DNA damage reflected in the evolution of G-runs in genomes
I.R. Grin, D.O. Zharkov

TL;DR
This paper explores how DNA damage, specifically 8-oxoguanine formation in G-runs, influences genome evolution and mutagenesis patterns in prokaryotes and humans.
Contribution
The study reveals how G-run properties and flanking nucleotide patterns correlate with DNA oxidation and mutagenesis in vivo.
Findings
G-runs are shorter and less likely to be elongated compared to A-runs.
T is more frequent in the 5’-flanking position of G-runs in aerobic organisms, suggesting G→T substitutions via 8-oxoG.
Long G-runs in the human genome show increased C in the 5’-flanking position and elevated 5’-G→A substitutions in telomeres.
Abstract
DNA oxidation is one of the main types of damage to the genetic material of living organisms. Of the many dozens of oxidative lesions, the most abundant is 8-oxoguanine (8-oxoG), a premutagenic base that leads to G→T transversions during replication. Double-stranded DNA can conduct holes through the π system of stacked nucleobases. Such electron vacancies are ultimately localized at the 5’-terminal nucleotides of polyguanine runs (G-runs), making these positions characteristic sites of 8-oxoG formation. While such properties of G-runs have been studied in vitro at the level of chemical reactivity, the extent to which they can influence mutagenesis spectra in vivo remains unclear. Here, we have analyzed the nucleotide context of G-runs in a representative set of 62 high-quality prokaryotic genomes and in the human telomere-to-telomere genome. G-runs were, on average, shorter than…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
