] with the parameters --genus Wolbachia --compliant --rfam --addgenes --usegenus was used for annotation. Next, Pseudofinder v1.1.0 [50] with parameters -ce and -db with a database containing all Wolbachia proteins from NCBI (2022-03-02), was used to mark potential pseudogenes. Additionally, hmmsearch as implemented in pfam_scan.pl was used for domain prediction with the PFAM database [51]. The Illumina reads used for the assemblies were mapped back to each genome with BWA-mem to verify frameshifts and nonsense mutations. All marked pseudogenes were inspected manually and annotated as pseudogenes if the start or stop codons were missing, they contained nonsense or frameshift mutations or were truncated by, for example, an IS element insertion.
The draft assemblies were annotated the same way, except that (1) Pseudofinder was run with length_pseudo set at 0.8 and the protein database only included Wolbachia genomes wAu, wHa, wMa, wNo, wSan, wTei, wYak, wPauO11, wProPE2 and wWilP98 that were manually annotated either in this or previous studies [40, 52], (2) only pseudogene calls in CDS sequences were inspected manually, (3) pseudogenes that we identified in the complete genomes (wPauO11, wProPE2 and wWilP98), but were not found by pseudofinder in the draft genomes, were transferred to the draft genome annotation if they matched with high similarity and extensive coverage.
For identifying repeated sequences, nucmer from the MUMmer 4.0 package [53] was used with flags --maxmatch --nosimplify to align each genome to itself. The positions of non-overlapping repeat bases were then extracted using a perl script. ISEScan v 1.7.2.3 [54] was used to identify IS elements. Bar plots of the percentage of repeats and IS elements in each genome were made in R [55, 56]. Phage WO regions were annotated by blast searches to Wolbachia genomes (wMel, wAu, wHa, wMa, wNo, wSan, wTei, wYak) with previously identified phage WO regions. All genomic regions that contained similarities to known phage WO genes were manually inspected to verify their borders based on the information in [25].
MLST phylogeny of Wolbachia genomes
Genomes that contained the keyword “Wolbachia” in the taxon name, were downloaded from NCBI using the command line tool “datasets” [57] on May 6th, 2022. tblastn of the Blast v2.7.1 + suite [58] with the parameters --max_hsps 1 --evalue 1e-10 was used to search for the five wMel MLST proteins and Wolbachia surface protein (Wsp) (GenBank accession numbers AAS13898.1, AAS14036.1, AAS14201.1, AAS14415.1, AAS14719.1, AAS14884.1) in 1504 downloaded genomes. Genomes with blast hits for all six genes were kept for phylogenetic inference. Nucleotide sequences of the identified genes were extracted from each genome using samtools faidx, and translated, aligned with MAFFT v6.850b using the linsi algorithm [59], pruned to remove gap sites present in more than 50% of aligned sequences and then backtranslated to nucleotides. A phylogeny of the six genes from 1262 genomes was then constructed using RaxML v8.2.9 [60] with parameters –f a –m GTRGAMMA –x 12,345 –p 12,345 -# 100. The resulting phylogenetic tree and all other figures presenting phylogenetic trees were made using FigTree v1.4.4 [61].
Protein clustering and strain phylogeny
At the time of downloading Wolbachia genomes, the ones from [8] were not yet available. Hence, to include them in our analysis, we downloaded the genomes classified as supergroup A by the authors. We then annotated the genomes from all strains classified as supergroup A Wolbachia in the MLST phylogeny and in [8] as described above for our draft assemblies, used their proteomes for clustering and inferred a phylogeny from single-copy genes. Clustering was done with Orthofinder v2.5.4 [62] with -I 3 and filtered all-against-all blastp results as input. Blastp hits between two matching proteins were filtered so that the size of one should not be smaller than 60% of the size of the other, the e-value should be lower than 10^− 5^ and the alignment length should be at least 80% of the size of the smaller protein. The nucleotide sequences of genes from orthogroups with single-copy proteins were extracted, translated, aligned with mafft-linsi, pruned to remove gap sites present in more than 50% of the aligned sequences and backtranslated to obtain codon-based nucleotide alignments.
To ensure that the genes used in the phylogeny did not show signs of recombination, we ran Phipack [63] with the -p 100,000, -o, -w 50 on all alignments. Phipack runs three tests to detect recombination, the Phi test, the max χ2 test and the NSS test. For a gene to be considered as recombined, at least two of the three recombination tests needed to have a p-value lower than 0.05. Additionally, we searched for signs of intergenic recombination by analysing the topology of the single-copy gene trees. We did this by inferring the gene trees with IQTREE v2.2.0 [64] and calculating pairwise Robinson-Fould (RF) distances between all single-copy genes using the TreeDist package v2.9.2 [65]. The RF distances were used to search for topology clusters, reasoning that non-recombined single-copy genes will form one main cluster, and recombined genes, with alternative topologies, will form smaller clusters. To estimate how clustered the tree topologies were, we calculated Silhouette coefficients for hierarchical, k-means and k-medoids clustering, for 2–10 clusters. Single-copy gene alignments without recombination (which were all, since we found no recombination) were then concatenated with geneStitcher.py from Utensils [66].
IQ-TREE v2.2.0 [64] was then used for phylogenetic inference of the species tree using the concatenated alignments of the single-copy genes, with one partition per gene, and the parameters -m MFP, -B 1000 -T 20 --seed 12,345. Individual gene trees were inferred using the same parameters. The phylogenetic trees were visualized using FigTree v1.4.4. To test if the data fit better with the placement of wPau according to the host nuclear phylogeny rather than our strain phylogeny, we followed [67] and used IQ-TREE v1.6.12 with the same parameters as in the strain phylogeny together with the -z flag to add the two topologies in Newick format and the -wsl and -wpl flags to get the likelihood for each site and gene. The differences in the gene likelihoods between the two topologies were calculated and plotted in R [55, 56]. To measure the gene (gCFs) and site (sCFs) concordance factors, we used the same pruned concatenated phylogeny as in the log-likelihood differences and produced pruned gene trees including the same strains with preserved branch lengths using a Python script and the ete3 library [68]. To calculate the gene and site concordance factors, the pruned species tree was then provided to IQ-TREE with the flag -t, the fasta alignments with -s, the partition file with -p, the gene trees were provided with --gcf and --scf 100 was used.
