Effect of the [Fe(salen)]2‑μ-oxo Catalyst Electronic Structure on Reductive Hydroamination
Emily Pocock, Nathan J. Buxton, Martin Diefenbach, Andrew D. Bond, Simon E. Lewis, Vera Krewald, Ruth L. Webster

TL;DR
This paper studies how the electronic structure of a specific iron catalyst affects its performance in a chemical reaction called reductive hydroamination.
Contribution
The study introduces three electronically distinct [Fe(salen)]2-μ-oxo complexes and shows how ligand electronics influence catalytic efficiency.
Findings
Modulating the salen ligand electronics significantly impacts product distribution and catalytic efficiency.
The complex with para-CF3 substituents outperformed other analogues in various olefin reactions.
DFT calculations and experimental data support a mechanism involving iron-hydride intermediates and hydrogen atom transfer.
Abstract
Salen ligands are privileged scaffolds in transition metal catalysis due to their electronic tunability and capacity to stabilize diverse oxidation states. Herein, we report the synthesis and comparative study of three electronically differentiated [Fe(salen)]2(μ-oxo) complexes and their application in catalytic reductive hydroamination (HA) of nitroarenes with alkenes. A mechanistic framework involving iron-hydride intermediates and hydrogen atom transfer (HAT) was developed, revealing that modulation of the salen ligand electronics significantly impacts product distribution and catalytic efficiency. Systematic investigation of substrate LUMO energies and precatalyst UV–vis spectroscopy, cyclic voltammetry, along with DFT calculations on the key HAT step, was undertaken. Notably, the complex bearing para-CF3 substituents outperformed its analogues across a range of olefin partners.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1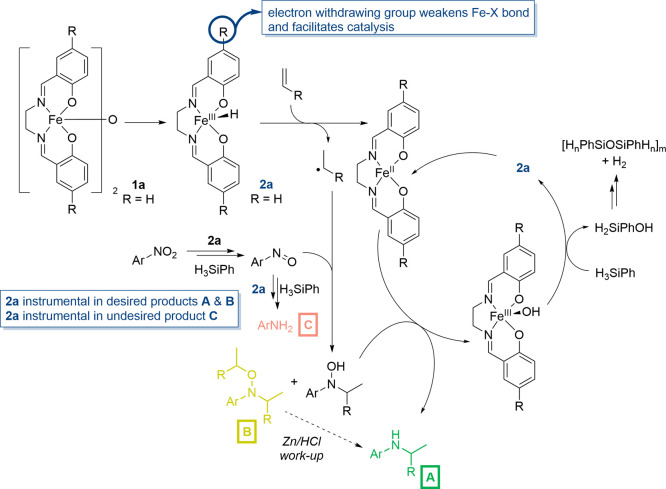 2
