Unmasking Isolated Glucocorticoid Deficiency: Clinical Insights From 2 Cases
Ayushi Singhal, Jayakrishnan C Menon, Subhash Chandra Yadav, Ayush Gupta, Gunna Sriharsha, Eesh Bhatia

TL;DR
This paper presents two cases of rare familial glucocorticoid deficiency, highlighting genetic causes and clinical features for better diagnosis.
Contribution
The study identifies novel genetic variants in AFF2 and CYP11A1 associated with familial glucocorticoid deficiency.
Findings
Whole-exome sequencing revealed a biallelic variant in the melanocortin 2 receptor gene in one patient.
Compound heterozygous variants in the CYP11A1 gene were found in the second patient.
Both patients improved with glucocorticoid replacement therapy.
Abstract
Familial glucocorticoid deficiency (FGD) is a rare autosomal recessive disorder characterized by unresponsiveness to adrenocorticotropin (ACTH) with preserved mineralocorticoid secretion. We describe 2 patients who presented with FGD. The first patient was born to a third-degree consanguineous marriage, and suffered from global developmental delay and recurrent seizures since childhood. He presented to us at age 22 years with severe hyponatremia. Laboratory investigations showed subclinical hypothyroidism, low cortisol, high ACTH, and normal plasma renin activity. After exclusion of common causes of primary adrenal insufficiency (PAI), we undertook whole-exome sequencing (WES) that revealed 2 variants—a hemizygous deletion involving exons 10 to 21 of the AFF2 gene known to cause X-linked intellectual developmental disorder-109 and a biallelic variant in the melanocortin 2 receptor gene…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
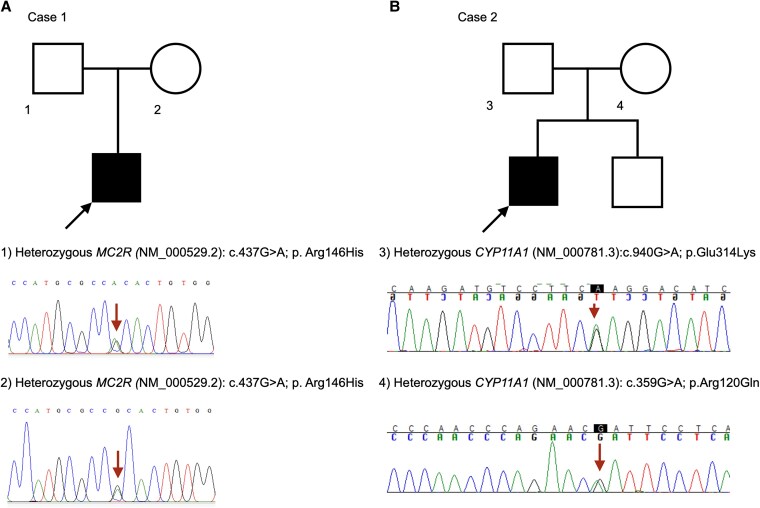 Figure 1
Figure 1| Gene symbol | Gene name | FGD subtype | Inheritance pattern | OMIM No. | Key features |
|---|---|---|---|---|---|
|
| Melanocortin 2 receptor | FGD type 1 | Autosomal recessive | 202200 | Most common (25% of cases); typically presents in early childhood [ |
|
| Melanocortin 2 receptor accessory protein | FGD type 2 | Autosomal recessive | 607398 | Accounts for 20% of cases of FGD. Often severe and presents in infancy [ |
|
| Nicotinamide nucleotide transhydrogenase | FGD type 4 | Autosomal recessive | 614736 | Associated with increased oxidative stress [ |
|
| Thioredoxin reductase 2 | FGD type 5 | Autosomal recessive | 617825 | Rare; mitochondrial redox pathway defect [ |
|
| Steroidogenic acute regulatory protein | FGD-like | Autosomal recessive | 600617 | Usually associated with lipoid congenital adrenal hyperplasia [ |
|
| Cytochrome P450 family 11 subfamily A member 1 | FGD-like | Autosomal recessive | 613743 | May present later; phenotype variable [ |
|
