ACTN4 p.Ile150Met Causes FSGS With Validation in Primary Fibroblasts and Immortalized Podocytes
Melanie Grosch, Jan René Haak, Cathiana Kolb, Izabela Plogmann, Mara Sanches Guaragna, Antje Wiesener, Francesca Pasutto, Florian J. Wopperer, Lena Pollinger, Felix B. Engel, Michael S. Wiesener, Mario Schiffer, Tilman Jobst-Schwan

Abstract
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
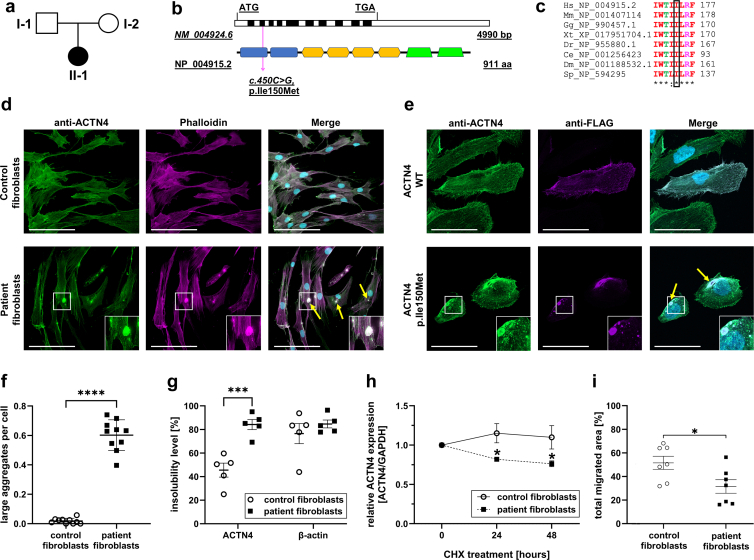 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsComplement system in diseases · Renal Diseases and Glomerulopathies · Immunodeficiency and Autoimmune Disorders
Introduction
Podocytes are highly specialized, terminally differentiated epithelial cells forming an essential part of the glomerular filtration barrier. Through foot processes, they surround capillaries and maintain the selective permeability of the glomerular filter. This architecture relies on a tightly regulated actin cytoskeletonS1; dysregulation causes podocyte injury, detachment, and progression to end-stage renal disease. Multiple actin-regulating proteins, when mutated, are known to cause podocytopathies.1 The α-actinin (ACTN) family comprises 4 actin filament–crosslinking proteins.S2^,^S3 ACTN4 (OMIM: 604638S4) is the predominant paralogue in podocytes,S5 where it preserves cytoskeletal integrity.2^,^S6 Pathogenic ACTN4 variants cause podocyte injury and focal segmental glomerulosclerosis (FSGS1, OMIM: 603278S4).3, 4, 5, 6^,^S7^,^S8 Reported mutations cluster within the actin-binding domain, increasing actin affinity, and generating actin/ACTN4 aggregates4 (Supplementary Table S1 and Figure S1). Functional validation of ACTN4 variants has typically relied on transient or stable overexpression in immortalized podocytes.
Human dermal fibroblasts (HDFs) provide an alternative; they are obtained by skin biopsy, retain the patient’s genetic background, and have supported the functional validation of novel renal disease genes.S9–S11 However, HDFs have not been applied to the functional assessment of ACTN4 variants.
Here, we identified a novel ACTN4 variant, NC_000019.10: g.38704986C>G, c.450C>G, p.Ile150Met (NM_004924.6). We show that patient-derived HDFs reproduce the key pathogenic features of ACTN4 variants—abnormal actin binding, aggregate formation, reduced protein stability, and impaired motility—supporting their utility for functional characterization of novel ACTN4 variants. All material and methods used are described in detail in the Supplementary Material and Methods.