2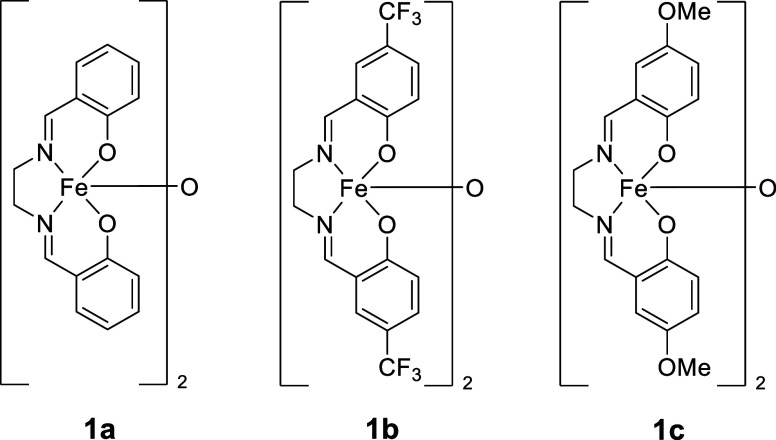 1
1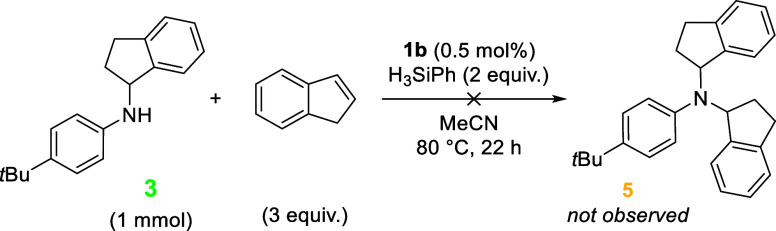 3
3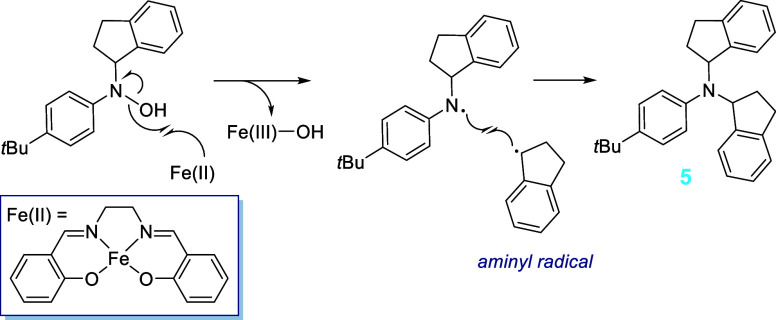 4
4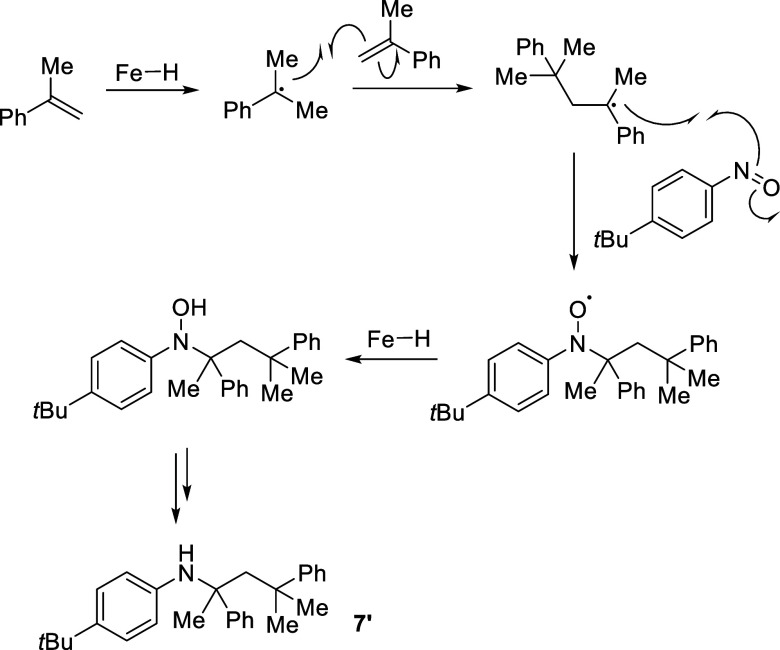 5
5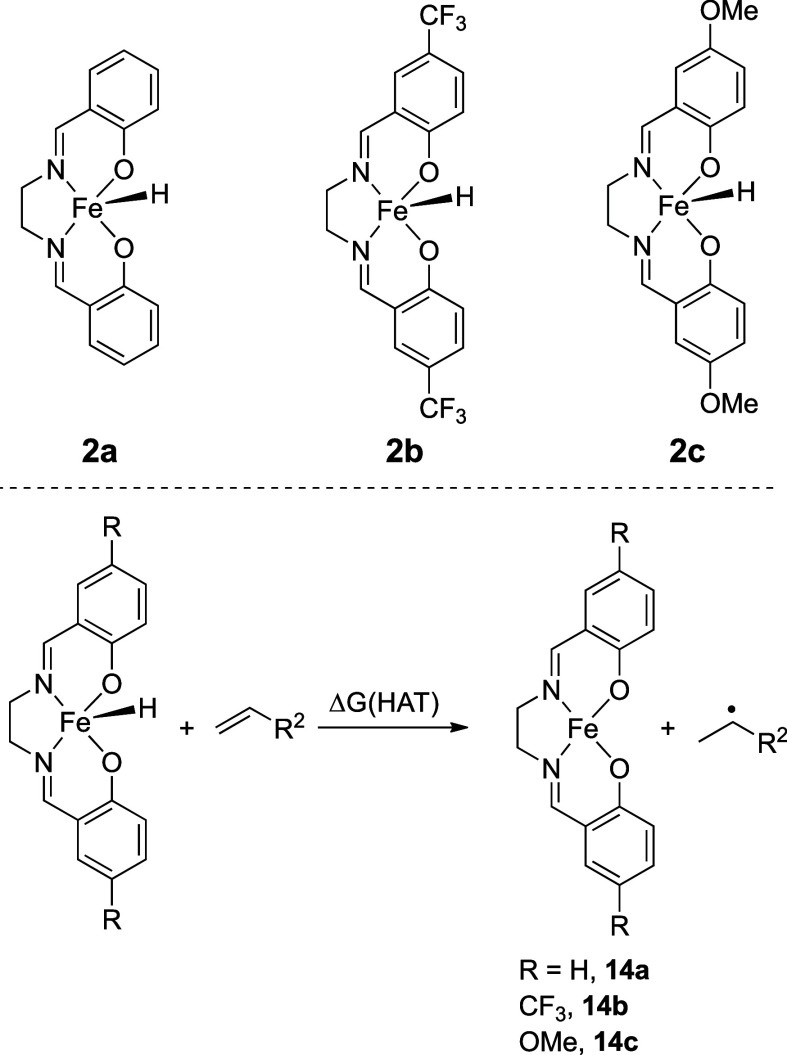 2
2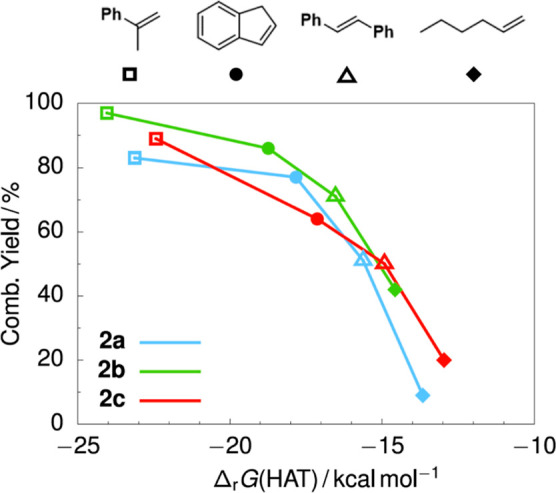 3
3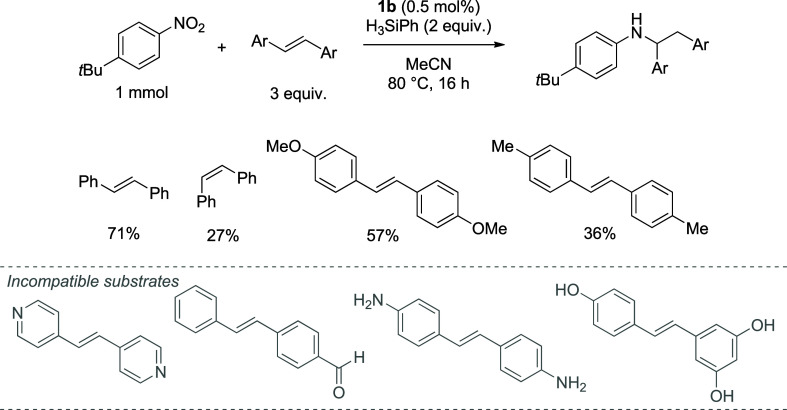 4
4| entry | alkene | A (%) | B (%) | C (%) | LUMO (eV) |
|---|---|---|---|---|---|
| 1 | 1-hexene | 5 | 0 | 87 | +0.21 |
| 2 | Indene | 77 | 0 | 0 | –0.91 |
| 3 | α-methylstyrene | 0 | 83 | 0 | –1.16 |
| 4 |
| 37 | 14 | 44 | –1.84 |
| complex | λmax (nm) | E1/2 (V vs Fc+/Fc) |
|---|---|---|
|
| 480 | –0.95 |
|
| 346 | –1.23 |
|
| 524 | –0.98 |
| 1a | 1b | 1c | |
|---|---|---|---|
| Combined Yield, % | 77 | 86 | 72 |
|
| 77 | 72 | 46 |
|
| 0 | 0 | 14 |
|
| 0 | 14 | 4 |
|
| 0 | 0 | 8 |
| 1a | 1b | 1c | |
|---|---|---|---|
| combined yield, % | 83 | 97 | 89 |
|
| 0 | 21 | 14 |
|
| 83 | 76 | 75 |
|
| 0 | 0 | 0 |
| 1a | 1b | 1c | |
|---|---|---|---|
| combined yield, % | 96 | 84 | 78 |
|
| 5 | 33 | 12 |
|
| 4 | 9 | 0 |
|
| 87 | 42 | 58 |
|
| 0 | 0 | 8 |
| 1a | 1b | 1c | |
|---|---|---|---|
| combined yield, % | 95 | 86 | 82 |
|
| 37 | 71 | 48 |
|
| 14 | 0 | 2 |
|
| 44 | 15 | 32 |
| spin state | ⟨ |
|
|
|---|---|---|---|
|
| |||
| HS | 8.762 | 0.00 | 0.00 |
| IS | 3.819 | 7.11 | 8.13 |
| LS | 0.801 | 1.90 | 5.15 |
|
| |||
| HS | 8.761 | 0.00 | 0.00 |
| IS | 3.869 | 6.25 | 9.16 |
| LS | 0.8 | 1 | 5.65 |
|
| |||
| HS | 8.763 | 0.00 | 0.00 |
| IS | 3.814 | 7.74 | 9.49 |
| LS | 0.802 | 0.49 | 4.05 |
| spin state | ⟨ |
|
|
|---|---|---|---|
|
| |||
| HS | 6.033 | 0 | 0 |
| IS | 2.035 | 6.41 | 7.67 |
| LS | 0 | 44.12 | 46.39 |
|
| |||
| HS | 6.032 | 0 | 0 |
| IS | 2.035 | 6.66 | 7.94 |
| LS | 0 | 45.8 | 47.68 |
|
| |||
| HS | 6.036 | 0 | 0 |