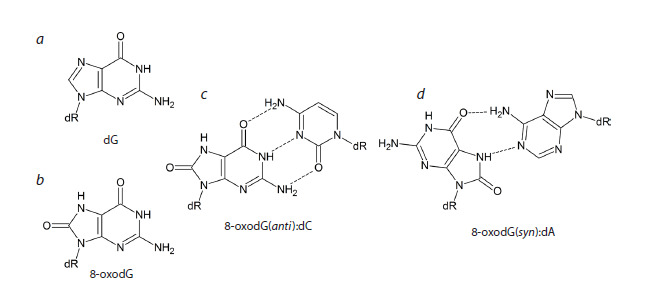 Fig. 1
Fig. 1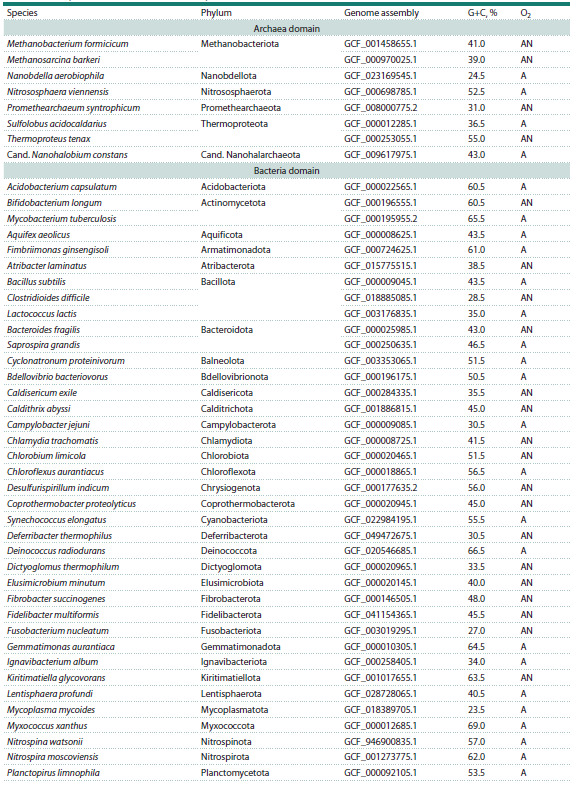 Table 1
Table 1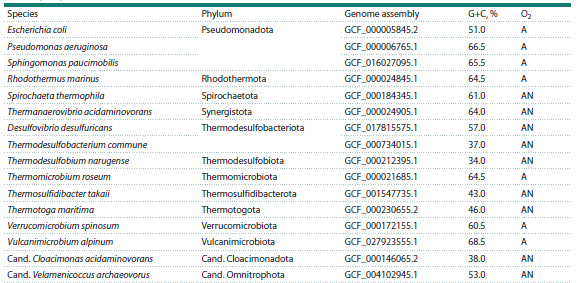 Table 1end
Table 1end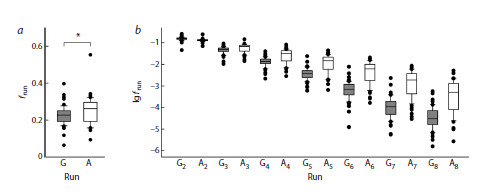 Fig. 2
Fig. 2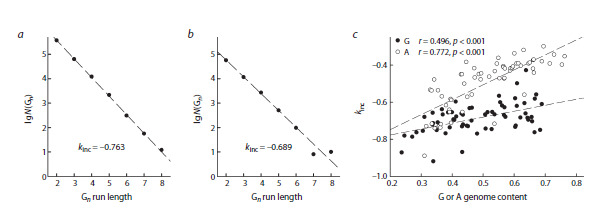 Fig. 3
Fig. 3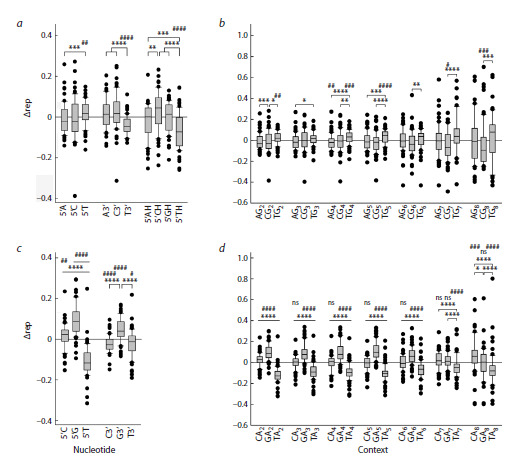 Fig. 4
Fig. 4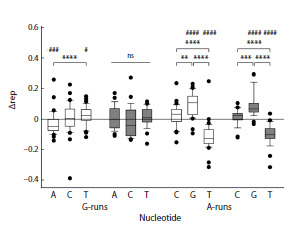 Fig. 5
Fig. 5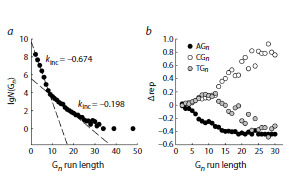 Fig. 6
Fig. 6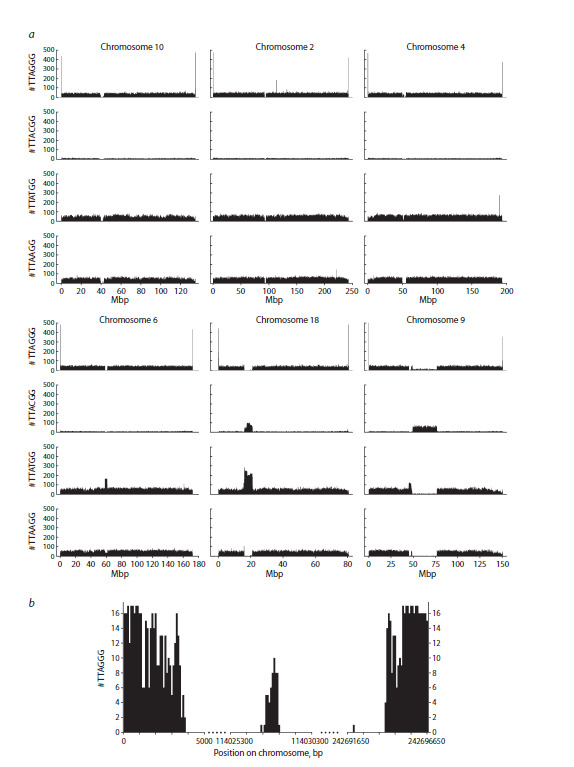 Fig. 7
Fig. 7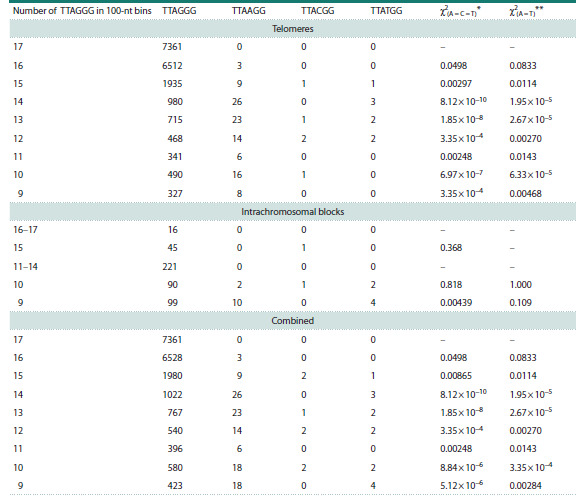 Table 2
Table 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDNA Repair Mechanisms · DNA and Nucleic Acid Chemistry · Carcinogens and Genotoxicity Assessment
Introduction
Oxidative DNA damage is an inevitable consequence of respiration, which relies on the oxidation of organic compounds with molecular oxygen and has been the basis of energy metabolism in the vast majority of living organisms for over two billion years (Prorok et al., 2021). Damaged nucleotides are generally quickly repaired; however, some of them may remain in DNA until replication, which is one of the main sources of mutations (Liu et al., 2016; Chatterjee, Walker, 2017; Tubbs, Nussenzweig, 2017). Based on our understanding of the molecular mechanisms of DNA polymerase errors, it has now become possible to identify characteristic patterns of mutations caused by various types of genotoxic stress or even by specific damaged bases (Alexandrov et al., 2013; Koh et al., 2021).
