A QIIME2-based workflow for multi-amplicon 16S rRNA profiling
Armando G. Licata, Marica Zoppi, Chiara Dossena, Federico Rossignoli, Davide Rizzo, Luca Bergamaschi, Olga Nigro, Stefano Chiaravalli, Maura Massimino, Loris De Cecco

TL;DR
This paper introduces a QIIME2-based workflow for analyzing 16S rRNA sequencing data from multiple amplicon regions.
Contribution
The novel contribution is a validated open-source pipeline that matches proprietary tools in accuracy and depth for multi-amplicon 16S rRNA sequencing.
Findings
The workflow achieves a taxonomic F1-score of 0.875 when benchmarked against a mock community.
Multi-region sequencing outperforms single amplicon approaches in taxonomic resolution.
The pipeline is robust for semiconductor-based sequencing data.
Abstract
We present an open-source QIIME2 pipeline for 16S multi-amplicon sequencing. Benchmarked against proprietary software with a mock community, our workflow demonstrates comparable sequencing depth and taxonomic accuracy (F1-Score=0.875). The multi-region approach outperforms single amplicons, validating our pipeline as a robust alternative for semiconductor-based sequencing data.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
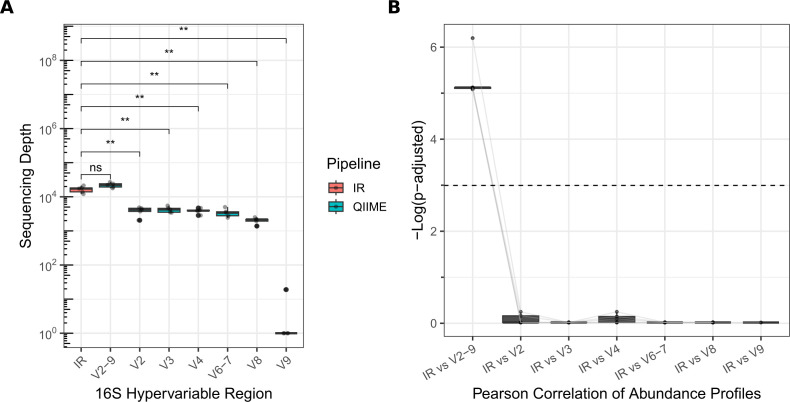 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Phylogenetic Studies · Chromosomal and Genetic Variations · Microbial Community Ecology and Physiology