| IS | 2.039 | 6.48 | 7.75 |
| LS | 0 | 40.92 | 43.53 |
- —AstraZeneca10.13039/100004325
- —Engineering and Physical Sciences Research Council10.13039/501100000266
- —Leverhulme Trust10.13039/501100000275
- —Yusuf Hamied Department of Chemistry, University of CambridgeNA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Hydrogenation and Catalysis · Metal-Catalyzed Oxygenation Mechanisms · Catalytic C–H Functionalization Methods
Introduction
1
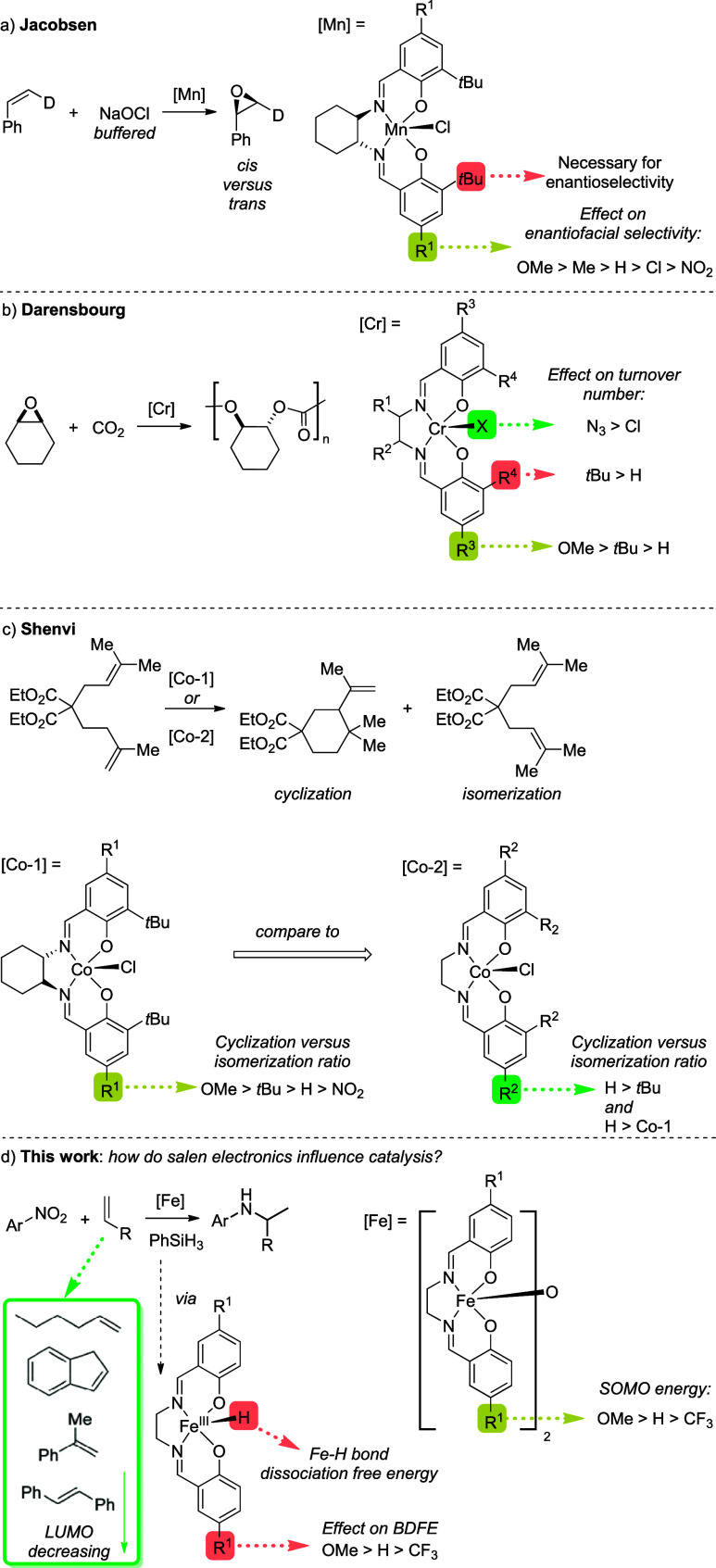
Salen complexes are ubiquitous in a wide range of catalytic transformations. This is largely due to the ability of the pro-ligand to form complexes with a large number of transition metals and to stabilize a range of oxidation states. ?,? Salen ligands are also inexpensive and easy to synthesize, and their sterics and electronics are readily tuned. These often-subtle changes to the ligand have, in many cases, been reported to bear a significant effect on reaction outcome. ?,?
A principal example of a salen ligand being key to reactivity can be seen in the use of Mn(salen) complexes in enantioselective epoxidations.? During Jacobsen’s studies into ligand effects, steric bulk in the ortho-position of the phenyl ring was crucial to obtain high enantioselectivities (Schemea). ?,? Alongside this, electron-donating substituents in the para-position also led to increased asymmetric induction, with the opposite being true for electron-withdrawing substituents. Other key examples come from Darensbourg and co-workers, who investigated how variations to the readily available (salen)CrX complex (where X = Cl or N_3_) could influence the rate of CO_2_/cyclohexene oxide copolymer production;? the use of a modified salen complex containing electron-donating groups on the phenoxy fragment led to an increased rate of