Of all DNA structural elements, the guanine base has the lowest redox potential (Cadet et al., 2008, 2017; Fleming, Burrows, 2022). The most common product of its oxidation, 7,8-dihydro-8-oxoguanine (8-oxoG), occurs in DNA at the background level of ~1/106 guanines, and this level increases significantly under oxidative stress of various origins (ESCODD et al., 2005; Dizdaroglu et al., 2015; Chiorcea- Paquim, 2022; Fig. 1a, b). The presence of an oxygen atom at C8 in 8-oxoG sterically hinders the regular anti conformation of its nucleoside, 8-oxo-2′-deoxyguanosine (8-oxodG), and the syn conformation becomes energetically favorable (Cho et al., 1990; Fig. 1c, d ). Consequently, in the absence of Watson– Crick bonds with cytosine, which additionally stabilize the anti conformation, 8-oxodG preferentially adopts the syn conformation, in which it can form a Hoogsteen-type pair with adenine (Kouchakdjian et al., 1991; McAuley-Hecht et al., 1994; Lipscomb et al., 1995). Because of this, DNA polymerases incorporate dAMP opposite 8-oxoG in the DNA template with high frequency (Shibutani et al., 1991; Miller, Grollman, 1997; Maga et al., 2007; Yudkina et al., 2019).
Structures of 2’-deoxyguanosine (a), 8-oxo-2’-deoxyguanosine (b), Watson–Crick 8-oxodG(anti ):dC pair (c) and Hoogsteen 8-oxodG(syn):dA pair (d).
In the living cell, the outcome of primary DNA oxidation events can be influenced by numerous additional factors and DNA repair systems that remove damaged bases from the genome. Even so, 8-oxoG exhibits relatively high mutagenicity in vivo, characterized by a spectrum dominated by G→T transversions mostly independent of the surrounding nucleotide context (Wood et al., 1992; Moriya, 1993). Such mutations are frequently found in human tumors and form the basis of the SBS18 and SBS36 mutational signatures (Alexandrov et al., 2013; Pilati et al., 2017; Viel et al., 2017; Kucab et al., 2019). Guanidinohydantoin and spiroiminodihydantoin, the products of further oxidation of 8-oxoG, also significantly contribute to mutagenesis, predominantly causing G→C transversions (Fleming, Burrows, 2017; Kino et al., 2020).
The stacked π system of DNA has considerable hole conductivity (Giese, 2002; Genereux, Barton, 2010). Numerous experiments and quantum mechanical calculations show that a positive charge resulting from one-electron oxidation of DNA can migrate along the π system over significant distances, and its final acceptors are the G bases, which are mainly oxidized to 8-oxoG. In this case, the G bases located in the first 5′-position in runs of several Gs are especially sensitive to oxidation (Sugiyama, Saito, 1996; Saito et al., 1998; Kurbanyan et al., 2003; Adhikary et al., 2009).
Although the mechanism of positive charge migration and preferential oxidation of guanines at the 5′-end of G-runs is generally accepted today, all experimental data supporting were obtained in relatively simple in vitro systems. The mutagenesis spectra caused by the appearance of 8-oxoG in this context have not yet been studied. If preferential conversion of G to 8-oxoG does indeed occur at the 5′-end of G-runs, it can be expected that the mutagenic properties of 8-oxoG at these positions will result in an increased frequency of G→T mutations, which should be reflected in an increased frequency of T before G-runs. In this study, to test this hypothesis, we analyzed the occurrence of nucleotides flanking G-runs from the 5′-side in prokaryotic and human genomes.
Materials and methods
The T2T-CHM13v2.0 human genome assembly, which includes full-length telomeres and highly repetitive regions (Nurk et al., 2022), and the prokaryotic genomes listed in Table 1 were used for the analysis.
Prokaryotic genomes used for the analysis
Table 1end.Note. Assembly ID in the RefSeq database (O’Leary et al., 2016). A, aerobes and facultative anaerobes; AN, anaerobes.