Results
Clinical and Genetic Findings
We evaluated a 32-year-old woman with severe childhood-onset steroid-resistant nephrotic syndrome and no family history of renal disease (Figure 1a). Extrarenal manifestations included sensorineural hearing impairment and asymmetric breast development. At age 5, kidney biopsy revealed minimal change glomerulonephritis, progressing to end-stage renal disease by the age of 8 years. Native kidney tissue was unavailable for histological review.Figure 1. Genetic analysis of the patient with the ACTN4 c.450C>G, p.Ile150Met variant and functional analysis in cell culture experiments. (a) Pedigree of the index patient. A novel variant in ACTN4 was detected by whole exome sequencing. Sanger sequencing confirmed absence of the variant in the patient mother (I-2) and presence in the patient (II-1) at NC_000019.10:g.38704986. The patient’s father was not available for genetic testing. (b) Graphical visualization of the mRNA (exons in alternating colors) and the protein structure (blue: calponin homology domains; orange: spectrin repeats; green: EF-hand motif) of ACTN4 and localization of the patient variant in Exon 4/the first CH domain (magenta arrow). (c) Alignment with orthologues of different species shows the high conservation of the affected amino acid residue in ACTN4 (black box). (d) Primary human fibroblasts were stained for ACTN4 (green) and F-actin (phalloidin; magenta). In patient fibroblasts, expressing the ACTN4 p.Ile150Met variant, actin/ACTN4 aggregates are visible (yellow arrows), but not in healthy control fibroblasts (n = 3). Scale bars correspond to 100 μm. (e) Immortalized human podocytes were electroporated and transfected with FLAG-tagged ACTN4 WT or ACTN4 c.450C>G variant. After transfection, cells were fixed and stained for FLAG-tag (green) and with phalloidin (magenta) and DAPI (blue). Cell imaging was performed using a Zeiss confocal microscope at 20× magnification. Control transfected cells did not show actin/FLAG-tagged ACTN4 WT aggregates (upper panel). Contrary to that, FLAG-tagged ACTN4 p.Ile150Met forms actin/ACTN4 aggregates (lower panel; yellow arrows), similarly to the patient fibroblasts. Scale bars correspond to 50 μm. (f) Particle analysis script in Fiji ImageJ was used to quantify aggregates in fibroblasts. Each microscopic field of view was investigated for the number of large aggregates (size > 0.97 μm^2^) related to cell numbers (based on nuclei numbers). The graph represents 2 technical replicates per group with 5 microscopic fields of view (20×) per replicate (control: 37 aggregates/1714 nuclei in total, patient: 939 aggregates/1647 nuclei; mean and SEM; ∗∗∗∗P < 0.0001). (g) Healthy control fibroblasts and patient fibroblasts were lysed in Triton X-100 containing lysis buffer and centrifuged to obtain Triton X-100 insoluble (TI) and soluble (TS) fractions. TI fractions contain large cytoskeletal structures including F-actin, whereas TS fractions contains G-actin. Densitometry of fractionation experiments showed significantly higher insolubility level of ACTN4 (displayed as relative expression levels TI/[TI+TS]) in patient fibroblasts (ACTN4 p.Ile150Met) compared with control fibroblasts (ACTN4 WT). In contrast, the distribution of actin is comparable between control and patient fibroblasts. (Results of n = 5 experiments, mean and SEM, ∗∗∗P < 0.001). (h) Control and patient fibroblasts were treated with 100 μg/ml CHX for up to 48 hours. Proteins were isolated and subjected to immunoblotting. Densitometry of CHX pulse chase assay experiments (normalized by GAPDH) displayed a faster degradation of ACTN4 p.Ile150Met (patient fibroblasts) compared with ACTN4 WT (control fibroblasts). Results of n = 3 experiments are displayed (mean and SEM, ∗P < 0.05). (i) Control and patient fibroblasts were plated in an ibidi 2-chamber silicone insert. Insert was removed and cell migration was monitored in real time. Migration of patient fibroblasts (ACTN p.Ile150Met) is significantly reduced compared with control (ACTN4 WT) fibroblasts. Total migrated area in % was manually outlined and measured using ImageJ. Results of n = 7 experiments are displayed (mean and SEM; ∗ P < 0.05). CHX, cycloheximide; WT, wild type.
She underwent her first kidney transplant at the age of 10 years, which failed because of recurrent pyelonephritis. A second transplant, from her mother at the age of 31 years was initially complicated by chronic active T-cell–mediated rejection (BANFF IA) after azathioprine substitution for mycophenolate mofetil to enable pregnancy. Graft function stabilized after mycophenolate mofetil reintroduction alongside tacrolimus and prednisolone. Whole-exome sequencing with analysis of the abnormal renal glomerulus morphology panel (HP_0000095, October 2019) identified a heterozygous ACTN4 c.450C>G, p.(Ile150Met) variant, located in exon 4 within the first calponin-homology domain (Figure 1b). No additional variants appeared in an extended 726-gene abnormal renal physiology panel (HP_0012211, May 2024). Screening of 149 genes associated with hearing impairment (https://hereditaryhearingloss.org) was negative. Only one other ACTN4 variant (p.Trp59Arg) has been associated with both childhood end-stage renal disease and hearing impairment, suggesting an expanded extrarenal phenotype.S7^,^S12
Segregation analysis showed absence of the variant in the healthy mother (Supplementary Figure S2). Paternal DNA from the father and other relatives was unavailable, but no family history of renal disease was reported. The variant was absent from gnomADS13 and ClinVarS14. Cross-species alignment confirmed conservation of Ile150 across orthologues (Figure 1c, Table 1). In silico prediction tools (SIFTS15, MutationTasterS16, PolyPhen-2S17) classified the variant as deleterious.Table 1. Genetic data of a novel ACTN4 variantGeneACTN4Accession numberNM_004924.6hg38 positionchr19:38704986Nucleotide changec.450C>GAmino acid changep.Ile150MetExon (zygosity)4 (heterozygous)gnomADnot applicableClinVarnot applicableSIFTdeleteriousMutation Tasterdisease causingPolyPhen-21ACMG classificationlikely pathogenic (PS3, PM1, PM2, PP3)Amino acid conservation to speciesSchizosaccharomyces pombeACMG, American College of Medical Genetics.Whole exome sequencing was performed using a blood sample of a female patient with primary focal segmental glomerulosclerosis using a lllumina HiSeq 2500 (Illumina, San Diego, CA). Subsequent analysis using the HP_0000095 gene panel (abnormal renal glomerulus morphology) revealed a heterozygous nucleotide change at position 450 from cytosine to guanine, leading to an amino acid change at position 150 from isoleucine to methionine. The affected amino acid is highly conserved across different species. The amino acid change is predicted to be deleterious and disease-causing by different algorithms (SIFTS15 MutationTasterS16 PolyPhen-2S17). The variant is not present in gnomAD or in ClinVar.ACMG classificationS24: PS3: well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product; PM1: located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation; PM2: absent from controls in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium; PP3: multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.).