copolymerization of 65.6 h^–1^ using para-OMe from 35.5 h^–1^ using para-* ^t^ *Bu. ?,? This report indicates the impact on the metal center (and thus overall reactivity) that can occur from simple changes to ligand electronics (Schemeb). ?,? Shaver, Storr, and co-workers explored a series of Co(II) salen complexes with varied para-ring substituents for the reversible-termination organometallic-mediated radical polymerization of styrene, methyl methacrylate, and vinyl acetate. The change of electronics in this position has a significant influence on the electron density at the metal center, suggesting that cobalt–carbon bond strength varies with the ligand substitution.? More recently, Shenvi and co-workers reported cyclization and alkene retrocycloisomerization using a Co(salen) complex (Co(salen^ t ^Bu,^ t ^Bu)Cl). The mechanism is believed to be a radical isomerization via a reversible HAT.? When investigating the efficiency of cyclodimerization, the group reported that reaction efficiency seems proportional to the persistence of an intermediate carbon-centered radical.? To explore this further, a variety of Co(salen) complexes were synthesized with varying electronics on the ligand (Schemec). Electron-deficient ligands led to poor conversion and a low ratio of cycloisomerization; electronically neutral ligands showed moderate selectivity for cyclization, while the use of an electron-rich ligand significantly increased the desired cyclization reaction. These observations were consistent with those reported for the equivalent changes in reversible HAT and metal–carbon radical collapse in polymerizations mediated by Co(salen) complexes. Electron-deficient complexes appear to terminate the carbon-centered radical, while these radicals are stabilized by electron-rich complexes. ?,?