| Minichromosome maintenance complex component 4 | FGD-like | Autosomal recessive | 609981 | Associated with natural killer cell deficiency [ |
| Laboratory tests | Patient 1 | Patient 2 | Normal range |
|---|---|---|---|
| Hemoglobin | 13.6 gm/dL | 13.0 gm/dL | 12-16 gm/dL |
| 136 g/L | 130 g/L | 120-160 g/L | |
| Total leukocyte count | 10.1 × 10⁹/L | 7.2 × 10⁹/L | 4-11 × 10⁹/L |
| Platelet count | 152 × 10⁹/L | 200 × 10⁹/L | 150-400 × 10⁹/L |
| Serum creatinine | 1 mg/dL | 0.8 mg/dL | 0.5-1.6 mg/dL |
| 88.4 µmol/L | 70.7 µmol/L | 44-141 µmol/L | |
| Serum sodium | 120 mEq/L | 130 mEq/L | 135-145 mEq/L |
| 120 mmol/L | 130 mmol/L | 135-145 mmol/L | |
| Serum potassium | 4.5 mEq/L | 3.9 mEq/L | 3.5-5.5 mEq/L |
| 4.5 mmol/L | 3.9 mmol/L | 3.5-5.5 mmol/L | |
| Blood glucose | 82 mg/dL | 77 mg/dL | 70-100 mg/dL |
| 4.6 mmol/L | 4.3 mmol/L | 3.9-5.6 mmol/L | |
| Serum osmolarity | ND | 263 mOsmol/kg | 280-300 mOsmol/kg |
| Urine osmolarity | ND | 301 mOsmol/kg | 500-800 mOsmol/kg |
| Urinary spot sodium | ND | 30 mEq/L | 20-301 mEq/L |
| 30 mmol/L | 20-301 mmol/L | ||
| TSH | 8.3 mIU/L | 6.7 mIU/L | 0.4-4.0 mIU/L |
| Total T4 | ND | 9.4 µg/dL | 4.5-12.5 µg/dL |
| 120.6 nmol/L | 58.0-162.0 nmol/L | ||
| Free T4 | 1.0 ng/dL | ND | 0.9-1.9 ng/dL |
| 13.0 pmol/L | 12-24 pmol/L | ||
| TPO antibody | 30 IU/mL | 21 IU/mL | <35 IU/mL |
| Total testosterone | 340 ng/dL | 384 ng/dL | 250-835 ng/dL |
| 11.8 nmol/L | 13.3 nmol/L | 8.6-29.0 nmol/L | |
| 8 | 705.0 pg/mL | 1963.0 pg/mL | 7.3-63.0 pg/mL |
| 155.3 pmol/L | 432.0 pmol/L | 1.6-13.9 pmol/L | |
| 11 | ND | 141 pg/mL | <50 pg/mL |
| 31 pmol/L | <11 pmol/L | ||
|
| <1.0 µg/dL | < 0.2 µg/dL | 3.0-25.0 µg/dL |
| <27.6 nmol/L | <5.0 nmol/L | 83.0-690.0 nmol/L | |
| Stimulated cortisol | ND | 0.3 µg/dL | >14.5 µg/dL |
| 8.8 nmol/L | >400.0 nmol/L | ||
| DHEAS | 22.1 µg/dL | 7.4 µg/dL | 66.3-405.3 µg/dL |
| 0.6 µmol/L | 0.2 µmol/L | SI: 1.8-11.0 µmol/L | |
| Aldosterone | ND | 14.4 ng/dL | 7.0-34.0 ng/dL |
| 0.4 nmol/L | 0.2-0.9 nmol/L | ||
| PRA | 1.2 ng/mL/h | 0.9 ng/mL/h | 0.5-4.9 ng/mL/h |
| 1.2 µg/L/h | 0.9 µg/L/h | 0.5-4.9 µg/L/h |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHormonal Regulation and Hypertension · Adrenal Hormones and Disorders · Regulation of Appetite and Obesity
Introduction
Autoimmune adrenalitis accounts for the majority of cases of primary adrenal insufficiency (PAI) in Western countries, and infections such as tuberculosis and HIV cause most of the cases in countries where they are common [1]. Among children, adolescents, and young adults, monogenic forms of PAI are also frequently encountered, and their detection can have important implications for therapy and genetic counseling. The most common monogenic form of PAI is classic congenital adrenal hyperplasia due to CYP21A2 mutations, followed by X-linked adrenoleukodystrophy [1].