Substitution rates
Pairwise synonymous substitution rates were calculated with codeml from the PAML v4.8 package [69] using the alignments of the 620 single-copy Wolbachia genes from the ortholog clustering, as well as the alignments of 692 BUSCO genes and 13 mitochondrial genes from Drosophila (the same as in [39]) generated the same way as the Wolbachia alignments (see above). For substitution rate estimates from MCMCTree, we calibrated our phylogeny based on estimates from [70] on the willistoni-saltans group divergence at around 14.2–20.6 million years and the D. willistoni-D. tropicalis divergence at around 4.2–6.1 million years. A rough estimate of 19–21 million years for the age of the tree and a time unit of 100 million years gives a beta value of 156 for the gamma distribution of rates. We used alpha parameters of 1, 5, and 10 for the shape of the distribution to describe scenarios of low, medium, and high mutation rates. The whole alignment of single-copy genes as one partition was given as input (ndata = 0, seqtype = 0), and the GTR substitution model (model = 8) was used with independent rates (clock = 2). Each of the three rate scenarios was run two times to verify convergence of estimates for the chains, for 20,100,000 generations (burnin = 100,000, sampfreq = 1,000, nsample = 20,000).
Gene order phylogeny
Syntenic blocks between the complete genomes listed in Table 1 were obtained using SynChro_linux (January 2015) [71] with a delta value of 2. The output from SynChro was then used for phylogenetic reconstruction with PhyChro [72, 73].
IS4 element and Undecim cluster copy numbers in draft wPau genomes
To estimate the number of IS4 elements and Undecim cluster copies in the draft wPau genomes, we first masked all copies of the IS4 element and one copy of the Undecim cluster in the wPauO11 genome. To do that, we created an IS4 element consensus sequence by blasting (blastn) one IS4 element from wPauO11 against the wPauO11 genome, extracting the sequences for all matches, aligning them with MAFFT v7.490 with flags “--maxiterate 1000” and “--localpair” and building a consensus with Jalview v2.11.4.0 [74]. The consensus IS4 element sequence was then blasted against the wPauO11 genome using blastn and all hits were masked. The Undecim cluster copies were first identified by homology to the Undecim cluster genes in the wMel genome. One of the Undecim cluster copies, at positions 898,960-913,092 in wPauO11, was then manually masked. The Illumina reads used to assemble the draft wPau genomes were then mapped to the masked wPauO11 genome and the IS4 element consensus sequence using BWA-mem v0.7.17-r1198-dirty. Repeated areas were identified from the annotation (see above) and non-repeated regions of the wPauO11 genome were identified using “bedtools complement” and the repeated regions as input. Coverage over the IS4 element consensus sequence and the unmasked Undecim cluster copy was calculated with “samtools coverage” and the coverage of the non-repeated regions of the genome was calculated by getting the average per-base coverage in these regions by using “samtools depth”. The copy numbers of the IS4 element and the Undecim cluster were calculated by dividing the average coverage over these regions by the average coverage over the non-repeated regions of the genome.
SNP phylogeny of wPau genomes
To get a more resolved relationship between the wPau strain variants, we called SNPs for all wPau strain variants from the Illumina reads mapped to the masked wPauO11 genome (see above). Reads with MQ < 1 were filtered out and bcftools was used to call SNPs using the command “bcftools mpileup --threads 6 -Ou -d 1000 -f $genome -b bam.file.list.txt | bcftools call --threads 6 -cv --ploidy 1 -Ob -o variants_cflag.bcf”. To filter low-quality variants, the command “bcftools view --types snps -i ‘QUAL > 30 & INFO/DP > 10 & MQ > 30’” was used. The variants located in non-repeated areas of the wPauO11 genome were extracted using “bcftools view” with the coordinates identified as described above and “bcftools consensus” was used to create pseudo-chromosomes for each strain. The sequence for each pseudo-chromosome was put into a multi-fasta file and used as input for IQ-TREE v2.2.0 using the same parameters as in the Wolbachia strain phylogeny. The phylogeny was visualized using FigTree v1.4.4.
IS4 element insertion dynamics
To detect differences in IS4 insertions between the wPau genomes, we identified improperly mapped read pairs in the Illumina reads that were mapped to the masked wPauO11 genome (see above). If an IS4 element is present in the genomes with mapped reads, the insert size of paired reads in this region is 0, as one read of the pair would map on the masked Wolbachia genome and the other read would map on the consensus IS4 sequence. To get such improperly paired reads, we filtered with samtools view using parameters -F2, MQ > 0 and -e “tlen = = 0”. Reads that mapped within 750 bases upstream of the start or 750 bases downstream of the end of an IS4 element in wPauO11 (as specified in a provided bed file) are from an IS4 element shared with wPauO11. Reads mapping at other places are associated with IS4 insertions not present in wPauO11, and they were filtered by using samtools view with the -U -L flag and the same bed file as above. If an IS4 element is present in wPauO11 but absent in the genome with mapped reads, the reads from a pair are further apart than expected, specifically the insert size would appear to be larger than the ca. 1500 bases of the IS4 element, and both reads of the pair should map on the masked Wolbachia genome. To get such improperly paired reads, reads mapping 750 bases upstream and 750 bases downstream of IS4 elements (as above) and that had an insert size larger than 1500 bases but shorter than 5000 bases were filtered using samtools view -e “(tlen > 1500 && tlen < 5000) || (tlen>−5000 && tlen < −1500)”. The filtered read alignments of improper pairs were visualized in IGV (v2.16.0) to identify unique IS4 element insertions across the different wPau strain variants. The wPau whole genome SNP phylogeny was then used to infer where and when IS4 copies were inserted in the wPau strain variants. Because complete deletions of IS4 elements occur rarely, differences were explained by insertions, rather than deletions.
Additionally, we inferred the number of IS4 element copies present at each node of the tree using complete Wolbachia genomes. We identified the shared IS4 elements across the wAu-like strains, wMel and wCin2USA1 by blasting (blastn v2.7.1+) genomes to each other and visualizing the regions flanking IS4 elements, identified by ISEscan, with the Artemis comparison tool (ACT) [75]. An IS4 element was considered absent from a genome if a region had the same gene order as an IS4 element-containing region of another genome but didn’t contain an IS4 element or if an IS4 element-containing region in other genomes shared no synteny with the target genome.
Gene content variation between genomes wAu-like Wolbachia
To compare gene content across the different Wolbachia genomes, we used the gene count output of Orthofinder to produce a hierarchical clustering of the genomes based on gene presence/absence. First, we excluded all single-copy genes and then we assigned the count of multi-copy genes to 1 so that we could cluster based on presence and not copy-number. Then, we used the “agnes” function for agglomerative clustering with the “average” method from the cluster v2.1.4 package in R [76].
Additionally, the variability in gene content between the wAu-like_ws_ genomes was investigated by identifying orthogroups where only one or several of the lineages, wPau, wTro/wWil and wPro/wSpt, were present and one or several lineages were absent. Unless otherwise stated, presence was counted if a gene was present in the complete genome of the lineage (wPauO11, wWilP98 and wProPE2) and at least one draft genome of the same lineage.