a) Jacobsen’s Seminal Work on Enantioselective Epoxidation Showed That Salen Electronics Influenced Selectivity; (b) Darensbourg’s Variation of Sterics and Electronics Led to Vast Changes in Turnover Number (TON) and Turnover Frequency; (c) Co-Salen Catalysts Synthesized by Shenvi and Co-Workers for the Investigation of How Ligand Electronics Affects Isomerization; (d) In This Work, We Show That Changes to Electronics Affect SOMO Energy and Bond Dissociation Free Energy (BDFE) of a Postulated Intermediate, with a Range of Outcomes Observed Based on the Electronics of the Olefin Coupling Partner
Our own studies on [Fe(salen)]2-μ-oxo complexes have focused on the simplest ligand system. The resultant precatalyst (1a, see Schemesd and ?) is highly active in hydrophosphination, hydroboration, and alkyne and phosphaalkyne cyclotrimerization. ?−? ? ? ? It is important to note that steric bulk in the ortho-position, for our catalytic reactions, often results in very low rates of reaction.? More recently, we extended this set of transformations to include the reduction of nitro-compounds.? An extensive mechanistic study was conducted, which revealed the reaction is likely to proceed via a nitroso intermediate and the generation of an on-cycle iron-hydride (2a, Scheme). Observations made in the seminal work reported by Baran and co-workers,? along with there being similar intermediates investigated in analogous studies with iron precatalysts, ?−? ? ? ? ? ? ? ? ? meant that we were also able to extend our catalyst system to reductive hydroamination (HA) reactions using nitro compounds and alkenes. Using a nitro compound in HA allows us to bypass a separate reduction step to form an amine (cf. classical HA methodology) and, in theory, allows the use of substrates containing both nitro and amine functionality without functionalization of the latter. However, reductive HA of nitro compounds comes with a wide variety of challenges based on the postulated mechanism: the nitro-compound must be partially reduced to a reactive nitroso intermediate and then trapped by the alkyl radical; the alkyl radical is formed by the same iron hydride species via a HAT reaction. Therefore, the relative rates of both of these transformations should be controlled to selectively generate HA product (A, Scheme) or an alkyl-hydroxylamine species (B) while avoiding over-reduction to aniline (C) and other undesired radical reactions such as radical (alkene) polymerization.
Simplified Hypothetical Catalytic Cycle for the [Fe(salen)]2-μ-oxo Catalyzed HA Reaction, Which Is the Basis of the Experimental and Theoretical Studies Described Herein
In our original study,? we linked HA reactivity to the magnitude of the alkene’s lowest unoccupied molecular orbital (LUMO) energy. Little to no HA reactivity was observed when alkenes with a positive LUMO eigenvalue were employed in the reaction. For example, 1-hexene did not undergo the desired HA reaction, and the reduction to aniline C dominated, indicating the energy gap between the alkene LUMO and the Fe–H SOMO (which is the alkyl radical forming reaction) is too large (Table, entry 1). In contrast, when indene was employed, the HA reaction dominated, generating a large amount of desired product A (entry 2). This is also true for α-methylstyrene, where a large amount of product B was observed, which is indicative of the HA reaction taking place (entry 3). However, when the LUMO energy becomes significantly more negative, such as when trans-stilbene was employed, a loss of selectivity for the HA reaction was observed (entry 4). We therefore envisioned that the HA reaction could be further explored by varying the ligand’s electronics to modulate the key HAT step and therefore change the product distribution.
1: Summary of LUMO Energy and HA Product Distribution
Results and Discussion
2
Synthesis and Analysis of Precatalysts
2.1
Three [Fe(salen)]2-μ-oxo complexes (1a to 1c, Figure) were synthesized in good yields by varying the salicylaldehyde used in the ligand formation step.?
Fe(salen) complexes synthesized to investigate how varied ligand electronics could affect product distribution in reductive HA.
We initiated studies by probing the precatalysts to see if we could make a link between the electronic nature of 1a to 1c and the outcomes of HA. Using UV–visible spectroscopy, the band of greatest interest is the ligand-to-metal charge transfer (LMCT) band observed between 400 and 530 nm. ?−? ? Altering the functional groups in the para-position of the phenolate units (complexes 1a, 1b, and 1c) results in a significant change of the energy of the LMCT bands (Table and the Supporting Information). Complex 1c has a band within the in-plane LMCT region present at 524 nm; thus, it has the lowest energy gap for the LMCT transition. 1b proved challenging to analyze using UV–visible spectroscopy, but there may be a high-frequency band below 400 nm (tentatively assigned at 346 nm), which would be consistent with a significantly larger SOMO-LUMO gap. Consistent with these data, 1a has an intermediate absorption band at 480 nm.
