Clinical outcomes of exclusive enzyme therapy (laronidase) in a cohort of patients with mucopolysaccharidosis type I
Nathalie Guffon, Magali Pettazzoni, Nicolas Pangaud, Nathalie Reynes, Eliane Le Peillet Feuillet, Pierre Journeau, Alain Fouilhoux

TL;DR
This study examines the long-term effects of enzyme replacement therapy in patients with mucopolysaccharidosis type I, showing improvements in disease markers and quality of life.
Contribution
The study provides a detailed retrospective analysis of exclusive enzyme therapy's effectiveness in both severe and attenuated MPS I phenotypes over a decade.
Findings
ERT reduced GAG levels by 88% in severe and 71% in attenuated MPS I patients.
Cognitive development was normal in 84% of attenuated patients at last follow-up.
Early ERT and multidisciplinary care slowed disease progression and improved quality of life.
Abstract
Mucopolysaccharidosis type I (MPS I), is an autosomal recessive disorder caused by a deficiency in the enzyme α-L-iduronidase (IDUA), leading to the accumulation of glycosaminoglycans (GAGs) in tissues. Early diagnosis and treatment [i.e., bone marrow transplantation and/or enzyme replacement therapy (ERT) with laronidase] are essential to prevent irreversible damage. The long-term effectiveness of exclusive ERT has been primarily described in attenuated phenotypes, while only a few cases have been reported in severe phenotypes. This study is a retrospective analysis summarising the collective experience of disease progression in 48 patients with severe and attenuated MPS I who were treated exclusively with laronidase over a median of 10 years at the Lyon Reference Centre for Hereditary Metabolic Diseases in France. Patients were categorised by genotype and further stratified by age at…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
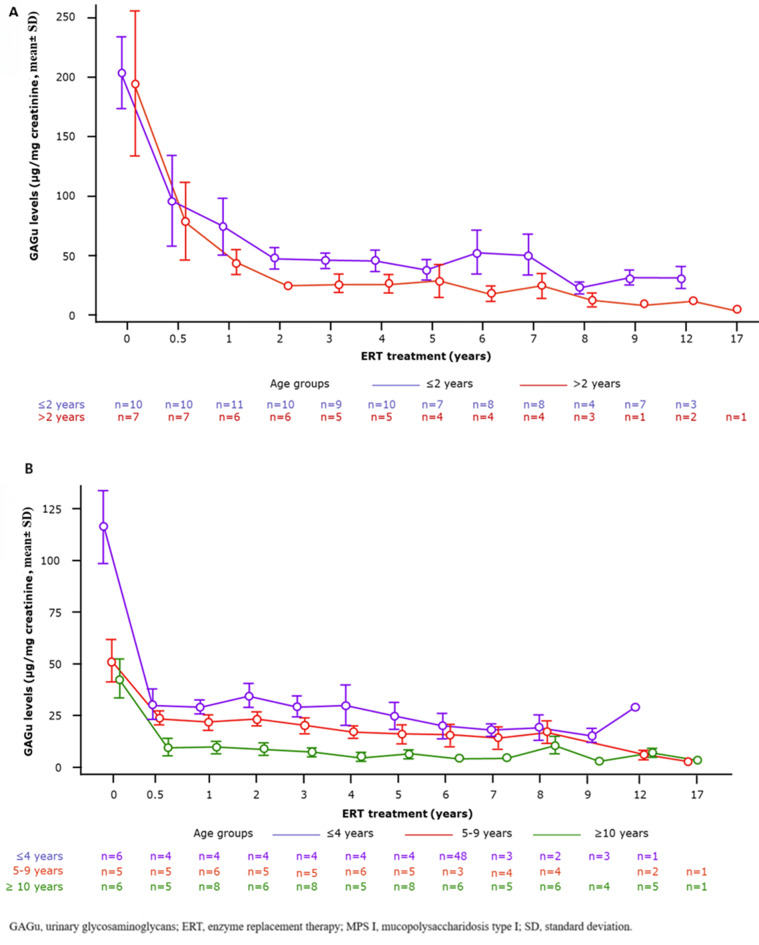 Figure 1
Figure 1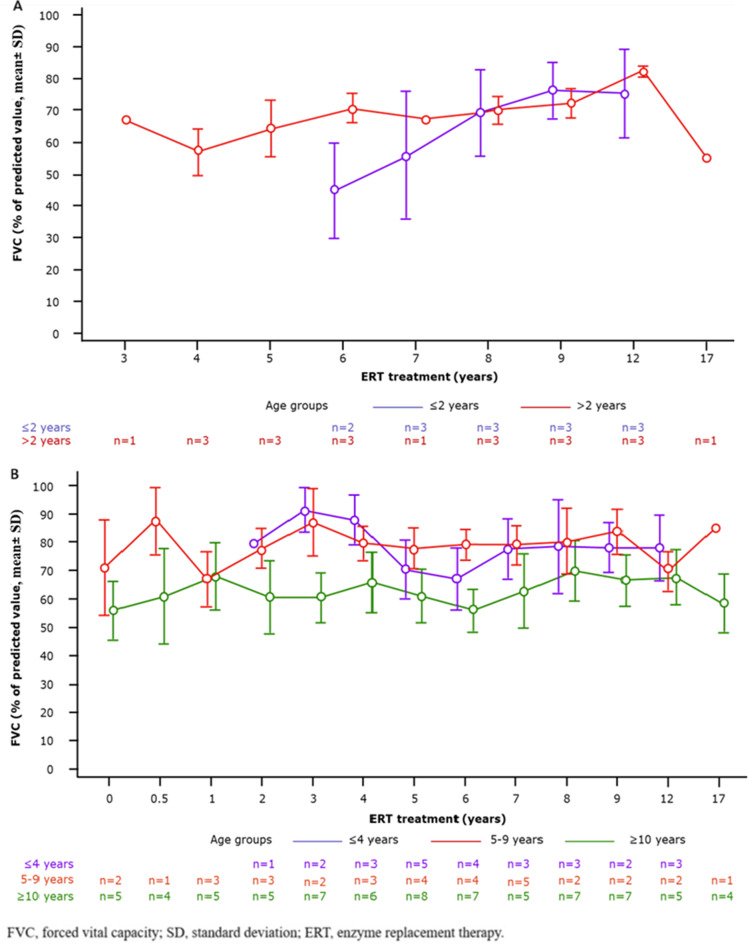 Figure 2
Figure 2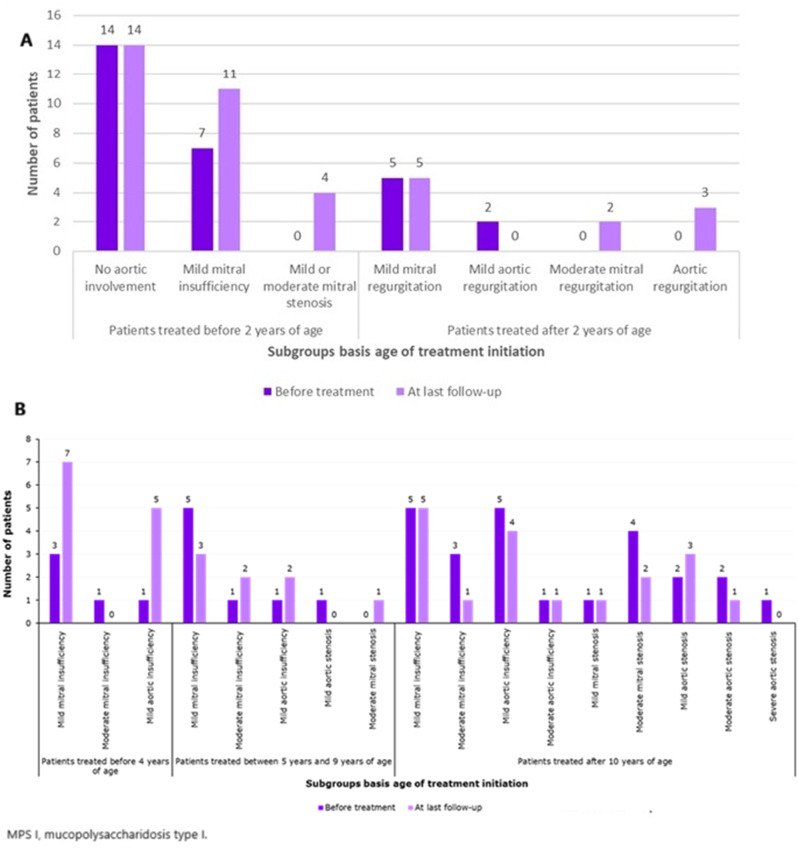 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Glycogen Storage Diseases and Myoclonus · Amyloidosis: Diagnosis, Treatment, Outcomes