UGENE v37.0 software package (Okonechnikov et al., 2012) and custom-written bash scripts were used to extract nucleotide frequencies at given positions. The expected frequency of nucleotides in the flanking positions before and after Gn (or An) runs in prokaryotic genomes was calculated based on the total number of A, C, and T (or C, G, and T) in a given genome as pA = NA/(NA+NC+NT), where pA is the expected representation (in this case, for A), and NA, NC, and NT are the numbers of A, C, and T in both strands of the genome, respectively. For the human genome, due to the well-known underrepresentation of the CG dinucleotide, the expected frequency was calculated in a similar way but based on the number of AG, CG, and TG dinucleotides. Statistical analysis was performed using SigmaPlot v11.0 (Grafiti, USA), DATAPLOT (National Institute of Standards and Technology, USA), and RStudio v1.2 (Posit PBC, USA). Dunn’s correction was used for all multiple comparisons and test series to adjust the significance level.
Results and discussion
To analyze the nucleotide distribution in prokaryotic genomes, a sample of 54 bacterial and 8 archaeal genomes was compiled, maximally reflecting the taxonomic diversity in these domains of life (Table 1). Only high-quality genomes classified in the RefSeq database (O’Leary et al., 2016) as reference genomes were included. The sample taxonomic representation was one genome per phylum, with the exception of Methanobacteriota and Thermoproteota for Archaea, and Actinomycetota, Bacteroidota, and Thermodesulfobacteriota for Bacteria with a representation of 2 genomes from different orders per phylum, as well as Bacillota and Pseudomonadota (3 genomes from different orders per phylum). The G+C content in the studied genomes ranged from 23.5 to 69 % (Table 1). The parameters of archaeal genomes did not differ significantly from those of bacterial ones, so the representatives of both domains were considered as a single group of prokaryotes.
Since the prokaryotic genomes mostly consist of proteincoding sequences, mutations in which can be subject to natural selection, we have first assessed the possible impact of all 16 potential amino acid substitutions resulting from G→A, G→C and G→T nucleotide substitutions in the first position of G-runs (codon changes HHG→HHH, HGG→HHG, GGG→HGG, where H is A, C or T). Two independent metrics were used for this purpose: the conservation index Cn, calculated on the basis of partition distances in a set of physicochemical properties of amino acid residues (Taylor, 1986; Livingstone, Barton, 1993), and the weights of amino acid substitutions in the BLOSUM62 matrix, compiled from several hundred groups of homologous proteins (Henikoff S., Henikoff J.G., 1992). Although G→A substitutions generally caused smaller changes in the properties and occurrence of amino acid residues, as expected for class-conserving point mutations, the difference from G→C and G→T substitutions was not statistically significant (Kruskal–Wallis test with Dunn’s correction for multiple comparisons, p > 0.05).
All genomic sequences were searched for the HGnH and BAnB runs and the corresponding complementary-strand DCnD and VTnV runs (H = A, C or T; B = C, G or T; D = A, G or T; V = A, C or G) with the length n ≥ 2. The frequency of polypurine runs in the genomes was higher than that expected from a random nucleotide distribution with the same G+C composition (one-sample Wilcoxon test, p < 0.001), indicating the functional importance of such sequences. An increased frequency of substitutions at the first position of G-runs should gradually lead to their shortening. Indeed, when comparing the lengths of G-runs and A-runs in prokaryotic genomes, adjusted for the content of the respective purine nucleotides, it turned out that G-runs are, on average, shorter (Fig. 2a). In this case, HGG trinucleotides were more common than BAA, but in longer repeats, the frequency of A-runs was higher (Fig. 2b).
Length of polypurine runs in prokaryotic genomes. a, the total fraction of G or A in runs of any length in the respective purine nucleotide content in the genome (frun). * p <0.05 (Mann–Whitney test). b, the fraction of G or A in the runs 2 to 8 nucleotides long in the respective purine nucleotide content in the genome. In all cases, the difference between G-runs and A-runs is significant at p < 0.001 (Mann–Whitney test).Here and below, the line in the box marks the median, the boundaries of the box correspond to the first and third quartiles, the whiskers, to the 10th and 90th percentiles, and the dots are outliers.