2: UV–Visible Spectroscopy and Cyclic Voltammetry Data for Complexes 1a to 1c
Cyclic voltammetry studies reveal that 1b has the lowest formal electrode potential at −1.23 V, while 1a (with no donating/withdrawing groups) has a formal electrode potential of −0.95 V, and 1c (with para-OMe groups) is similar at −0.98 V (Table and the Supporting Information). CF_3_ groups alter the formal electrode potential such that 1b is the most prone to oxidation, and accessing an on-cycle Fe(II) species may occur less readily, 1a and 1c are very similar, and they may behave similarly in catalysis. The complexes all appear to show reversible redox cycles, which are important for the HA reaction, which is believed to proceed via an Fe(III)/(II) cycle. The calculated Nicholson parameters indicate that the redox processes are reversible, as their values are approximately equal to 1 (1.14–1.33).? This is also consistent with the theory that there is no significant geometry change around the metal center with any change in oxidation state.? At this stage, we were ready to investigate whether trends in the electronic nature of these precatalysts would be borne out in discernible trends in reductive HA catalysis.
Study of Precatalysts 1a to 1c in Reductive
HA
2.2
The four olefins, 1-hexene, indene, α-methylstyrene, and trans-stilbene, with LUMO energies ranging from 0.21 to −1.84 eV, were employed in the HA reaction using our previously optimized conditions. The challenges associated with in situ analysis of these reaction mixtures mean that isolated yields are reported. It is also worth noting that there is very little unreacted nitro starting material observed in any of the reactions studied.
Indene was initially investigated as the olefin coupling partner because it was the most selective for the desired HA product when precatalyst 1a was used (3, Table). In comparison, 1b is less selective, with modest amounts of a double HA product forming (5, 14%) in addition to 72% 3. 1c shifts reactivity away from the formation of 3 (46%), and the indenyl-hydroxylamine forms in modest amounts (4, 14%) along with trace amounts of a new double HA product, 5, and aniline 6 (from the undesired over reduction of the nitro starting material).
3: Product Distribution from the Reaction of Indene and tert-Butyl-4-nitrobenzene Using Precatalysts 1a to 1c
The formation of compound 5 is unexpected. To investigate this, a test reaction was conducted where HA product 3 was subjected to the standard reaction conditions using catalyst 1b (Scheme). Monitoring the reaction by thin-layer chromatography and ^1^H NMR spectroscopy shows no further reaction of 3, indicating that this is not the route to formation of product 5.
Attempted Reaction of 3 under Standard Reaction Conditions Using Precatalyst 1b
Based on the proposed HA mechanism (Scheme), 5 could be forming from a side reaction of a hydroxylamine intermediate (Scheme). The hydroxylamine intermediate reacts with Fe^II^(salen) to form an aminyl radical. While these are reportedly short-lived species, in this specific case the radical is likely to be resonance stabilized due to the presence of the aromatic ring.? The resultant aminyl radical could then undergo a termination process by combining with the corresponding indenyl radical that has formed during the HAT step.
Proposed Radical Mechanism for the Formation of Product 5
Indene is a good substrate for this chemistry; we therefore turned our attention to less reactive HA substrates. α-Methylstyrene was investigated next because it is selective for the formation of alkyl-hydroxylamine 8 using 1a, and we questioned how precatalyst electronics could affect reaction selectivity (Table). In the case of 1b and 1c, the yield of 8 is reduced, and moderate amounts of the desired HA product are obtained (21% and 14% from 1b and 1c, respectively). As with 1a, aniline 6 is not formed.
4: Product Distribution from the Reaction of α-Methylstyrene and tert-Butyl-4-nitrobenzene Using Precatalysts 1a to 1c
Trace amounts of 7′ are also obtained using precatalyst 1c (Scheme). 7′ could form when the alkyl radical reacts with a second equivalent of α-methylstyrene. In turn, this resultant radical species could react with the nitroso radical, which can then re-enter the standard catalytic cycle. This is likely to be a disfavored process due to the longevity of the alkyl radicals and the reactive nature of the nitroso-intermediate, which provides an explanation as to why 7′ has only ever been isolated in trace amounts, and in this case only.? This result also highlights the propensity for oligomerization during this reaction and hence the reduction in the combined isolated yields reported.
Proposed Radical Mechanism for the Formation of Product 7′
The reaction of 1-hexene with 1a was unselective for HA, with a large amount of aniline 6 being generated. However, using 1b, the yield of 9 increases to 33% (compared to 5% using 1a, Table). The increase in the yield of 9 occurs concomitantly with a decrease in the yield of 6 (87% using 1a, 42% using 1b). Although the yield of 9 using 1b could be described as a moderate yield, this is a greater than 6-fold increase in yield compared to our previous report. 1c generates only 58% 6, 12% 9 (doubling the yield compared to 1a), and 8% of the azo compound 11. The reaction is clearly finely balanced, with different reaction pathways being switched on based on the change in catalyst. However, compounds 1a and 1c behave fairly similarly in terms of the quantity of compound 9 formed.
5: Product Distribution from the Reaction of 1-Hexene and tert-Butyl-4-nitrobenzene Using Precatalysts 1a to 1c
Finally, the HA reaction was examined by using trans-stilbene as the olefin coupling partner. When the reaction was conducted using precatalyst 1a, we noted that this reaction has the widest product distribution, highlighting a system that might be highly susceptible to catalyst-driven shifts (Table). The yield of 12 ostensibly doubles using 1b (71% using 1b versus 37% using 1a), while the formation of 6 is reduced by two-thirds (15% using 1b versus 44% using 1a). No alkylhydroxylamine 13 is obtained using 1b. Employing 1c seems to limit the formation of 13 (2% 13 and 48% 12), but the formation of 6 is a dominant reaction pathway (32%).