All figures presenting genome and gene comparisons were made with genoPlotR [77], after using blastn to search each genome against every other genome.
Analyses of the cif genes
To identify the cif genes, we looked for proteins that clustered with the CifA and CifB proteins from wMel in Orthofinder. Additionally, we searched for cif genes in all the wAu-like genomes that may have been missed in the annotation by using tblastn with CifA and CifB of Types II-V from Martinez [35] with -evalue 0.000001. The proteins in the same ortogroups as the Cif proteins in our clustered proteomes and the different Cif Types from Martinez et al. 2021 were aligned with MAFFT. The protein alignment was then backtranslated to nucleotides to produce a codon alignment. The nucleotide sequences of pseudogenes found by blast or annotation were then added to the codon alignment with MAFFT using the --add and --reorder flags. Phylogenies for cifA and cifB were inferred using IQTREE2 with -m MFP -B 1000 --seed 12,345. Finally, the Cif proteins from wPauO11 and wProPE2 were searched for protein domains using HHPred [78] with default parameters.
Prophage WO classification and gene module assignment
To investigate the prophage WO content in detail, we identified prophage WO regions in the complete wAu-like genomes, wPauO11, wProPE2, wWilP98, wInn, wAu, and in wMel, wCin2USA1. All proteins of prophage WO origin were re-clustered to orthogroups with OrthoFinder v2.5.4 with -I = 1 and custom blastp (as above), which put most phage WO paralogues into single orthogroups. We manually validated the orthogroups based on the functional annotation and gene order in complete genomes. The orthogroups containing proteins with the same functional annotation that were syntenic were manually merged. For each orthogroup, we inferred a phylogeny using IQ-TREE v2.2.0 as described for single-copy genes. Genes were assigned to WOa and WOb based on whether they formed a clade with either WOMelA or WOMelB genes and/or were syntenic with WOMelA or WOMelB. For orthogroups where a WOMelB gene was present, we followed the classification of [25]. Otherwise, we looked at what module the neighbouring genes belonged to and assigned the orthogroup to that module. If an orthogroup had no functional annotation and could not be assigned to a module using the above method, it was assigned to the EAM module if flanked by genes in a structural module on one side and an EAM gene on the other side.
Results
We assembled three complete (wPauO11, wProPE2 and wWilP98) and eleven draft genomes (Table 1, Table S3a) from Wolbachia strains infecting three different species of the Drosophila willistoni group: seven wPau strain variants from the Orinocan (OR) semispecies of D. paulistorum (as determined by the host phylogeny), three wWil strain variants from D. willistoni, one wTro strain variant from D. tropicalis, and two species of the Drosophila saltans group: two wPro strains variants from D. prosaltans and one wSpt strain variant from D. septentriosaltans. We refer to each strain variant by the Wolbachia strain name followed by the Drosophila line it was sequenced from, i.e., wPauO11 is the wPau strain variant from the D. paulistorum line O11. Strain variants infecting the same host species had low sequence variation, with average nucleotide identity (ANI) values from 99.85% for the wTro strains and up to 99.99% for two of the wWil strains (Table S3b).
Table 1. Genomic features in complete wAu-like and outgroup genomeswPauO11^a^wWilP98^a^wProPE2^a^wAu^b^wInn^c^wMel^d^wCin2^e^Genome size(bp)1,365,1981,268,5271,339,4661,268,4551,290,5871,267,7821,538,351GC (%)34.835.235.235.235.335.235.1CDSs100010161033996107310111473% Coding73.877.576.375.770.176.086.2Pseudogenes177123130122227111216Repeats (%)22.911.415.811.016.89.813.6Phage WO (%)8.810.812.610.69.09.524.3IS elements (%)15.37.59.47.69.97.07.6^a^Wolbachia genomes from Neotropical Drosophila sequenced in this study ^b^[52], ^c^[79], ^d^[80], ^e^[81]
Co-speciation between Wolbachia and their Neotropical Drosophila hosts
To determine if Wolbachia co-speciated with their willistoni and saltans group hosts, we started by producing a phylogeny of our new genomes and other closely related Wolbachia genomes. To achieve that, we downloaded all publicly available Wolbachia genomes and inferred a phylogeny using the five Multi-Locus Strain-Typing (MLST) genes [82] and the wsp gene. We determined 23 of the 1613 downloaded genomes to be closely related to ours (Table S4) and clustered their proteomes with the proteomes of our 14 genomes. The resulting 620 clusters with single-copy orthologous genes, all without detectable recombination, were used to produce a phylogeny.
The concatenated gene alignment contained only 0.55% parsimoniously informative sites (3,088 of 562,755 sites), showing the very close relationship between all 37 strains. Despite this, the inferred phylogeny had high support for a clade including the five Wolbachia strains from the willistoni and saltans Drosophila groups, wTro, wWil, wPau, wPro and wSpt (Fig. 1). This clade, which also contained Wolbachia strain wAu from the African host D. simulans, further included Wolbachia strains wInn, wBor and wInc, which infect Drosophila species of various, distantly related, American species groups (quinaria, virilis, and flavopilosa, respectively). Thus, from here on, these nine highly similar Wolbachia strains are what we refer to as wAu-like, and the strains infecting willistoni and saltans group flies are referred to as wAu-like_ws_. Finally, as eight of the nine Drosophila hosts are American species, we assume that wAu-like strains evolved in the Americas and have just recently infected the African host D. simulans following its human-mediated introduction to the continent.Fig. 1. Phylogeny of *Wolbachia *genomes with high similarity to wAu-like_ws_ strains. The branches with Wolbachia strains associated with Drosophila species from the willistoni and saltans groups, i.e., the wAu-like_ws_ Wolbachia strains, are coloured. The clade referred to as the wAu-like is indicated with an arrow. The tree was inferred from an alignment of 620 single-copy orthologs with IQ-TREE. Support values from 1000 ultrafast bootstrap replicates are displayed on the nodes of the tree. Only bootstrap values greater than 60 are shown
The concatenated gene alignment of these nine wAu-like genomes contained only 0.3% informative sites and 13.2% of the genes had no informative sites at all. Even so, inside the wAu-like clade, all strain and most strain variant relationships, except the position of wPau (bootstrap 62), were highly supported (Fig. 1).