For a more detailed analysis of the run length distribution, we have studied the variability of their lengths in each genome. The number of G-runs and A-runs in each genome decreased almost strictly exponentially in the length range from 2 to 5–6. At n > 5–6, deviations in either direction were observed in some cases due to the small number of such runs, especially in small genomes (Fig. 3a, b). Using the linear portion of the relationship between the log of the number of repeats and run length, one can determine the increment coefficient kinc, which indicates how easily a run can be extended by one nucleotide in a genome with a given nucleotide composition: the higher the kinc, the greater the proportion of longer runs in the genome. When comparing the dependence of kinc for G-runs and Aruns in genomes of different composition, we have found that G-runs grow more slowly with increasing G+C content than A-runs grow with increasing A+T content (Fig. 3c). Thus, in prokaryotic genomes, the balance of G-run elongation and shortening, determined by many factors, is shifted towards shortening compared to A-runs.
Dependence of the number of polypurine runs in prokaryotic genomes on the run length and the nucleotide composition of the genome. a, b, examples of the dependence of the number of G-runs N(Gn) on their length for the genomes of E. coli (a; genome size 4.64 × 106 bp, G+C content 51.0 %) and Ch. trachomatis (b; genome size 1.04 × 106 bp, G+C content 41.5 %). c, dependence of kinc on the nucleotide composition of the genome (G+C content for G-runs, A+T content for A-runs). Black dots, G-runs, white dots, A-runs; dashed lines show linear regressions with the regression coefficients indicated on the plot.
The lengths of polypurine runs can change in either direction due to DNA polymerase slippage during DNA synthesis (Kunkel, Bebenek, 2000) or selection based on the physicochemical properties of polypurine regions (Bansal et al., 2022), but these processes are independent of the nucleotides surrounding the run. In contrast, shortening of G-runs due to damage to the 5′-terminal base should be accompanied by a characteristic mutational spectrum determined by the properties of replicative DNA polymerases. Therefore, it was of interest to determine the extent to which the frequencies of 5′-flanking nucleotides differ from each other and from their overall abundance in the genome. To quantitatively characterize these differences, we have introduced the Δrep parameter representing the difference between the observed and expected frequency of each nucleotide. The frequency of T in the first position before G-runs was statistically significantly higher than expected and than the frequency of A and C (Fig. 4a). The frequency of A and C nucleotides in this position was slightly lower than expected, but this difference did not reach significance; their representation also did not differ from each other. T was more frequent than either A or C nucleotide at any G-run length, and its representation was higher than expected before G2, G4, G5, and G6 runs (Fig. 4b). A was underrepresented in this position only before G4 runs, and C was underrepresented before G5 and longer G-runs. In contrast, T was underrepresented both at the 3′-side of Gruns and at the second position from their 5′-side (Fig. 4a). Overall, these data support a model of preferential oxidation of the first G in the runs to 8-oxoG followed by G→T transversions.
Representation of different 5’- and 3’-flanking nucleotides in polypurine runs. a, c, deviation from the frequency of 5’- and 3’-flanking nucleotides for G-runs (a) and A-runs (c) of any length expected on the basis of the content of the respective nucleotide in the genome. b, d, deviation from the frequency of 5’-flanking nucleotides in G-runs (b) and A-runs (d) 2–8 nucleotides long.Difference from expected: # p <0.05, ## p < 0.01, ### p < 0.005, #### p <0.001 (one-sample Wilcoxon test with Dunn’s correction for multiple comparisons); ns, no significant difference. Differences between groups: * p < 0.05, ** p < 0.01, *** p < 0.005, **** p < 0.001 (Kruskal–Wallis test with Dunn’s correction for multiple comparisons).
Quite unexpectedly, the nucleotide distribution before A-runs was even more uneven than before G-runs. At this position, T was underrepresented, while C and G were overrepresented (Fig. 4c). For C, this deviation was explained primarily by overrepresentation of CAA trinucleotides, while for G, an increased frequency of occurrence was observed up to a run length of 6 nucleotides (Fig. 4d ). A decrease in the fraction of T also occurred in runs of any length (Fig. 4d ). After A-runs, the occurrence of C and T was lower than expected, while G was higher than expected (Fig. 4c). It is possible that these deviations can also be explained by DNA damage and subsequent DNA polymerases errors; however, the mechanistic reasons underlying such events remain unclear at present
The amount of 8-oxoG generated in the genome directly depends on the presence of reactive oxygen species in the intracellular environment (Halliwell, Gutteridge, 2015). Prokaryotes are exceptionally diverse in their energy metabolism pathways: some follow a strictly anaerobic lifestyle, while others are obligate aerobes or facultative anaerobes and are subject to more intense oxidative stress. We have compared the statistics of the occurrence of 5′-flanking nucleotides of G-runs in the genomes of these two groups (Table 1). In aerobic prokaryotes, T was found at this position with an increased frequency compared to the expected, and A, with a decreased frequency (Fig. 5). For anaerobic microorganisms, no significant difference in the occurrence of 5′-flanking nucleotides was found (Fig. 5). However, when comparing the abundance of A, C and T directly between the aerobic and anaerobic groups, the differences did not reach statistical significance, which is most likely due to insufficient sample size. For A-runs, the difference in the occurrence of 5′-flanking nucleotides in the genomes of aerobes and anaerobes was the same as in the combined group (compare Fig. 4c and Fig. 5). Thus, the reduced level of oxidative stress in anaerobic microorganisms may be associated with a less pronounced predominance of T at the position flanking the 5′-side of G-runs; however, further research is required to answer this question more definitively.
