Osteoporosis in Patients With Marfan Syndrome: A Narrative Review of Bone Health and Management
Abdul Waheed Bahir, Munir Ahmad Bahir, Qudratullah Bahir, Gu Shao, Xiong Ying

TL;DR
This paper reviews bone health issues in Marfan syndrome, highlighting the unique challenges of osteoporosis and the need for specialized treatments.
Contribution
The paper provides a comprehensive review of bone health in Marfan syndrome, emphasizing the need for skeletal-specific therapies.
Findings
Osteoporosis in Marfan syndrome occurs earlier and differs from general population patterns.
Current osteoporosis treatments do not fully address connective tissue defects in Marfan syndrome.
Pharmacologic agents for cardiovascular protection in Marfan syndrome have limited impact on bone health.
Abstract
Marfan syndrome is a hereditary connective tissue disorder that is caused by pathogenic variants in the FBN1 gene and is traditionally known to have cardiovascular and ocular presentations. There has been growing data showing that bone quality impairment and decreased bone mineral density are significant but undervalued factors of the illness. Osteoporosis in patients with Marfan syndrome can develop earlier, follow different pathways, and lead to an increased risk of fragility fractures in comparison with those in the general population. The present narrative review is a critical evaluation of the existing knowledge of bone health in Marfan syndrome and includes the underlying molecular pathways of skeletal fragility, the impact of skeletal abnormalities and biomechanical changes, and how growth-factor malregulation can drive bone remodeling. Diagnostic analysis has been difficult…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
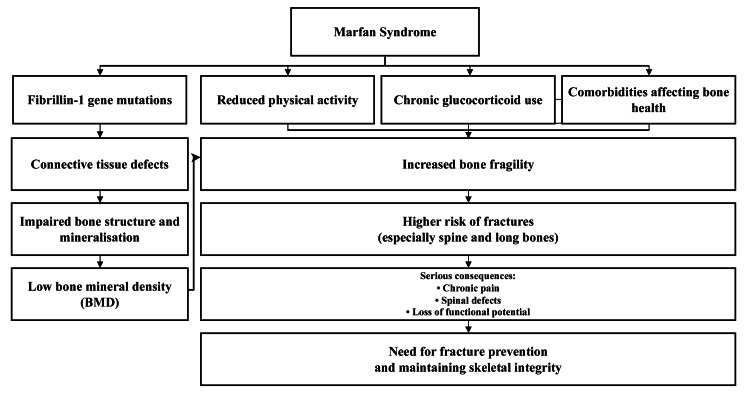 Figure 1
Figure 1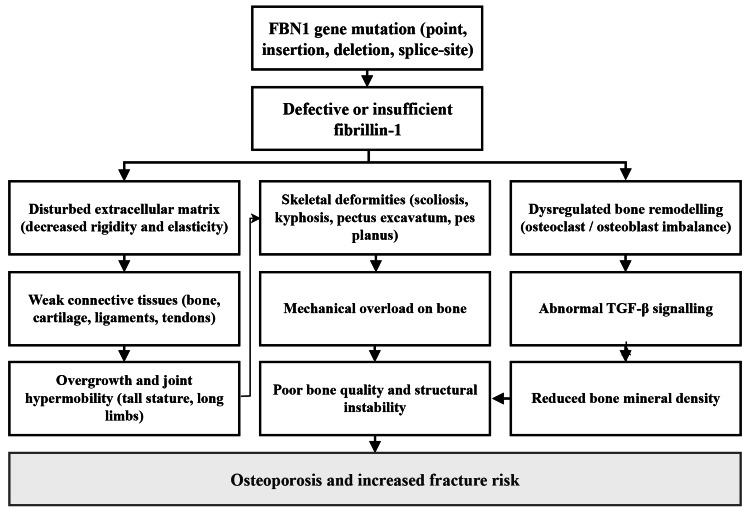 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective tissue disorders research · Tendon Structure and Treatment · Spine and Intervertebral Disc Pathology
Introduction and background
Osteoporosis is a bone disorder characterized by the loss of bone mass and bone microarchitecture deterioration, leading to increased bone fragility and a higher susceptibility to fractures [1]. Although commonly coupled with the aging process, osteoporosis is also seen in people with genetic connective tissue disorders, such as Marfan syndrome [2]. Marfan syndrome is an autosomal dominant syndrome that involves variants in the FBN1 gene pathogenic to biological activities that cause the synthesis of fibrillin-1, which is a fundamental structural protein involved in connective tissue integrity in all parts of the body [3]. These molecular defects play a role not only in the established cardiovascular and ocular morbidity of the syndrome but also in the skeletal development and bone disease [4]. Common approaches to the management of osteoporosis include lifestyle changes, nutritional supplementation, and pharmacological treatment [5]. The effectiveness of antiresorptive agents continues to lie in the foundation of osteoporosis management, having proven effective in decreasing the bone turnover and risk of fractures, although therapeutic responsiveness across skeletal sites can be difficult, and bone mineral density (BMD) increases do not always lead to an increase