Thus, to evaluate the position of wPau, we calculated the gene (gCF) and site concordance factors (sCF) for the internal branches of the subtree that only included one representative genome (wPauO11, wWilP98, wTro, wProPE2 and wSptPLR) of each wAu-like_ws_ strain, which are the focus of the co-speciation analysis. The concordance factors represent the percentage of gene trees, gCF, or informative sites, sCF, that support a split in the tree over all other possible splits. After removing single-copy genes without any informative sites, we found that almost all informative sites (99.6–100%) place wPauO11 between the wWil/wTro and wPro/wSpt clades and most of the genes (76.1–81.2%) also agree with this placement (Figure S1). Thus, the position of wPauO11 in our inferred phylogeny had strong support from informative sites and a majority of the individual genes. Furthermore, the low bootstrap value for the split between wPau and other strains only occurred when the wInc strain of D. incompta was included in our phylogenetic analysis. When wInc was excluded, the wPau strain variants were in the same position in the tree but with high bootstrap support (100) (Figure S2). Since the focus of this study is the topology of the wAu-like_ws_ Wolbachia strains, we also inferred phylogenies using only those genomes and an outgroup. The resulting phylogeny confirms that the position of wPau is supported (bootstrap 99) with respect to the other wAu-like_ws_ Wolbachia strains (Fig. 2, left part).Fig. 2. Phylogenetic comparison between wAu-like *Wolbachia *and *Drosophila *of the willistoni and saltans groups. A Maximum Likelihood Phylogenies of wAu-like Wolbachia on the left and 692 nuclear-encoded proteins of the host on the right (from [39]). The wAu-like Wolbachia phylogeny is a subset of the tree in Fig. 1. B Maximum Likelihood Phylogenies of wAu-like Wolbachia on the left (same as in A) and the whole mitochondrial genome of the Neotropical Drosophila host species on the right (from [39])
When comparing our inferred wAu-like_ws_ strain phylogeny to the nuclear phylogeny of the willistoni and saltans group hosts [39], we saw that the trees were congruent except for the position of wPau and D. paulistorum (Fig. 2A). Rather than being positioned inside the clade of the other two willistoni group strains, wWil and wTro, wPau is positioned between the willistoni and saltans group (wPro and wSpt) strains. However, full congruence was observed when comparing the Wolbachia tree with the phylogeny of mitochondrial genomes from the same Drosophila species (Fig. 2B) [39].
To test if our Wolbachia data fit better with the host nuclear or mitochondrial topologies, we calculated the log-likelihood difference per gene between topologies congruent with either the mitochondrial or the nuclear phylogeny. We found that most Wolbachia genes strongly supported the mitochondrial-like topology, and a few genes weakly supported the nuclear-like topology (Figure S3). Overall, our results indicate that the wAu-like_ws_ Wolbachia strains are highly congruent with the host mitochondrial phylogeny, and partly congruent with the nuclear phylogeny of the hosts.
For co-speciation to be true, the divergence times of the hosts and Wolbachia strains should also match (referred to as temporal congruence). To get an idea about temporal congruence, we calculated relative pairwise synonymous substitution rates of wAu-like_ws_ Wolbachia and host genes (Table S5) and compared them to previously described cases of Wolbachia co-speciation in Nasonia and Nomada [13, 14] and horizontal transmission in the D. suzukii subgroup [83]. The relative substitution rates ranged from 0.00025 for Wolbachia in the D. suzukii subgroup to 0.31 for Wolbachia of Nasonia (Table 2), with wAu-like_ws_ Wolbachia in between (0.0025–0.019).
Table 2. Evolutionary rates of WolbachiaCompared Wolbachia strainsdS ratioWolbachia/Host nuclearHost divergence time (MY)^a^Mutation rate (substitutions/synonymous site/year)wWil - wPau0.0194.35.7 × 10^− 10^wTro - wWil0.00365.11.2 × 10^− 10^wPro - wSpt0.0069--wPau - wPro0.002817.41.3 × 10^− 10^wWil - wPro0.002517.41.2 × 10^− 10^wSuz - wSpc^b^0.000254.07.6 × 10^− 12^wNlonB1 - wNgirB^c^0.310.49.0 × 10^− 9^wNleu - wNfla^d^0.0150.34.0 × 10^− 10^wNleu - wNpa^d^0.0520.47.0 × 10^− 10^wNfla - wNpa^d^0.0550.47.0 × 10^− 10^wNfe - wNleu^d^0.132.41.0 × 10^− 9^wNfe - wNfla^d^0.132.41.0 × 10^− 9^wNfe - wNpa^d^0.152.41.1 × 10^− 9^^a^Host divergence times of all Drosophila species are from schemeA in [70]. dS ratios from Wolbachia of the D. suzukii subgroup, ^b^[83], dS values and host divergence times of Nasonia from, ^c^[13], and Nomada from, ^d^[14]
We also calculated mutation rates for these Wolbachia strains by dividing the pairwise synonymous substitution rates by the divergence times of their hosts (if available). The estimated mutation rates for wAu-like_ws_ Wolbachia were between 1 × 10^− 10^ to 5 × 10^− 10^ synonymous substitutions per synonymous site per year, which is in the same range as some of the Wolbachia in Nomada, but again higher than in Wolbachia from the D. suzukii subgroup, where Wolbachia was suggested to be horizontally transmitted and lower than the ones in Nasonia, where Wolbachia is suggested to co-speciate with their hosts (Table 2, Table S5). Hence, the evolutionary rates of wAu-like_ws_ did not match those from previously suggested examples of Wolbachia co-speciation or horizontal transmission.
Additionally, we estimated the mutation rate of wAu-like_ws_ Wolbachia in a Bayesian framework using host divergence times for the willistoni-saltans groups and D. willistoni – D. tropicalis [70] as priors. This resulted in a mutation rate for wAu-like_ws_ Wolbachia of 2.6–6.8 × 10^− 11^ substitutions per site per year (Table S6), which is one order of magnitude smaller than the rate based on synonymous substitutions.
Repeat expansion and rearrangements in the genome of obligate strain wPau.
Contrary to the expectations for an obligate strain, the wPauO11 genome was larger than all other wAu-like Wolbachia genomes (Table 1), had a lower coding content and 25–50% more repeats than both the genomes of wAu-like Wolbachia and two closely related facultative outgroup strains, wMel and wCin2USA1 (Table 1; Fig. 3A). Specifically, we discovered a unique and extensive expansion of an IS4 element, which accounted for 9.9% of the total genome size of wPauO11 (Fig. 3B).
Fig. 3. Repeats and IS-elements in complete genomes of wAu-like strains. A Percentage of repeats in each genome. B Percentage of each IS family in the genomes, as identified by ISEscan
Since repeats act as templates for genomic rearrangements via intragenomic homologous recombination, we examined gene order differences between our closely related genomes. From a whole genome alignment, we saw that the wPauO11 genome exhibited many changes in gene order when compared to other wAu-like genomes (Fig. 4A). To quantify this, we identified pairwise syntenic blocks between all genomes and used this information to construct a phylogeny (Fig. 4B). In the resulting tree, wPauO11 was on the longest branch, indicating that it has experienced more genomic rearrangements than the other Wolbachia genomes.