Representation of 5’-flanking nucleotides in polypurine runs in the genomes of aerobic (white) and anaerobic (gray) microorganisms.Deviation from the expected representation is shown for G-runs and A-runs of any length. Difference from expected: # p < 0.05, ### p < 0.005, #### p < 0.001 (one-sample Wilcoxon test with Dunn’s correction for multiple comparisons). Differences between groups: ** p < 0.01, *** p < 0.005, **** p < 0.001 (Kruskal– Wallis test with Dunn’s correction for multiple comparisons); ns, no significant difference
Unlike those of prokaryotes, eukaryotic genomes are characterized by a large number of repetitive elements such as transposons, satellite and telomeric DNA, the precise sequences of which are inaccessible to traditional highthroughput sequencing methods (Richard et al., 2008; Liao et al., 2023). The advent of ultra-long sequencing (Oxford Nanopore, PacBio HiFi) has made it possible to fill these gaps. The recently published human genome read using a combination of methods with telomere-to-telomere (T2T) coverage and high quality (estimated telomeric error rate of ~ 4×10−8) (Nurk et al., 2022), provides the opportunity to analyze the context of G-runs without the distortions caused by a higher representation of unique sequences.
The significantly larger size of the human genome compared to prokaryotic ones allowed us to identify interesting patterns in the distribution of Gn runs size. For n = 2–8, their number decreased exponentially and was described by an increment coefficient kinc = −0.674, which is very close to the center of the distribution of kinc values for G-runs in prokaryotes (compare Fig. 6a and Fig. 3c; z = 0.141). For n = 9–16, the exponential dependence was preserved, but the rate of decrease in the number of runs decelerated: the kinc value increased to −0.198, which lies far outside the range of kinc values for prokaryotic genomes (compare Fig. 6a and Fig. 3c; z = 5.97). Runs of this size were absent in prokaryotic genomes or were present in a handful of cases, so it was impossible to detect this transition. Further increase in the length of G-runs was accompanied by an even greater deceleration of the rate of decrease in their number (Fig. 6a). Obviously, around n = 8–9 (the breakpoint value determined by the piecewise regression method: n = 8.72 ± 0.04), the balance of G-run shortening and elongation is shifted in favor of the latter; run growth due to DNA polymerase slippage during replication or repair becomes self-sustaining, as in the well-studied case of trinucleotide repeat runs (Mirkin, 2007; McMurray, 2010).
Dependence of the number of G-runs and the representation of the 5’-flanking nucleotides in the human genome on the run length. a, dependence of the number of G-runs N(Gn) on their length. The dashed lines correspond to the linear regression; the kinc values for n = 2–8 and n = 9–16 are shown on the plot. b, The dependence of Δrep of the 5’-flanking nucleotide (black, A, white, C, gray, T) on the run length.
An even more unexpected pattern emerged from the analysis of the frequency of 5′-flanking nucleotides. Since it is well known that the number of CG dinucleotides in the human genome is reduced due to their role in epigenetic regulation (Fazzari, Greally, 2004), the expected frequency was calculated based on the dinucleotide rather than the total nucleotide frequency. At n = 2, the nucleotide frequency closely matched the expected value, but then the Δrep values for A steadily decreased, while the representation of C and T, in contrast, increased at virtually the same rate (Fig. 6b). However, starting from n = 8–11 (the breakpoint value for Δrep(C)−Δrep(T), de0.6 termined by the piecewise regression method: n = 9.28 ± 1.10), the dependencies for C and T diverged sharply: the representation of T decreased, while the representation of C increased. One possible explanation for this phenomenon may be that longer G-runs serve as more effective traps for holes migrating along the DNA duplex leading to hyperoxidation of the 5′-terminal 8-oxoG to guanidinohydantoin and spiroiminodihydantoin with a corresponding switch in the preferential nucleotide substitutions from G→T to G→C.