Familial glucocorticoid deficiency (FGD) encompasses a spectrum of recessive disorders that solely affect adrenocorticotropin (ACTH) signaling and hence spares mineralocorticoid production [2]. Mutations in the melanocortin 2 receptor (MC2R) gene account for approximately 25% of cases (FGD type 1; OMIM No. 202200), while mutations in the melanocortin 2 receptor accessory protein (MRAP) gene, essential for MC2R trafficking and function, account for about 20% (FGD type 2; OMIM No. 607398) [3]. Other genes are less frequently implicated, as summarized in Table 1. The diagnosis of FGD is of practical significance since mineralocorticoid replacement is not indicated in these cases. Given its autosomal recessive inheritance, genetic counseling is crucial for early identification of at-risk individuals and informed reproductive decisions. In this report, we describe the clinical course of 2 unrelated individuals with isolated glucocorticoid deficiency.
Case Presentation
Case 1
This male patient was born of a third-degree consanguineous marriage (parents were first cousins). His perinatal period was normal and his birth weight was 2.6 kg. He had delayed milestones, speaking monosyllables by age 2 years and walking independently by age 3 years. He developed recurrent episodes of generalized tonic-clonic seizures since age 4 years. He was admitted with status epilepticus in a nearby hospital at age 8 years, where he underwent evaluation including lumbar puncture and brain imaging, and was diagnosed with encephalitis. No electrolyte abnormalities were documented. Seizures improved with antiepileptic therapy. He was admitted into a school for children with a disability at age 9 years, but was forced to drop out due to general ill health, fatigue, and poor scholastic ability. The parents give history of recurrent episodes of vomiting with frequent hospital admissions. He presented to our hospital at age 22 years with intractable seizures and severe hyponatremia, prompting referral to the endocrinology department. Examination was unremarkable except for generalized hyperpigmentation. He had normal stature (163 cm at presentation), which was also appropriate for his mid-parental height.
Case 2
A 25-year-old man presented with recurrent episodes of dizziness, vomiting, and seizures over the past 5 years. Hyponatremia was documented on multiple occasions, with a nadir sodium of 105 mEq/L (SI: 105 mmol/L) (reference range, 135-145 mEq/L [SI: 135-145 mEq/L]). He was managed with hypertonic saline, fluid restriction, and oral salt on these occasions. Prior brain imaging and electroencephalogram were normal, and he was on brivaracetam 50 mg twice daily at presentation. He was born at term to a nonconsanguineous couple with a normal perinatal period and development. There was no history of genital ambiguity, significant past illness, or similar family history. Parents did not notice hyperpigmentation or dysphagia. On examination, height was 169 cm and weight 53 kg (body mass index, 18.6). Testicular volume was 20 mL bilaterally, pubic hair stage 5, and stretched penile length 11 cm. There was no evidence of alacrimia, and examination was otherwise unremarkable.