Fig. 4. Genomic rearrangements in wAu-like strains. A Whole genome alignments of complete Wolbachia wAu-like genomes and outgroup. For each genome, genes are visualized with blue and pseudogenes with green rectangles on either the forward or reverse strand (above and below the midline respectively). Similarities between genomes are visualized as red and blue ribbons, with red representing forward and blue reverse hits. B Gene order phylogeny of the same genomes as in A. Branch lengths written on each branch represent the estimated number of genomic rearrangements
These findings highlight that the wPauO11 genome differs significantly from the genomes of other wAu-like_ws_ strains in several ways, such as having a high proportion of non-coding DNA and repeats and frequent genomic rearrangements. Interestingly, a similar pattern, except for the large repeat expansion, is also seen in the genome of the wAu-like strain wInn from D. innubila.
IS4 expansion is still ongoing in the wPau genomes
Given the significant expansion of IS4 elements in wPauO11, we investigated whether these elements are still actively transposing in wPau genomes. By identifying all complete and incomplete copies in the wPauO11 genome, we estimated that it originally had at least 96 complete IS4 elements (Figure S4). To quantify IS4 elements in all other wPau genomes, we masked all but one copy in the wPauO11 genome, mapped the Illumina reads of each wPau strain to it and calculated the coverage over the IS4 element relative to non-repetitive regions. Applying this approach to Illumina reads from wPauO11, we estimated that this genome harbours 98 IS4 copies, which closely matched our observation of 96 copies. When extended to other wPau variants, the analysis revealed variability in copy numbers of the IS4 element across genomes (95–124 copies/genome) (Table S7). Additionally, by examining improper read pairs, we identified the positions of IS4 element insertions that occurred after the divergence of the wPau strain variants (Table S8). To get a better resolution of the wPau strain variant relationships and thus be able to calculate the number of insertions on each branch more correctly, we inferred a phylogeny based on whole genome SNPs between the wPau variants (Figure S5). Using this tree topology, we identified seven IS4 element insertion events that occurred after the divergence of the wPau strain variants (Table S8, Figure S6). Hence, both read mapping approaches indicated that the IS4 element is still actively transposing in these wPau genomes.
Next, we approximated the rate and timing of IS4 element insertions in all complete genomes of the wAu-like clade. Using copy number counts, synteny comparisons and our phylogeny, we estimated that the genome of the ancestor of all wAu-like strains had four IS4 elements (Table S9) and the number of IS4 element insertions in all non-wPau genomes to be 22 (Figure S6). Using branch lengths as an approximation of time, we calculated a rate of 6,571 insertions/(substitution/site) for the non*-w*Pau genomes. We inferred 88 IS4 element insertions on the branch leading up to the ancestor of the wPau variants by estimating a minimum of 93 IS4 element copies in the genome of the last common ancestor of wPau. This gave us a rate of 123,422 insertions/(substitution/site). The seven IS4 element insertions in the wPau strain variants gave us an estimated rate of 83,333 insertions/(substitution/site). The insertion rate of IS4 elements is thus 18.8 times greater in the branch leading to the wPau ancestor and 12.7 times greater in the current wPau than in the other analysed Wolbachia strains (Table S10). Our analyses thus showed that the IS4 element insertion rate was slightly higher in the wPau genome in the past than it is today, but expansion is still ongoing with a relatively fast rate.
Major loss of prophage WO genes, but retention of CI genes and duplication of the Undecim cluster in wPau
To explore which genes might be linked to phenotypic differences among wAu-like_ws_ strains, we clustered the genomes based on the presence and absence of genes. Our analysis revealed that the genomes of wPro and wSpt clustered together, as did those of wWil, wTro, and wAu, consistent with their phylogenetic relationships. However, the gene content of wPau was notably distinct from the other wAu-likews genomes (Fig. 5).
Fig. 5. Gene content clustering of Wolbachia wAu-like genomes. The cladogram on the left shows how the Wolbachia strains cluster based on the presence and absence of orthogroups in each genome, which is depicted in the plot to the right
We analysed the variation in gene content between the wAu-like_ws_ strains further, by identifying which genes were uniquely present or absent in the three lineages, wPau, wWil/wTro and wPro/wSpt. Our results showed that wPau (defined as wPauO11 and at least one wPau draft genome) lacked many genes compared to the other wAu-like_ws_ strains (Fig. 5, Table S11), rather than having unique ones (Table 3, Table S12). Most missing genes were from prophage WO regions (Table 3), as suggested by the lower proportion of prophage WO sequences in the wPauO11 genome (Table 1). Of the 39 missing prophage WO-associated genes in wPau, most encoded structural phage WO proteins, such as tail, head, or baseplate proteins, whereas only six belonged to the Eukaryotic Association Module (EAM).
Table 3. Gene content variation between wAu-like_ws_ Wolbachia. The numbers represent the presence or absence of an orthogroup in one lineage compared to the other two lineages in the tablewPauwWil/wTrowPro/wSptAbsent (total)661812Phage WO396-Ankyrin212Hypothetical933Dozen island-4- Present (total)
6
11
16 Phage WO-56Ankyrin1-1Hypothetical462Dozen island--6
Given the large number of missing prophage WO genes in wPauO11, we wanted to determine if they all came from the same prophage WO copy. The prophage WO regions identified in the complete wAu-like genomes were highly similar to either the WO-A or WO-B regions in wMel and are referred to here as WOa and WOb, respectively. By classifying the prophage WO proteins into WOa- and WOb-like, we found that WOa-like prophages are generally more degraded than WOb-like prophages in all genomes, with the Lysis, Tail, and Tail fiber modules of the former entirely missing in all wAu-like genomes as well as in wMel and wCin2USA1 (Fig. 6).Fig. 6. Prophage WO genes in complete wAu-like genomes and outgroup. Number of genes per module for the two prophage WO variants identified in wAu-like and related genomes. WOa genes were found in the same clade as genes from WO-A in wMel and WOb genes were found in the same clade as genes from WO-B in wMel when inferring single gene phylogenies. Annotations and assignment of orthogroups to prophage WO modules can be found in Table S13
However, in wPauO11, the WOa-like prophage was almost completely absent, except for a few genes within the Replication and Repair module. The WOb-like prophage also displayed a reduced gene contentin wPau, particularly in the Connector/Baseplate module. Conversely, the wInn genome, which also has a low proportion of prophage WO, retained all Baseplate/Connector genes in WOa but instead lost them in WOb. The presence of genes associated with both WOa and WOb in wPau and other complete wAu-like genomes suggests that intact copies were likely present in their common ancestor. Thus, wPau has predominantly lost genes from the WOa copy, but both prophage WO copies have experienced gene loss.