Telomeric DNA is a distinct class of highly repetitive DNA in eukaryotic genomes, represented in humans by multiple copies of the TTAGGG hexanucleotide. Telomeric repeats are known to serve as hotspots for DNA oxidation to form 8-oxoG (Billard, Poncet, 2019; Opresko et al., 2025). Telomere ends in germline cells are elongated by telomerase, a specialized DNA polymerase that uses telomerase RNA as a template, so changes in these regions are not associated with damage to genomic DNA. However, even in the presence of active telomerase, the bulk of telomere length is replicated by the usual semiconservative mechanism (Pfeiffer, Lingner, 2013; Higa et al., 2017; Bonnell et al., 2021), which can lead to the accumulation of mutations in them. Thus, the telomere sequence in human somatic cells (in the case of the T2T genome, the immortalized telomerase-expressing CHM13hTERT chorionic cell line) reflects both their recent elongation by telomerase in germline cells and mutagenesis events in past generations and in individual development
The distribution of TTAGGG repeats in chromosomes (calculated for both DNA strands) had a fairly expected pattern, with frequency peaks at the ends of the chromosomes and a dip in the pericentromeric region (Fig. 7a). The only exception was chromosome 8, for which, on the contrary, a slight increase in the number of these repeats was observed in the centromere region. On chromosome 2, a peak in the frequency of telomeric repeats was clearly visible in the region of the fusion of two ancestral hominid chromosomes that formed the evolutionarily young human chromosome 2 (Ijdo et al., 1991; Fig. 7a). However, a more detailed analysis of this region shows that it has already significantly degraded, keeping far fewer TTAGGG repeats than in true telomeres (Fig. 7b). Interestingly, similar peaks were found on chromosomes 15 and 22 in the introns of the active protein-coding genes ATP10A and MICAL3; they may represent remnants of translocated telomere fragments.
Examples of the distribution of TTAGGG, TTACGG, TTATGG, and TTAAGG repeats in human chromosomes. a, distribution of the repeats along the entire length of chromosomes 10, 2, 4, 6, 18, and 9. The number of repeats is calculated in 100-kb bins. b, distribution of TTAGGG repeats in telomeric regions and in the region of fusion of the ancestral telomeres on chromosome 2.The number of repeats is calculated in 100-bp bins.
TTAAGG, TTACGG, and TTATGG repeats were distributed across chromosomes without telomeric peaks. The overall frequency of TTACGG repeats was significantly lower than that of TTAAGG and TTATGG, consistent with the reduced abundance of CG dinucleotides in the human genome (Fig. 7a). Separate peaks in repeat frequency were observed on chromosome 2 for TTAAGG, chromosomes 8, 12, 17, and Y for TTACGG, and chromosomes 4 and 22 for TTATGG (Fig. 7a). A characteristic pattern of repeat distribution in the pericentromeric region with gaps in all TTANGG variants was observed for chromosomes 1–5, 7, 10–12, 16, 19, and 21. In other cases, one repeat type predominated in the centromere region, while others were depleted, with their combined deficiency compensating for the excess of the predominant type, as shown in Fig. 7a for chromosome 6. In chromosomes 6, 13–15, 22, and X, TTATGG was the predominant repeat; in chromosome 8, it was TTAGGG, and in chromosome 17, TTACGG. Chromosome 18 was distinguished by coinciding peaks in the distribution of two repeats, TTACGG and TTATGG (Fig. 7a). In the long arm of chromosome 9, in the region of constitutive heterochromatin adjacent to the pericentromeric region with an excess of TTATGG, there was a long stretch with a predominance of TTACGG.
Obviously, the cases of co-localization or oppositely phased localization of TTANGG repeats in non-telomeric regions are not due to point mutations in the TTAGGG repeat but reflect the presence of repeating elements containing one or two of these hexanucleotides in these loci. In contrast, point mutations in the first position of the G3-run of the telomeric repeat should be most obvious in the regions consisting mainly of TTAGGG, that is, in the telomeres proper and intrachromosomal blocks of telomere-like repeats. To analyze the frequency of substitutions in such regions, we have singled out the telomeric regions and intrachromosomal blocks where at least 100 copies of the TTAGGG repeat were found in 100-kb bins. They were divided into shorter 100-bp bins. A bin filled with only TTAGGG repeats corresponds to 16 or 17 copies (depending on the position of the first complete hexanucleotide in the bin). The bins containing at least 9 TTAGGG copies, accounting for more than half the bin length, were selected for analysis.