ACTN4 p.Ile150Met Forms Actin/ACTN4 Aggregates in Primary Fibroblasts and Immortalized Podocytes
To assess cellular effects, we established HDFs from the patient and an age- and sex-matched control. HDFs are well-suited for ACTN4 functional studies because they express ACTN4 robustly7 and circumvent limitations of podocyte culture.S18
In control fibroblasts, ACTN4 colocalized with filamentous actin (Figure 1d). In patient fibroblasts, actin/ACTN4 aggregates were observed (Figure 1d and f). For validation, we transfected immortalized human podocytes with FLAG-tagged ACTN4 wild type or ACTN4 p.Ile150Met. Aggregates occurred only in cells expressing ACTN4 p.Ile150Met (Figure 1e). To confirm aggregate formation, we performed actin fractionation of control and patient fibroblasts, separating Triton X-100 insoluble (TI) and soluble (TS) fractions. The TI fraction contains large structures, including cytoskeletal aggregates, whereas soluble components are contained in the TS fraction.2^,^S20 In the patient fibroblasts, significantly more ACTN4 was detected in the TI fraction than in the control, whereas no significant differences were observed in the TS fraction (Figure 1g, Supplementary Figure S2). Actin distribution between TI and TS fractions did not differ between groups (Figure 1g, Supplementary Figure S3). These findings suggest increased actin affinity of ACTN4 p.Ile150Met in both patient fibroblasts and transfected podocytes compared with the wild type.
Reduced Stability of ACTN4 p.Ile150Met
Actin binding is mediated by the tandem N-terminal calponin-homology domains.S21 Variants in these domains reduce ACTN4 stability.5 Five computational algorithms predicted destabilization of ACTN4 p.(Ile150Met), consistent with analogous substitutions in ACTN1 and ACTN3 (Supplementary Table S2).
Cycloheximide chase assaysS22 support this prediction; ACTN4 levels in control fibroblasts remained stable over 48 hours, whereas patient fibroblasts showed significant decline after 24 hours and further loss at 48 hours (Figure 1h, Supplementary Figure 4A and B).
Impaired Fibroblast Motility
ACTN4 mutations forming aggregates are known to impair cell motility.2^,^5 To assess this, we performed migration assays. At 24 hours, control fibroblasts covered a significantly larger area than patient fibroblasts (Figure 1i, Supplementary Figure 4C/D), indicating impaired motility and cytoskeletal reorganization associated with ACTN4 p.Ile150Met.7^,^S23
Conclusion
Pathogenic ACTN4 variants were first linked to familial focal segmental glomerulosclerosis 2 decades ago.3 Since then, > 20 variants have been described, most within the actin-binding domain. Our patient with early-onset steroid-resistant nephrotic syndrome and end-stage renal disease harbored the novel variant, ACTN4 c.450C>G; p.Ile150Met. Functional assays demonstrated all major hallmarks of pathogenic ACTN4 variants, namely aggregate formation, increased actin affinity, reduced protein stability, and impaired cellular motility.
Feng et al.4 outlined 4 criteria to establish pathogenicity of ACTN4 variants, namely segregation, localization to a conserved domain, abnormal aggregates, and increased actin binding. Our variant fulfils 3 of these 4; segregation was not possible because of lack of paternal DNA. Under the American College of Medical Genetics criteria,S24 ACTN4 c.450C>G, p.Ile150Met is likely pathogenic (PS3, PM1, PM2, PP3).