Apart from prophage WO genes, nine ankyrin repeat domain-containing genes (ANKs) and nine hypothetical proteins were absent from all wPau strain variants but present in the wWil*/wTro and wPro/w*Spt lineages. Of the remaining 11 protein clusters absent in all wPau, functions varied and included two non-WO phage proteins, a Fic protein, Type IV secretion system protein VirB6, Malonyl-CoA decarboxylase, Cytochrome d ubiquinol oxidase, subunit I and Exopolysaccharide synthesis ExoD-related protein. All missing protein clusters in wPau were found in the genomes of one or several of the non-wAu-like_ws_ strains, indicating that they have likely been lost in wPau rather than gained in the other wAu-like_ws_ lineages.
Much fewer protein clusters were uniquely absent in the other two wAu-like_ws_ lineages. Twelve clusters were missing from the wPro and wSpt genomes, and 20 were absent in wWil and wTro (Table 3). Noteworthy, the wWil/wTro lineage lacks the prophage WO-associated genes responsible for CI, cifA and cifB, as previously observed [35]. All other wAu-like Wolbachia carry Type I cif genes with high similarity to wMel (Figure S7 and S8). However, we found that the cifB gene contains a frameshift mutation in the wSpt genome, likely rendering it non-functional (Figure S7). Furthermore, the wBor and wInn strains have identical frameshift mutations in cifB and non-sense mutations in cifA, while wInc has a non-sense mutation in cifB but an intact cifA [35]. Apart from these Type I genes, no other cif genes were found in the wAu-like_ws_ strains, but Type V-like genes were found in wBor and wInn. However, none of those contain the protein domains conserved across cifs from known CI-inducing strains [35]. Thus, the only wAu-like strains that potentially can induce CI are wPro and wPau. These strains carry cif genes encoding proteins differing in only four and nine aa’s, respectively, compared to the proteins in wMel, and both strains also contain the domains previously detected in functional Type I cif genes. Since highly similar Type I cif genes are found in a syntenic region in all wAu-like genomes except in one clade (wTro/wWil/wAu), the common ancestor of wAu-likes likely carried Type I cif genes (Figure S7B). Hence, our results indicate that the cif genes have been lost or pseudogenized independently multiple times in the wAu-like clade (Figure S7).
Only six protein-coding genes, encoding four hypothetical proteins, one ANK and a ComF family protein, were present in wPauO11 (and at least one other draft wPau genome) but absent in the other wAu-like_ws_ strains (Table 3). Potentially, one of the hypothetical proteins and the ANK gene were acquired by wPau, since no homologs were found in any other wAu-like genome. Additionally, one of the hypothetical proteins is homologous to WD0462 in wMel, which correlated with CI induction in another set of Wolbachia strains [52]. However, since the gene is pseudogenized in wPro, a strain known to induce CI, the correlation was not seen among our strains. Finally, the ComF family protein, with a potential role in DNA uptake, is pseudogenized in the other wAu-like_ws_ genomes.
More genes are uniquely present in the wWil/wTro and the wPro/wSpt lineages. For example, homologs of WD0463, the neighbouring gene of WD0462, are only intact in wPro/wSpt. This genomic region has additional variability between wAu-like genomes, with wPau and wPro carrying an extra gene between WD0462 and WD0463 (Figure S9). Hence, this region is highly variable between Wolbachia genomes [52], even among these very close relatives, suggesting that it might be under diversifying selection. Additionally, we noted that six genes in wPro and five in wSpt from the Dozen Island, a genomic region likely stemming from an integrated plasmid [52, 84], were not present in wPau or wWil/wTro. An additional four genes of the Dozen Island were found in wPro/wSpt and wPau but were lost from the wWil/wTro lineage.
Finally, although the loss of prophage WO genes appeared to be frequent in the wPau genome, we identified a large and unique duplication of a prophage WO-associated region in wPauO11 containing ten (of which one is pseudogenized in one copy) of the eleven genes in the Undecim cluster [25] (Fig. 7).
Fig. 7. Duplication of the phage WO-associated genes in the Undecim cluster in wPauO11. Similarities between genomes are visualized with grey bands and the duplication is marked in yellow. Genes in orange are part of the Undecim cluster. Mobile elements are dark grey and pseudogenes green, with the border colour indicating Undecim cluster or mobile element
To determine if the duplication was present in the other wPau strain variants, we estimated the copy number by mapping reads from those genomes to the wPauO11 genome with one copy masked. Coverage over the Undecim cluster was twice as large as coverage of non-repeated sequences for wPauFG111, wPauPOA1 and wPauTP37 (Table S7), indicating duplication of Undecim in those genomes. For wPauFG103, wPauFG295 and wPauRP, coverage over the Undecim cluster was more like non-repeated sequences. However, for those lines, the standard deviation of coverage for non-repeats was as much as 30% of the mean, compared to 10% for the other lines. This suggested that non-repeat coverage may be noisy for some samples, and we thus plotted the coverage of reads from all strains around and over the Undecim cluster of wPauO11 (Figure S10). Since the coverage was approximately doubled compared to the surrounding single-copy regions for all wPau strain variants, the same genomic region is likely duplicated in all sequenced wPau genomes.
Discussion
Neotropical Drosophila species of the willistoni and saltans groups were previously shown to carry closely related Wolbachia strains with high similarity to the wAu strain from D. simulans [32]. Using 14 new Wolbachia genomes from five Neotropical Drosophila species, we confirm that these strains are highly similar and belong to a monophyletic clade including the strain wAu – i.e., the wAu-likes.