in bone strength [6]. An example is that bowing alendronate could enhance the quality of the bone matrix with osteopenia and lordosis, but it has no effect on the evolution of aortic aneurysms, one of the cardiovascular outcomes that are the most detrimental manifestations of Marfan syndrome [7]. On the other hand, losartan, an agent that suppresses TGF 2 mounting and strengthens the aortic wall in severe cases of Marfan syndrome, does not prevent bone insurgency, thus highlighting the need for skeletal-specific treatment strategies in this group [8]. Osteoporosis is often referred to as a silent thief because it has an insidious progression until the occurrence of a fracture [9]. Even though it can be frequently seen among post-menopausal women and older men [10], this syndrome poses a specific clinical problem in patients with Marfan syndrome. Typical skeletal phenotypes such as long extremities, spinal defects, and defects in the chest wall, combined with impaired bone quality, could increase the rate of fracture and may contribute to skeletal and cardiovascular adverse events [4]. This study underscores the need to note osteoporosis as a comorbidity that has not been given serious consideration in Marfan syndrome and the need to promote early assessments and tailored interventions. To support this narrative review, a thorough literature search was conducted using PubMed, Google Scholar, and Cochrane Library databases. Among the keywords used were osteoporosis, bone density, fracture risk, Marfan syndrome, FBN1 mutation, and connective tissue disorder, preparing and combining them in varying formulations. Publications in the English language that covered bone health in relation to Marfan syndrome were prioritized. This methodological approach helped in the synthesis of the heterogeneous findings, hence giving an overall picture of osteoporosis in patients with Marfan syndrome.
Review
Pathophysiology of Marfan syndrome
Marfan syndrome is a hereditary connective tissue disorder mostly due to mutations in the FBN1 gene, encoding fibrillin-1, a major component of the extracellular matrix [11]. The mutations destabilize the structure and functioning of fibrillin-1, leading to impaired microfibril formation and disordered connective tissue organization. Fibrillin-1 plays a primary role in supporting the structure and elasticity of the skeletal, cardiovascular, and ocular systems [12]. Such widespread distribution of fibrillin-1 is the result of the systemic nature of Marfan syndrome. In the musculoskeletal system, there are abnormalities of connective tissue that tend to bring about long limbs, scoliosis of the spine, and pectus excavatum of the chest wall [13]. All of these skeletal distortions point to a structural weakness in people that puts them at risk of having slack joints, postural abnormalities, and a high risk of fractures. FBN1 mutations are associated with the cardiovascular system, aortic wall weakening resulting in dilation of the aortic root, development of aneurysms, and potentially fatal aortic dissection. The ocular symptoms, such as dislocation of the lens and myopia, are additional examples of the widespread effect of fibrillin-1 deficiency and the support of the systemic activity of connective tissues in Marfan syndrome [14].