Diagnostic Assessment
Case 1
Laboratory findings at presentation are summarized in Table 2. The combination of clinical features, low serum cortisol (Am cortisol <1 µg/dL [SI: <27.6 nmol/L]; reference range, 3-25 µg/dL [SI: 83-690 nmol/L]), and elevated Am ACTH (705 pg/mL [SI: 155.3 pmol/L]; reference range, 7.3-63 pg/mL [SI: 1.6-13.9 pmol/L]) confirmed PAI. Serum DHEAS (dehydroepiandrosterone sulfate) was low (22.1 µg/dL [SI: 0.6 µmol/L]; reference range, 66.3-405.3 µg/dL [SI: 1.8-11.0 µmol/L]). He was started on glucocorticoid and mineralocorticoid replacement despite normal plasma renin activity (PRA) (1.2 ng/mL/hour [SI: 1.2 µg/L/hour]; reference range, 0.5-4.9 ng/mL/hour [SI: 0.5-4.9 µg/L/hour]). Vomiting, fatigue, seizures, and hyponatremia resolved with treatment, and his longstanding skin hyperpigmentation improved.
Evaluation for etiology revealed no alacrimia, dysphagia, or autonomic dysfunction. Computed tomography (CT) showed atrophic adrenals. X-linked adrenoleukodystrophy was excluded by normal urinary very long-chain fatty acids. 21-Hydroxylase (21-OH) antibody estimation (using an in-house immunoprecipitation assay using in vitro transcribed and translated 21-OH [4]) was negative.
Given the early age of onset and parental consanguinity, an inherited form of PAI was suspected. Exome sequencing using the Illumina HiSeq X platform (Illumina) identified 2 variants: a monoallelic deletion involving exons 10 to 21 of the AFF2 gene, which is implicated in X-linked intellectual developmental disorder-109 (MRX109) (OMIM No. 309548) [5], and a biallelic missense variant in the MC2R gene (NM_000529.2: c.437G > A; p. Arg146His), previously reported in FGD1 [6-9]. The MC2R variant was absent in the 1000 Genomes database and had a minor allele frequency of 0.002% in gnomAD (v 4.1.0). Parental testing confirmed heterozygosity by Sanger sequencing (Fig. 1). The variant was classified as pathogenic as per American College of Medical Genetics and Genomics (ACMG) 2015 criteria (PS3 PM1 PM2 PM3 PP3 PP4) [10].
Details of family screening for genetic variants. Square—male, circle—female, black—affected, white—unaffected. A shows the pedigree chart for case 1. Sanger sequencing of parents revealed that they were heterozygous for the variant (MC2R [NM_000529.2]: c.437G > A; p. Arg146His). B shows the pedigree chart for case 2. The proband was compound heterozygous for 2 variants in the CYP11A1 gene. Sanger sequencing of the parents revealed that he inherited the CYP11A1 (NM_000781.3): c.940G > A; p. Glu314Lys variant from his father and the CYP11A1 (NM_000781.3): c.359G > A; p. Arg120Gin variant from his mother.
Case 2
Laboratory investigations at presentation (see Table 2) were consistent with PAI (Am cortisol <1 µg/dL [SI: <27.6 nmol/L]; Am ACTH 1963pg/mL [SI: 432 pmol/L]). Serum DHEAS was low (7.4 µg/dL [SI: 0.2 µmol/L]). Plasma aldosterone and PRA (0.9 ng/mL/hour [SI: 0.9 µg/L/hour]) were within reference range. CT revealed bilaterally atrophic adrenals. Suspecting FGD, we performed exome sequencing using the Illumina HiSeq X platform (Illumina), which identified 2 CYP11A1 variants in trans (confirmed by parental testing; see Fig. 1). The first variant, CYP11A1 (NM_000781.3):c.940G > A (p.Glu314Lys) in exon 5, affects the cytochrome P450 domain, has minor allele frequencies of 0.05% in 1000 Genomes, and 0.39% in gnomAD (v4.1.0), and has been shown to cause aberrant splicing and FGD [11-14]. It was classified as “likely pathogenic” (PS3, PM3) per ACMG 2015 criteria [10]. The second variant, CYP11A1 (NM_000781.3):c.359G > A (p.Arg120Gln) in exon 2, also lies in the cytochrome P450 domain, is extremely rare (0.001% in gnomAD; absent in 1000 Genomes), and has been previously reported in PAI [14, 15]. In silico analyses (PolyPhen-2, SIFT, MutationTaster2021) predicted it to be damaging. This variant was likewise classified as “likely pathogenic” (PM2, PM3, PP3, PP5) [10].