The wAu-like Wolbachia show patterns of co-speciation and horizontal transmission
To investigate the possibility of co-speciation between the wAu-like Wolbachia strains infecting willistoni and saltans group flies, wAu-like_ws_ strains, and their hosts, we inferred a phylogeny and compared it to the nuclear and mitochondrial phylogenies of the hosts [39]. We found that the phylogeny of wAu-like_ws_ strains was congruent with the nuclear phylogeny of the host species, except for the Wolbachia strain wPau and its host D. paulistorum. Complete congruence was, however, found between the topologies of the two maternally transmitted genetic entities, Wolbachia and mitochondria. Hence, based on phylogenetic congruence, it seems plausible that a wAu-like strain was present in the ancestor of the willistoni and saltans groups at their divergence ca. 20 million years ago [70, 85] and that this strain has co-diverged with the current hosts ever since. However, phylogenetic congruence can be achieved in the absence of co-speciation, a phenomenon referred to as pseudo-co-speciation, which is especially common if host switching preferentially occurs between closely related species [86]. Thus, to infer true co-speciation, temporal congruence between the two compared entities should be observed. However, since no reliable estimate of the mutation rate exists for any Wolbachia strain, dating the nodes in our tree will be highly unreliable. Nevertheless, to attempt to investigate temporal congruence, we calculated the synonymous substitution rate of wAu-like_ws_ strains relative to that of their hosts and compared that to previously reported cases of Wolbachia-host co-speciation. We found the relative rates in our dataset to be quite constant between different Wolbachia-host pairs, with D. paulistorum - D. willistoni and their Wolbachia having the highest ratio, around 3–7 times higher than the others. Even so, all rates were between one and two orders of magnitude lower than in previous examples of co-speciation [13, 14, 83]. Thus, if all suggested co-speciation examples were true, the wAu-like_ws_ strains either have evolved more slowly than other co-speciating Wolbachia strains or the willistoni and saltans group flies have evolved faster than estimated in previous studies. However, we don’t know if the earlier reported cases represent true examples of co-speciation, since neither has proven temporal congruence. Additionally, mutation and substitution rates as well as generation times can vary widely between different organisms, even between very closely related bacterial and animal species. Hence, we also calculated mutation rates of Wolbachia, based on the synonymous substitution rates and the substitution rate in single-copy genes, assuming that Wolbachia divergence times are congruent with host divergence times. Using synonymous substitutions, we got mutation rates between 10^− 10^–10^− 8^ substitutions per site per year and using all substitutions in single-copy genes, we got even lower estimates of the mutation rate of 3–7 × 10^− 11^ substitutions per site per year. Both rates are lower than the estimated mutation rate in other bacteria [87] as well as in Wolbachia strain wMel, where the mutation rate was estimated from population data to be between 2.88 × 10^− 9^ – 1.29 × 10^− 8^ substitutions per site per year [88]. However, Richardson et al. [88], note that the wMel rate is biased, as it includes slightly deleterious mutations only observed at the population level, and not at the species level. Consequently, for our calculated rates to match the generational mutation rates of bacteria (ranging between 10^− 10^–10^− 8^ synonymous substitutions per synonymous site per generation [89]), all Wolbachia strains analysed here, except the ones in Nasonia, must have a generation time of almost a year. Since this is even longer than the generation times of the hosts, our estimated mutation rates are likely incorrect. There are indeed many potential sources of error for our mutation rate estimates, for example, synonymous substitutions could be under strong selection, or the recombination rate could be extremely high and eliminate diversity between strains, both of which could yield underestimated mutation rates. Additionally, the host divergence times could be wrong, which, depending on whether they are over- or under-estimated, would result in either lower or higher mutation rates. At this point, we can’t completely rule out any of these factors, although selection seems unlikely given that the effective population size of Wolbachia is probably low, and it is hard to understand how recombination between the wAu-like_ws_ strains could be so rampant if our Wolbachia strains are only vertically transmitted. Moreover, the divergence times of the hosts are likely a smaller problem, as they would have to be off by orders of magnitude for our estimated rates to be comparable to the rates of wMel and other bacteria. Alternatively, for co-speciation to be believable, most of the Wolbachia strains analysed here must have a very low mutation rate or long generation time, or a combination thereof. If not, neither the wAu-like_ws_ strains nor the Wolbachia strains from Nomada and Nasonia have co-speciated with their hosts but may instead have been horizontally transmitted into their hosts via introgression or alternative mechanisms, as suggested in other similar systems [90, 91].
It is unlikely that Wolbachia could have been recently transmitted between the relatively distantly related species of the willistoni and saltans group via introgression, but the topological congruence between mitochondria and Wolbachia could suggest that introgressions have facilitated at least some horizontal transmission events. As previously reported, D. paulistorum has two mitotypes, α and β, which were probably both introgressed from an unknown species within the willistoni group [39]. Hence, it is possible that wPau was introgressed into D. paulistorum from a willistoni group species together with one of the mitotypes. Potentially, the β mitotype, since it is most common among D. paulistorum semispecies and all D. paulistorum lines in this study carry it. As wPau has complete and thus likely functional cif genes, the spread of wPau and the replacement of the ancestral D. paulistorum mitochondrion could potentially have been driven by CI. However, to fully answer this question, data from additional D. paulistorum semispecies, especially the ones carrying the α-mitotype, are needed.
Additionally, at least four horizontal transmission events of wAu-like Wolbachia, i.e., wInn, wBor, wInc, and wAu, between distantly related Drosophila species have likely occurred relatively recently, as previously suggested [32, 92–94], through mechanisms other than introgression. We can only speculate about how these wAu-like Wolbachia strains infected and spread in these hosts, but CI is not a likely mechanism for the spread since none of them carry what appear to be functional cif genes. The well-studied strain wAu has long been known to not induce CI in its native host, D. simulans [95]. However, it does provide high antiviral protection in its host [34], a potentially beneficial phenotype that might have facilitated its spread. Nothing is currently known about the phenotypic effects wInc has on its host, but both wInn and wBor cause male-killing, which could have contributed to their spread. Furthermore, wInn was also seen to protect the host against RNA viruses [96]. It is also possible that wAu-like strains provide yet unknown beneficial host effects.
In conclusion, more research is required to determine whether Wolbachia switches hosts so frequently that pseudo-co-speciation is observed independently in multiple clades and to identify mechanisms that facilitate the transfer and spread of Wolbachia infections.
The genome of the obligate Wolbachia strain wPau is expanded and has some genomic features similar to recently host-associated symbionts
When free-living bacteria become host-associated, many of their genes become superfluous in the new nutrient-rich, stable environment of the host and are hence lost or pseudogenized due to lack of selection [2]. In addition, when symbionts transition from free-living to host-associated, they experience a reduction in effective population size that increases the effect of genetic drift [4, 97]. Consequently, their genomes tend to accumulate slightly deleterious mutations, including many IS element insertions [5]. However, over time, often after becoming obligate for their hosts, symbiont genomes generally shrink further in size and lose repeated sequences and pseudogenes [2, 5], leading to a conserved gene order. This stage of the genome reduction process can, for example, be observed in obligate mutualistic Wolbachia of nematodes that have small genomes [21, 23] with few repeats and mobile elements [22] compared to most facultative Wolbachia strains.
To our knowledge, wPau is the only Wolbachia strain found in the willistoni and saltans group of flies that is obligate for the host [36]. Hence, wPau is expected to have a smaller genome than its close facultative relatives. Contrary to the expectation, we found that wPauO11 has the largest genome among the wAu-like Wolbachia strains with complete genomes. Genome size increase in Wolbachia is generally associated with a higher proportion of prophage WO [8, 22, 81, 98], but in wPau, we found a reduction of prophage WO content. Instead, we attribute the increase in genome size in wPau to a still ongoing expansion of an IS4 element that makes up almost 10% of the genome and has greatly affected the gene order. The extensive IS4 element proliferation, high rate of genomic rearrangements and reduced coding percentage in the wPau genome are genomic features that are more common in recently host-associated facultative symbionts than evolutionary persistent obligate symbionts. All of them, however, suggest a large influence of genetic drift, potentially caused by a reduction in effective population size. We can think of at least three possible reasons why the effective population size of wPau could have decreased compared to its close relatives.