Skeletal manifestations
The skeletal abnormalities are key characteristics of the Marfan syndrome and the central clinical phenotype [15]. Patients commonly have disproportionate limb lengthening and long arms, legs, fingers, and toes, which can be explained by laxity of the connective tissue and other changes in the biomechanical forces applied to the developing bone mass [13]. Popular spinal defects include scoliosis, kyphosis, and severe lumbar lordosis, which can lead to pain, post-spinal instability, and, in extreme cases, erode pulmonary functionality [16]. Anatomy Discontinuities such as pectus excavatum and pectus carinatum are manifestations of structural weakness of the costal cartilage and sternum; they can cause cosmetic issues and, in a few cases, result in cardiopulmonary restrictions [17]. Other common aspects include joint hypermobility, which is a result of ligamentous laxity; excessive range of motion increases the risk of instability, recurrent subluxations or dislocation, and long-term musculoskeletal pain, thus hindering mobility and general quality of life [18]. Importantly, osteoporosis and fragility fractures are associated with a high risk in Marfan syndrome, probably because of inherent bone defects, low physical activity, and as a complication in some instances, because of chronic exposure to glucocorticoids [19]. These combinations are synergistic and decrease the strength of the bones and increase the risk of fractures.
Osteoporosis in Marfan syndrome
Osteoporosis is a systemic bone skeletal disease characterized by bone mass and microarchitectural bone destruction that leads to increased bone fragility and fracture. In the framework of Marfan syndrome, osteoporosis has become a relevant complication woven into connective tissue defects and the genetic factors [2]. People affected by Marfan syndrome usually have low BMD compared to average people, and hence this predisposes them to fractures after low-energy mechanical injuries [20]. Epidemiological data further support this increased skeletal vulnerability. Studies in children with Marfan syndrome demonstrate height-adjusted whole-body and lumbar spine BMD Z-scores reduced by approximately 0.8 SD compared with matched controls, indicating a mild but generalized deficit in bone mass [2]. In addition, a nationwide Danish cohort study involving 406 patients with Marfan syndrome reported that 21.9% had experienced at least one fracture, compared with 18.9% of matched controls, and that 10.3% met composite criteria for osteoporosis, compared with 3.3% in the reference population [19]. These findings highlight that reduced BMD and fracture risk are both clinically meaningful and disproportionately elevated in this population.
The resultant structural compromise, particularly in the vertebral column and long bones, is a consequence of affected bone quantity, in addition to the quality. Deformed bone structure and mineralization deficiency are mutations of the fibrillin-1 gene, which weaken the bones and increase their susceptibility to fractures [21]. Additional contributory factors include a reduction in physical activity, persistent intake of glucocorticoids, and supportive comorbidities, which adversely affect skeletal health [22]. A fracture of the spine and long bones in patients with Marfan syndrome can cause critical consequences, such as chronic pain, spinal defect, and loss of functional potential. Therefore, maintenance of skeletal integrity and fracture prevention are among the main goals in the management of osteoporosis in this group (Figure 1) [23].
Osteoporosis Pathway in Marfan Syndrome
Genetic links and impact on bone health
The FBN1 gene is the main causative agent of Marfan syndrome, and its mutations cause quantitative or qualitative deficiencies in fibrillin-1 [24]. These pathologic changes include a point mutation, insertions, deletions, and splice-site mutations, each of which could disrupt the biosynthesis or functional integrity of fibrillin-1. Due to the central structural position of fibrillin-1 in the extracellular matrix of connective tissue, its distortion leads to under-rigidity and decreased elasticity of tissues across the organism. Fibrillin-1 plays a critical role in maintaining the mechanical stability and integrity of connective tissues that maintain skeletal structures, such as bone, cartilage, ligaments, and tendons [12]. Connective tissues are also very sensitive to overstretching and deformation when the fibrillin-1 functions are impaired. This phenomenon is clinically manifested in the form of an increase in stature, overgrowth of limbs, joint hypermobility, and a distinctive body habitus of the Marfan syndrome. Dysregulated bone remodeling is also a threat to bone health. Normative bone remodeling also requires a balanced relationship between osteoclastic-mediated resorption and osteoblast-mediated formation [25]. In Marfan syndrome, this balance could be disrupted by perturbations in fibrillin-1 and other signaling cascades, such as TGF-β, and ends with disrupted bone formation, diminished BMD, and increased likelihood of fractures [26]. Fibrillin-1 also plays a role in bone development and mineralization under normal conditions. Its activity is disrupted, affecting coordinated ontogenetic events in development, resulting in skeletal deformities such as scoliosis, kyphosis, pectus excavatum, and pes planus [27]. Not only do these skeletal deformities modify external morphology, but they also subject the bone to aberrant mechanical loads even greater to reduce bone quality and structural stability [28]. The combination of genetic aberrations, disturbed remodeling processes, and mechanical forces creates a biological and biomechanical environment that predisposes patients to osteoporosis and fracture in Marfan syndrome (Figure 2).