Treatment
Case 1
The genetic diagnosis of FGD1 prompted a review of the initial presentation, underscoring the relevance of normal PRA and normokalemia. Consequently, mineralocorticoid therapy was discontinued, while glucocorticoid replacement was continued.
Case 2
The patient was started on glucocorticoid replacement at 10 mg/kg/m^2^ hydrocortisone.
Outcome and Follow-up
Both patients have remained well over nearly 2 years of follow-up, without recurrence of seizures, vomiting, or hyponatremia. Blood pressure, aldosterone, and PRA levels have remained normal, and the families received genetic counseling.
Discussion
We describe 2 cases of FGD with distinct genetic etiologies—one with biallelic MC2R mutations and the other with compound heterozygous CYP11A1 variants. Both patients presented in their 20s with severe hyponatremia and features of glucocorticoid deficiency. Normal PRA and aldosterone levels were key clues indicating preserved mineralocorticoid function.
The first patient had FGD1, diagnosed at age 22 years, although symptoms of adrenal insufficiency were probably present earlier. His developmental delay, intellectual disability, and initial seizures were probably due to the AFF2 gene mutation causing X-linked intellectual developmental disorder-109, as hyponatremia was not documented during the initial evaluation. This makes it difficult to determine the precise age of onset of FGD.
FGD1 typically presents later than FGD2, with the largest published cohort reporting a median age of onset of 2 years (range, 0-16 years) compared to 0.08 years (range, 0-1.6 years) for FGD2 [3]. Isolated cases with even later presentation have been described, with the oldest patient diagnosed at age 30 years [16]. This has been attributed to the severity of the mutation. Most MRAP mutations are splice-site or nonsense variants that abolish protein expression, resulting in complete retention of MC2R within the endoplasmic reticulum (ER). In contrast, most MC2R mutations are missense variants (as in our case), causing varying degrees of receptor misfolding and impaired trafficking from the ER to the cell surface, leading to reduced receptor expression (20%-100% of wild-type) and diminished ACTH signaling [9].
A feature of FGD1 that is being increasingly recognized is hypothyroidism [2]. The case presented here had subclinical hypothyroidism at presentation, while previous reports show that this can range from transient neonatal hypothyroidism to overt hypothyroidism. The exact mechanism for this has not been elucidated. Another frequently reported but variable phenotypic feature of FGD1 is tall stature, which is thought to result from the stimulatory effects of markedly elevated ACTH levels on growth plate chondrocytes [17]. This feature was not observed in our patients.
Severe loss-of-function mutations in CYP11A1 (encoding the cholesterol side-chain cleavage enzyme, P450scc) cause primary adrenal failure with 46, XY sex reversal. However, partial inactivating mutations in CYP11A1 have been implicated in the causation of isolated glucocorticoid deficiency [13]. While severe enzyme deficiency presents within the first few days of life, partial loss-of-function variants in CYP11A1 are associated with later onset of PAI ranging from 6 months to 16 years [13]. Patient 2 also had mild subclinical hypothyroidism at presentation (see Table 1), which normalized after glucocorticoid replacement, suggesting a transient, nonthyroidal thyrotropin elevation. An association between thyroid disease and CYP11A1 variants has not been reported to date. The minimal increase in serum cortisol levels following ACTH stimulation likely reflects partial preservation of enzyme activity, which may have contributed to the delayed clinical presentation.