Firstly, under the assumption of horizontal transmission, there might have been a reduction in the effective population size of wPau since only a few Wolbachia cells likely established the infection in D. paulistorum initially. However, we don’t observe an expansion of IS elements in other wAu-like strains with complete genomes that likely also shifted host recently, i.e., wInn and wAu. The wInn genome shares other features with the wPau genome, including many rearrangements and low coding density. Thus, possibly, wInn had more repeats at one point, but lost them over time due to deletions. Although these similarities with wInn might indicate that horizontal transmission could initially cause a higher effect of genetic drift, we do not believe this to be the main reason for the sustained high levels of genetic drift in wPau.
Secondly, the transition from facultative to obligate host association could have yielded a reduction in the effective population size of wPau. Several similar genome features can be found in the genome of the mutualistic Wolbachia strain wCle infecting bedbugs [99], including an expansion of IS elements (with over 100 copies of IS5) and reduced prophage WO content [22]. Potential IS element expansions are also recorded in other obligate symbiont genomes, for example, the co-obligate Serratia symbiotica strain Ct from the aphid Cinara tujafilina [100], where several IS element families have expanded [101] and in some species of the genus Sodalis [102–106]. However, since no genome sequences of closely related facultative strains are available in any of those cases, the IS element expansion can’t be linked to a transition to obligate host association.
Thirdly, a reduction in the effective population size of wPau could be a consequence of a reduction in the effective population size of its host and might be linked to the fact that D. paulistorum is a superspecies consisting of multiple fully or partly reproductively isolated semispecies. Currently, no data about the effective population sizes of any species in the willistoni or saltans groups exist; however, indirect evidence indicates a lower effective population size in D. paulistorum compared to other willistoni and saltans group species. Large eukaryotic genomes with a high proportion of transposable elements often occur in species with low effective population sizes [107], due to a high influence of genetic drift allowing mobile elements to accumulate. Since D. paulistorum has the largest genome with the highest repetitive content of the sequenced willistoni group species [108, 109], we might speculate that D. paulistorum has a lower effective population size than other willistoni and saltans group species. If so, this could explain not only the high rate of IS4 element insertions before the divergence of the wPau strain variants but also the continued high rate that seems to be ongoing in wPau even after its likely introgression and transition from facultative to obligate.
Prophage WO genes may be important for the obligate symbiosis of wPau
Prophage WO is known to play an important role in facultative Wolbachia host associations, given that it may carry genes directly responsible for reproductive manipulation [26–28, 110], infection titer and virulence [29, 30]. For the obligate wPau, we demonstrate loss of prophage WO genes compared to closely related facultative strains. Specifically, wPau has lost almost an entire prophage WO copy (WOa) and some parts of the other (WOb), suggesting a general lack of selection on prophage WO-associated genes. In contrast, nine of the eleven genes of the prophage WO-associated Undecim cluster are instead duplicated in wPauO11, as well as the other wPau strain variants. Although the functions of the Undecim cluster genes are currently unknown, they are thought to play a role in Wolbachia-host interactions, as they, among other functions, encode proteins potentially involved in exopolysaccharide and/or lipopolysaccharide biosynthesis [25]. Previous work has also shown that the Undecim cluster has been horizontally transferred between several unrelated insect endosymbionts and that the genes are constitutively highly expressed in Wolbachia strain wMel during the whole life cycle of D. melanogaster [111]. The fact that this prophage WO-associated set of genes is duplicated in wPau, while many other prophage WO genes are deleted, suggests that the region could encode proteins potentially important for the obligate symbiosis between wPau and its host. If true, prophage WO might contribute genes for the persistence of a Wolbachia infection, even in the absence of CI or other reproductive manipulations.
Conclusions
We conclude that the ancestor of Neotropical Drosophila in the willistoni and saltans groups may have been infected with a wAu-like Wolbachia and co-speciated with their hosts, except for the wPau strain in D. paulistorum, which was likely introgressed from another species within the willistoni group together with one of the two mitotypes. However, such an interpretation suggests that Wolbachia must have a very low mutation rate. Since the real mutation rate of any strain of Wolbachia is still unknown, further research is needed to determine if co-speciation is a possible interpretation.
We also found that, contrary to expectations, the genome of the obligate wPau strain was larger than its facultative relatives due to the ongoing proliferation of an IS4 element, typically associated with genomes of recently host-associated symbionts. A reduced coding percentage and more pseudogenes are also hallmarks of the early stages of genome reduction, all of which are likely due to a reduction in the effective population size.
In general, the effective population size can be reduced multiple times after a bacterium becomes host-associated. Thus, the genome reduction process might take place in a punctuated manner, as episodes of effective population size reduction can temporarily lead to an increased genome size via repeat expansion, and a decrease in the proportion of coding content, only to be reversed by homologous recombination at such repeats over time. Consequently, only when mobile elements and repeats are lost from a genome altogether can this cycle be broken.
Supplementary Information
Supplementary Material 1
Supplementary Material 2
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Russell SL, Chappell L, Sullivan W. A symbiont’s guide to the germline. Current topics in developmental biology. Elsevier; 2019:315–51. 10.1016/bs.ctdb.2019.04.007.10.1016/bs.ctdb.2019.04.00731155362 · doi ↗ · pubmed ↗
- 2Ryan JA, Ulrich JM, quantmod. Quant Financial Modelling Framew. 2022. https://CRAN.R-project.org/package=quantmod
- 3R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2018. https://www.r-project.org.
- 4Stamatakis A. R Ax ML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. 10.1093/bioinformatics/btu 033.10.1093/bioinformatics/btu 033PMC 399814424451623 · doi ↗ · pubmed ↗
- 5Rambaut A. Fig Tree v 1.4: Tree Figure Drawing Tool. 2009. http://tree.bio.ed.ac.uk/software/figtree/
- 6Utensils/gene Stitcher.py. at master · ballesterus/Utensils. Git Hub. https://github.com/ballesterus/Utensils/blob/master/gene Stitcher.py. Accessed 2 Sept 2025.
- 7Phy Chro | CHR Onicle. Phy Chro: phylogenetic reconstruction based on synteny block and gene adjacencies. 2023. https://www.lcqb.upmc.fr/phychro 2/10.1093/molbev/msaa 114PMC 747504532384156 · doi ↗ · pubmed ↗
- 8Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K. cluster: Cluster Analysis Basics and Extensions. 2021. https://CRAN.R-project.org/package=cluster