The mechanism by which ACTN4 variants cause podocyte injury remains incompletely understood. Some variants destabilize the cytoskeleton, whereas others form aggregates that may act through a toxic gain-of-function,2^,^4^,^5^,^8^,^S25 Our findings suggest that early-onset variants, such as p.Ile150Met, act through aggregate toxicity, consistent with previous studies on ACTN4 p.Met240Thr.6 Analogous to protein aggregation in neurodegenerative diseases such as Huntington’s or Parkinson’s,S26^,^S27 ACTN4 aggregates may disrupt cellular homeostasis.S28
Our study highlights the potential of primary HDFs as a functional assay system. Compared with immortalized podocytes, HDFs are easy to obtain,S29 maintain proliferative capacity for multiple passages,S30 and preserve the native genetic background.9 Importantly, we demonstrated hallmark features of pathogenic ACTN4 variants directly in patient-derived fibroblasts, avoiding immortalization or artificial overexpression.S31 Limitations of our study include donor age effects,S32^,^S33 passage-dependent changes,S34 and absent expression of some disease-relevant genes, though CRISPR-based activation strategies may overcome this.S35 However, reliance on the patient’s genetic background can be a drawback because the absence of an isogenic control line makes it difficult to exclude contributions from other genetic factors.
Previous studies using murine fibroblasts or immortalized human fibroblasts suggested this approach8^,^S28^,^S36; here we show, for the first time, that primary HDFs can directly validate a novel ACTN4 variant. This expands the toolkit for functional genomics of kidney diseases and strengthens the case for using patient-derived fibroblasts in personalized medicine.
Clinically, recognition of pathological ACTN4 variants is critical. For our patient, the ACTN4 variant indicates a structural podocytopathy rather than an immune-mediated disease. Knowledge of the genetic etiology could have prevented unnecessary long-term immunosuppression during childhood, reducing risks for infection and malignancy. Furthermore, recurrence of the primary disease in the kidney allograft can be excluded, with important implications for transplantation management.
In conclusion, we identified a novel ACTN4 c.450>G, p.Ile150Met variant in a patient with early-onset steroid-resistant nephrotic syndrome. Functional studies in patient-derived fibroblasts and podocytes confirm its deleterious effects on actin binding, protein stability, and cellular motility. Our data establish this variant as likely pathogenic and demonstrate that primary HDFs represent a powerful, accessible in vitro model for the characterization of novel ACTN4 variants. This approach can support genetic diagnosis, inform patient management, and advance personalized medicine in glomerular diseases.
Disclosure
All the authors declared no competing interests.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kopp J.B.Anders H.J.Susztak K.Podocytopathies Nat Rev Dis Primers 620206810.1038/s 41572-020-0196-732792490 PMC 8162925 · doi ↗ · pubmed ↗
- 2Michaud J.L.R.Chaisson K.M.Parks R.J.Kennedy C.R.Kennedy C.R.J.FSGS-associated α-actinin-4Kidney Int 7020061054106110.1038/sj.ki.500166516837921 · doi ↗ · pubmed ↗
- 3Kaplan J.M.Kim S.H.North K.N.Mutations in ACTN 4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis Nat Genet 24200025125610.1038/7345610700177 · doi ↗ · pubmed ↗
- 4Feng D.Steinke J.M.Krishnan R.Birrane G.Pollak M.R.Functional validation of an alpha-actinin-4 mutation as a potential cause of an aggressive presentation of adolescent focal segmental glomerulosclerosis: implications for genetic testing P Lo S One 112016 e 016746710.1371/journal.pone.0167467 PMC 515818627977723 · doi ↗ · pubmed ↗
- 5Bartram M.P.Habbig S.Pahmeyer C.Three-layered proteomic characterization of a novel ACTN 4 mutation unravels its pathogenic potential in FSGS Hum Mol Genet 2520151152116410.1093/hmg/ddv 63826740551 · doi ↗ · pubmed ↗
- 6Odenthal J.Dittrich S.Ludwig V.Modeling of ACTN 4-based podocytopathy using drosophila nephrocytes Kidney Int Rep 8202231732910.1016/j.ekir.2022.10.02436815115 PMC 9939316 · doi ↗ · pubmed ↗
- 7Shao H.Wang J.H.C.Pollak M.R.Wells A.α-actinin-4 is essential for maintaining the spreading, motility and contractility of fibroblasts P Lo S One 52010 e 1392110.1371/journal.pone.0013921 PMC 297868021085685 · doi ↗ · pubmed ↗
- 8Ehrlicher A.J.Krishnan R.Guo M.Bidan C.M.Weitz D.A.Pollak M.R.Alpha-actinin binding kinetics modulate cellular dynamics and force generation Proc Natl Acad Sci U S A 11220156619662410.1073/pnas.150565211225918384 PMC 4450414 · doi ↗ · pubmed ↗