Maharaj et al [13] undertook exome sequencing in 77 individuals or family members with PAI of unknown etiology and found that compound heterozygosity for the c.940G > A CYP11A1 variant with a second disruptive variant accounted for 21% (16/77) of undiagnosed PAI and 4% (16/395) of all patients with adrenal insufficiency in their cohort. The majority of these patients had isolated glucocorticoid deficiency. The exact same defect was detected in our second patient, indicating that this variant is present and may account for a substantial proportion of PAI in our population as well. Maharaj and colleagues [13] demonstrated that the c.940G > A variant, which was previously predicted to be benign, caused mis-splicing, resulting in absent or dysfunctional protein.
Learning Points
FGD should be considered in the differential diagnosis of PAI presenting in children, adolescents, and young adults, as the age at presentation can be highly variable.History of consanguinity or endogamy in parents, normokalemia, and preserved mineralocorticoid function (normal aldosterone and PRA) should raise clinical suspicion for FGD.Early recognition of FGD is crucial, as mineralocorticoid replacement is usually unnecessary, affected individuals may exhibit characteristic phenotypic features, and genetic counseling plays an important role in family screening and risk assessment.
Contributors
All authors made individual contributions to authorship. J.C.M., S.Y., E.B., G.S., A.S., and A.G. were involved in the diagnosis and management of the patients. J.C.M. and A.S. prepared the initial draft of the manuscript. J.C.M., S.Y., E.B., A.G., and A.S. were involved in genetic testing and interpretation. All authors reviewed and approved the final draft.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hahner S, Ross RJ, Arlt W, et al Adrenal insufficiency. Nat Rev Dis Primer. 2021;7(1):19.10.1038/s 41572-021-00252-733707469 · doi ↗ · pubmed ↗
- 2Maharaj AV . Familial glucocorticoid deficiency: the changing landscape of an eponymous syndrome. Front Endocrinol. 2023;14:1268345.10.3389/fendo.2023.1268345 PMC 1077134138189052 · doi ↗ · pubmed ↗
- 3Chung TTLL, Chan LF, Metherell LA, Clark AJL. Phenotypic characteristics of familial glucocorticoid deficiency (FGD) type 1 and 2. Clin Endocrinol (Oxf). 2010;72(5):589‐594.19558534 10.1111/j.1365-2265.2009.03663.x PMC 2855830 · doi ↗ · pubmed ↗
- 4Falorni A, Nikoshkov A, Laureti S, et al High diagnostic accuracy for idiopathic Addison's disease with a sensitive radiobinding assay for autoantibodies against recombinant human 21-hydroxylase. J Clin Endocrinol Metab. 1995;80(9):2752‐2755.7673419 10.1210/jcem.80.9.7673419 · doi ↗ · pubmed ↗
- 5Zou D, Qin B, Wang J, et al AFF 2 is associated with X-linked partial (focal) epilepsy with antecedent febrile seizures. Front Mol Neurosci. 2022;15:795840.35431806 10.3389/fnmol.2022.795840 PMC 9006616 · doi ↗ · pubmed ↗
- 6Weber A, Toppari J, Harvey RD, et al Adrenocorticotropin receptor gene mutations in familial glucocorticoid deficiency: relationships with clinical features in four families. J Clin Endocrinol Metab. 1995;80(1):65‐71.7829641 10.1210/jcem.80.1.7829641 · doi ↗ · pubmed ↗
- 7Capalbo D, Moracas C, Cappa M, et al Primary adrenal insufficiency in childhood: data from a large nationwide cohort. J Clin Endocrinol Metab. 2021;106(3):762‐773.33247909 10.1210/clinem/dgaa 881 · doi ↗ · pubmed ↗
- 8Lin L, Hindmarsh PC, Metherell LA, et al Severe loss-of-function mutations in the adrenocorticotropin receptor (ACTHR, MC 2R) can be found in patients diagnosed with salt-losing adrenal hypoplasia. Clin Endocrinol (Oxf). 2007;66(2):205‐210.17223989 10.1111/j.1365-2265.2006.02709.x PMC 1859977 · doi ↗ · pubmed ↗