6: Product Distribution from the Reaction of trans-Stilbene and tert-Butyl-4-nitrobenzene Using Precatalysts 1a to 1c
To summarize the findings thus far: precatalyst 1b is generally a better catalyst than 1a and 1c, often generating more of the desired HA products and minimizing the reduction to 6.
Computational
Investigation into the Reaction Results
2.3
We next turned to simple density functional theory (DFT) calculations to determine whether there were trends in HA reactivity and catalyst energetics. Although operationally simple, our initial studies showed that a simple comparison of the SOMO energies of 2a to 2c (Figure) would provide an inadequate link to the synthetic data (see the Supporting Information for these calculations).
Structures of the Fe(II) hydrides (2a to 2c) formed from the activation of precatalysts 1a to 1c (top) and the isodesmic reaction enthalpies for complexes 2 reacting with alkene via a HAT process to generate an alkyl radical and the Fe(II) complexes 14a to 14c (bottom).
Thus, a more exacting approach was needed, and we calculated the isodesmic HAT values for the reaction of the iron hydride (2a to 2c) with the alkene (Figure). First, the different potential spin states were calculated for complexes 2 and 14 to determine the ground state among the three spin states, i.e., low-spin doublet (LS), intermediate-spin quartet (IS), and high-spin sextet (HS). From our previous studies, 2a was found to have an HS ground state at the DFT level PBE0-D3/def2-TZVP//PBE-D3/def2-SVP, which was benchmarked against HF-DLPNO-CCSD(T). The HS state is also the lowest in energy for 2b and 2c (Table) using PBE0-D3/def2-TZVP//PBE-D3/def2-SVP. The spin states of the corresponding Fe^II^(salen) complexes were determined (14a to 14c, Table) at the same level, which also confirmed the HS configuration as the ground state.
7: Spin Expectation Values and Relative Energies for 2a to 2c on the HS, IS and LS Surfaces Computed at the PBE0-D3/def2-TZVP//PBE-D3/def2-SVP Level
8: Spin Expectation Values and Relative Energies for Complexes 14a to 14c on the HS, IS, and LS Surfaces Computed at the PBE0-D3/def2-TZVP//PBE-D3/def2-SVP Level
A plot of the reaction enthalpies versus the combined yield of HA product and hydroxylamine shows an excellent trend where the combined yield increases with increasing HAT reaction enthalpies (see Figure).?
*Plot of reaction enthalpy for Fe–H + olefin → Fe(salen)
- alkyl radical versus combined yield of the HA product + hydroxylamine product.*
Reaction
of Stilbenes
3
Finally, based on the improvement in yield for the HA reaction using trans-stilbene, different stilbene derivatives were investigated in the reaction using catalyst 1b (Figure).
Reaction scope for the HA reaction of trans-stilbene derivatives using 1b as the precatalyst.
Stilbene derivatives have HA yields varying from 71% to 27%. Unfortunately, the optimized catalytic conditions could not be easily transferred to the trans-stilbene derivatives, with many exhibiting poor solubility under the reaction conditions, e.g., free amines or alcohols, which were highly insoluble at elevated temperatures and in multiple solvents. The reaction with a pyridine-based stilbene gave only unreacted nitro (90% recovered). In the case of the aldehyde functionalized derivative, although initially soluble, precipitation occurred after a few hours at the reaction temperature. This solid could be a result of reductive amination occurring between the aniline and the aldehyde.
Methods
4
Precatalyst Synthesis
4.1
Fe(OAc)2 (1 equiv) was weighed into a round-bottom flask and dissolved in EtOH, resulting in a dark brown solution. To this, a solution of proligand (1.2 equiv, see the Supporting Information for methods of synthesis) in EtOH was added, yielding a red solution. The resulting red solution was stirred at 80 °C for 2 h. The flask was then allowed to cool to room temperature before the resulting precatalyst (1a–1c) was isolated via vacuum filtration.
Electrochemistry Procedure
4.2
Electrochemical measurements were performed in a 0.2 M solution of tetrabutylammonium hexafluoride with a 0.2 mM concentration of complexes 1a to 1c and a 0.2 mM standard of ferrocene in acetonitrile. A three-electrode cell was used for the electrochemical experiments employing a separate working, counter, and reference electrode. Cyclic voltammograms were cycled three or four times to obtain steady-state conditions. The working electrode was a polished glassy carbon disc (3 mm diameter) electrode. The electrode was polished and cleaned between each separate experiment. A platinum (Pt(0)) wire was used as the counter electrode. A silver (Ag(0)) wire was used as the reference electrode. The reference and counter electrodes were cleaned with isopropanol and flamed between each experiment. All electrochemical measurements were run under an inert nitrogen atmosphere, where the solutions were degassed by sparging with nitrogen for 15 min. Between measurements, the solutions were stirred briefly to renew the layer of solution near the surface of the electrodes.