How Marfan Syndrome Leads to Weak Bones and Fractures
Diagnostic considerations in Marfan-related osteoporosis
In patients with Marfan syndrome, osteoporosis needs to be carefully diagnosed after careful exclusion of symptoms due to deformities and actual bone fragility. The connective tissue defects caused by fibrillin-1 contribute to skeletal malformation (i.e., lengthening of the limbs, kyphosis, and deformity of the chest wall) [29], and osteoporosis results in fractures of the bone, bone pains, and defects. An in-depth examination should include all the clinical results, imaging, and assessment of the risk factors. Musculoskeletal instability and frequent joint injury are also associated with joint hypermobility, which is typical of Marfan syndrome and is caused by laxity of the connective tissues [30]. Medical instruments such as the Beighton score may be used to measure hypermobility and differentiate between pathological laxity of the joint and benign ones [31]. Osteoporosis does not cause joint hypermobility, but the subsequent instability and lack of physical activity have the indirect effect of deteriorating bone health. FBN1 mutations are genetic tests that can play an essential role in the diagnosis of Marfan syndrome, especially in patients with either overlapping or atypical phenotypes [28]. Identification of a pathogenic variant assists in differentiating Marfan syndrome from any other hereditary connective tissue disorder and helps in the early surveillance and preventive control. Imaging research is important for determining bone health. The most common form of measuring BMD in the diagnosis of osteoporosis or osteopenia is dual-energy X-ray absorptiometry (DXA) [32]. Further radiographic visuals could capture vertebral fractures or defects, which otherwise may be treated in a clinical manner. Skeletal, ocular, cardiovascular, and genetic characteristics are combined in clinical diagnostic models such as the Ghent criteria, which identify a diagnosis of Marfan syndrome and direct its management [33]. A careful physical examination is vital, and the proportions of the limbs, spinal position, chest wall structure, joint stability, and any evidence of cardiovascular compromise, including murmurs or aortic root dilation, are considered [34]. Simultaneously, laboratory tests, such as serum calcium, vitamin D, and bone-specific alkaline phosphatase, are used to evaluate bone metabolism and determine the correctable factors of low BMD. Among these, serum tartrate-resistant acid phosphatase 5b (TRACP-5b) is a highly specific marker of osteoclastic bone resorption, correlates strongly with osteoclast number, and is not influenced by renal function. Including TRACP-5b can therefore enhance the evaluation of bone turnover in Marfan-related osteoporosis and assist in monitoring therapeutic response [35]. Bone turnover measurements and, in some cases, sophisticated genetic measurements could help in stratifying the risk and monitoring therapy response [36].