Synthetic Procedure for Reductive Hydroamination
4.3
A mixture of precatalyst (0.005 mmol) and p-tert-butylnitrobenzene (1.0 mmol, 1 equiv) in dry acetonitrile (0.5 mL) was added to an ampule with a magnetic stirrer bar under nitrogen. Olefin (3 mmol, 3 equiv) was added to the solution, followed by phenyl silane (2 mmol, 2 equiv). The reaction mixture was then stirred at 80 °C under a positive flow of nitrogen for 22 h. After the reaction had gone to completion, the solvents were removed under vacuum. The resulting crude product was then purified by flash column chromatography on silica using hexane/dichloromethane.
Computational Methods
4.4
Molecular geometries were optimized at the DFT level employing the generalized gradient approximation (GGA) via the PBE? functional in conjunction with the D3 atom-pairwise dispersion correction without damping? and an implicit polarizable continuum solvent model? utilizing acetonitrile as the solvent. The split-valence double-ζ def2-SVP basis set? was used together with the corresponding auxiliary Coulomb-fitting basis set of Weigend.? At this GGA DFT level, abbreviated as PBE-D3(PCM)/def2-SVP, frequency calculations were performed on the optimized stationary points to characterize minima and transition structures and to extract thermal contributions to enthalpies and Gibbs energies at 298.15 K. For improved relative energies, single-point energy calculations were performed at the hybrid DFT level with 25% admixture of Fock exchange via the PBE0 functional ?,? employing the triple-ζ valence polarized def2-TZVP basis set? and the same dispersion and solvent corrections as above. The final relative Gibbs energies and orbital eigenvalues are thus reported at the PBE0-D3(PCM)/def2-TZVP//PBE-D3(PCM)/def2-SVP level.
Summary
5
Investigations showed that variations in the electronics of a salen ligand could result in a shift in product distribution for an iron-catalyzed HA reaction. The precatalysts were examined using UV–vis spectroscopy and cyclic voltammetry, and this showed that para-OMe gave a more reducible metal center and higher energy SOMO. A para-CF_3_ group had the opposite effect on the electronics at iron.
Each new precatalyst was examined in the HA reactions using tert-butyl nitrobenzene and the olefin coupling partners 1-hexene, indene, α-methylstyrene, and trans-stilbene. These HA reactions led to the discovery that the presence of an electron-withdrawing group (precatalyst 1b) improved the selectivity for HA reactions compared to the standard unsubstituted system.
To further explore the trends observed in reactivity, computational analysis was performed on the various iron hydride species to determine both the SOMO value and the BDFE of the intermediate iron hydrides. While these results cannot fully explain all changes in the reactivity profiles that were observed, they do act as a guide to help understand the difference in the reactivity of these new catalysts. Based on the complexity of this reaction, where there are three interlinked catalytic cycles (one being the unproductive nitroso to aniline process), it is clear that a deeper study of the mechanism, including kinetics of the individual cycles, is needed to develop an efficient set of catalysts that span a range of alkene substrates. This work is ongoing in our laboratories.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cozzolino M.Leo V.Tedesco C.Mazzeo M.Lamberti M.Salen, salan and salalen iron(III) complexes as catalysts for CO/epoxide reactions and ROP of cyclic esters Dalton Trans.20184737132291323810.1039/C 8DT 03169 J 30179239 · doi ↗ · pubmed ↗
- 2Chiang L.Allan L. E.Alcantara J.Wang M. C.Storr T.Shaver M. P.Tuning ligand electronics and peripheral substitution on cobalt salen complexes: structure and polymerisation activity Dalton Trans.201443114295430410.1039/C 3DT 51846 A 24048446 · doi ↗ · pubmed ↗
- 3Whiteoak C. J.Salassa G.Kleij A. W.Recent advances with π-conjugated salen systems Chem. Soc. Rev.201241262263110.1039/C 1CS 15170 C 21842040 · doi ↗ · pubmed ↗
- 4Kleij A. W.Nonsymmetrical Salen Ligands and Their Complexes: Synthesis and Applications Eur. J. Inorg. Chem.200919320510.1002/ejic.200800936 · doi ↗
- 5Jacobsen E. N.Zhang W.Muci A. R.Ecker J. R.Deng L.Highly Enantioselective Epoxidation Catalysts Derived from 1,2-Diaminocyclohexane J. Am. Chem. Soc.1991113187063706410.1021/ja 00018 a 068 · doi ↗
- 6Palucki M.Finney N. S.Pospisil P. J.Güler M. L.Ishida T.Jacobsen E. N.The mechanistic basis for electronic effects on enantioselectivity in the (salen)Mn(III)-catalyzed epoxidation reaction J. Am. Chem. Soc.1998120594895410.1021/ja 973468 j · doi ↗
- 7Darensbourg D. J.Mackiewicz R. M.Phelps A. L.Billodeaux D. R.Copolymerization of CO 2 and epoxides catalyzed by metal salen complexes Acc. Chem. Res.2004371183684410.1021/ar 030240 u 15612673 · doi ↗ · pubmed ↗
- 8Darensbourg D. J.Mackiewicz R. M.Rodgers J. L.Fang C. C.Billodeaux D. R.Reibenspies J. H.Cyclohexene Oxide/CO 2 Copolymerization Catalyzed by Chromium(III) Salen Complexes and N-Methylimidazole: Effects of Varying Salen Ligand Substituents and Relative Cocatalyst Loading Inorg. Chem.200443196024603410.1021/ic 049182 e 15360252 · doi ↗ · pubmed ↗