Clinical implications and future directions
The coexistence of Marfan syndrome and osteoporosis is a matter of significant diagnostic and treatment concerns. Early indicators of bone frailty may be obfuscated by overlapping skeletal abnormalities, and this is where proactive screening and routine BMD evaluation are essential in this potential high-risk group [19][37]. This necessitates an interdisciplinary solution, and the interdisciplinary team to use is cardiology, orthopedics, endocrinology, genetics, and rehabilitation medicine [38,39]. One-to-one management ought to consider age, sex, severity of diseases, fracture history, cardiovascular status, as well as the tolerability of treatment. The main aspects of long-term care include lifestyle changes, an exercise regimen tailored to the cardiovascular impairments, and a cautious pharmacologic approach. Prospective treatments that are aimed at bone remodeling and signaling pathways, for example, anti-sclerostin treatment or other new bone-active drugs, can bring future benefits to patients with Marfan-related osteoporosis but have to be evaluated thoroughly in clinical trials [40,41]. However, because anti-sclerostin antibodies such as romosozumab have shown higher rates of serious cardiovascular adverse events in phase III trials compared with control treatments [42,43] and regulatory agencies have issued cardiovascular warnings based on these findings [44], and because no dedicated safety data exist for individuals with Marfan syndrome, their use in this population should be considered theoretical and approached with caution until adequately studied. Imaging and age-related developments in bone assessment methods could also improve early detection, risk classification, and monitoring of the response. Continuous cooperation between academic centers, clinicians, and patient groups will also be critical to enhance evidence-based interventions and implement research findings into more promising results for patients with Marfan syndrome and osteoporosis.
Conclusions
Marfan syndrome refers to an inherited connective tissue disorder in which skeletal integrity is affected by the FBN1 defects and is prone to osteoporosis and fragility fractures. Skeletal deformities are associated with reduced bone mineral density, abnormal bone architecture, and mechanical stress, all of which increase the risk of fracture and impairment. Diagnosis relies on clinical criteria, imaging, genetic testing, and biochemical evaluation to accurately diagnose the disease. This narrative review has demonstrated the significance of osteoporosis as an important complication of Marfan syndrome and the necessity to treat it thoroughly in a personalized manner, which presupposes lifestyle changes, the use of pharmacologic treatment in case of need, and continuous multidisciplinary care. Existing interventions to maintain bone health, such as early detection and intervention, can mitigate the nightmares of bone fractures and enhance quality of life in this susceptible group.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Osteoporosis and its complications Med Clin North Am Varacallo MA Fox EJ 8178319820142499405410.1016/j.mcna.2014.03.007 · doi ↗ · pubmed ↗
- 2Assessment of bone mineral status in children with Marfan syndrome Am J Med Genet A Grover M Brunetti-Pierri N Belmont J 22212224158 A 20122288773110.1002/ajmg.a.35540 PMC 3429634 · doi ↗ · pubmed ↗
- 3Engineered mutations in fibrillin-1 leading to Marfan syndrome act at the protein, cellular and organismal levels Mutat Res Rev Mutat Res Zeyer KA Reinhardt DP 71876520152628176510.1016/j.mrrev.2015.04.002 · doi ↗ · pubmed ↗
- 4Cardiac complications in Marfan syndrome: a review Cureus Singh J Wanjari A 014202210.7759/cureus.29800 PMC 962202736340521 · doi ↗ · pubmed ↗
- 5Unique features of cardiovascular involvement and progression in children with Marfan syndrome justify dedicated multidisciplinary care J Cardiovasc Dev Dis Baban A Parlapiano G Cicenia M 411202410.3390/jcdd 11040114 PMC 1105018138667733 · doi ↗ · pubmed ↗
- 6New horizons in treatment of osteoporosis Daru Tabatabaei-Malazy O Salari P Khashayar P Larijani B 22520172817385010.1186/s 40199-017-0167-z PMC 5297185 · doi ↗ · pubmed ↗
- 7The spectrum of pediatric osteoporosis Pediatric Bone Bianchi ML Glorieux FH 978Amsterdam, Netherlands Elsevier 10162012
- 8Effectiveness of combination of losartan potassium and doxycycline versus single-drug treatments in the secondary prevention of thoracic aortic aneurysm in Marfan syndrome J Thorac Cardiovasc Surg Yang HH Kim JM Chum E van Breemen C Chung AW 30531214020102018919310.1016/j.jtcvs.2009.10.039 · doi ↗ · pubmed ↗