Counting the occurrence of TTAAGG, TTACGG, and TTATGG in the studied regions revealed clear significant enrichment of G→A substitutions at the first position of the G3-run compared to G→C and G→T substitutions (Table 2). In comparison with G→A, the total number of G→C and G→T changes was fivefold lower, and their frequencies did not differ significantly from each other. Thus, although telomeric repeats serve as preferential sites of guanine oxidation, this is not reflected in the increased frequency of G→T point mutations. The difference between the representation of A and C+T at the 5′-flanking position of GG dinucleotides between telomeric repeats and the rest of the genome may indicate the existence of a mutational process in telomeres that is distinct from G oxidation at the 5′-position of GGG.
Representation of TTANGG in telomeres and intrachromosomal blocks of telomere-like repeats χ2 values for the null hypothesis of equal representation of A, C and T. ** χ2 values for the null hypothesis of equal representation of A and T.*
Conclusion
In conclusion, the analysis of the nucleotide context of Gruns in a set of 62 complete prokaryotic genomes and in the human T2T genome revealed that the representation of T at the position adjacent to G-runs is generally increased, which is consistent with the model of G oxidation at the 5′-position of the runs followed by G→T mutations. Other patterns in the distribution of 5′-flanking nucleotides were also identified: uneven nucleotide frequency at the position adjacent to A-runs, increased representation of C at the 5′-side of long G-runs in the human genome, and the predominance of G→A substitutions at the 5′-position in telomeric repeats. The hypothesis that G-run elongation may lead to a shift in the specificity of single-nucleotide mutations from G→T to G→C due to a change in the nature of the precursor lesion can be tested experimentally. The characteristic mutation spectrum in telomeric repeats may be caused by their tendency to fold into G-quadruplex structures, which hinder the movement of DNA polymerases (Pfeiffer, Lingner, 2013; Higa et al., 2017; Bonnell et al., 2021), but this proposal requires a detailed study of the fidelity of human replicative DNA polymerases on intact and damaged templates of this structure. For A-runs, the existence of preferential sites of DNA damage is not known; given that A-runs are longer than G-runs (Fig. 2), the difference in the relative representation of C, G, and T in the 5′-flanking position may not be associated with the mutational process. The explanation of all these identified patterns requires further research.
Conflict of interest
The authors declare no conflict of interest.
References
Adhikary A., Khanduri D., Sevilla M.D. Direct observation of the hole protonation state and hole localization site in DNA-oligomers. J Am Chem Soc. 2009;131(24):8614-8619. doi 10.1021/ja9014869
Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., … Campo E., Shibata T., Pfister S.M., Campbell P.J., Stratton M.R. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415-421. doi 10.1038/nature12477
Bansal A., Kaushik S., Kukreti S. Non-canonical DNA structures: diversity and disease association. Front Genet. 2022;13:959258. doi 10.3389/fgene.2022.959258
Billard P., Poncet D.A. Replication stress at telomeric and mitochondrial DNA: common origins and consequences on ageing. Int J Mol Sci. 2019;20(19):4959. doi 10.3390/ijms20194959
Bonnell E., Pasquier E., Wellinger R.J. Telomere replication: solving multiple end replication problems. Front Cell Dev Biol. 2021;9: 668171. doi 10.3389/fcell.2021.668171
Cadet J., Douki T., Ravanat J.-L. Oxidatively generated damage to the guanine moiety of DNA: mechanistic aspects and formation in cells. Cadet J., Davies K.J.A., Medeiros M.H.G., Di Mascio P., Wagner J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free Radic Biol Med. 2017;107:13-34. doi 10.1016/j.freerad biomed.2016.12.049
Chatterjee N., Walker G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58(5):235-263. doi 10.1002/em.22087
Chiorcea-Paquim A.-M. 8-oxoguanine and 8-oxodeoxyguanosine biomarkers of oxidative DNA damage: a review on HPLC–ECD determination. Molecules. 2022;27(5):1620. doi 10.3390/molecules 27051620
Cho B.P., Kadlubar F.F., Culp S.J., Evans F.E. 15N nuclear magnetic resonance studies on the tautomerism of 8-hydroxy-2′-deoxyguanosine, 8-hydroxyguanosine, and other C8-substituted guanine nucleosides. Chem Res Toxicol. 1990;3(5):445-452. doi 10.1021/ tx00017a010
Dizdaroglu M., Coskun E., Jaruga P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic Res. 2015;49(5):525-548. doi 10.3109/10715762.2015. 1014814
ESCODD (European Standards Committee on Oxidative DNA Damage), Gedik C.M., Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study. FASEB J. 2005;19(1):82-84. doi 10.1096/ fj.04-1767fje
Fazzari M.J., Greally J.M. Epigenomics: beyond CpG islands. Nat Rev Genet. 2004;5(6):446-455. doi 10.1038/nrg1349
Fleming A.M., Burrows C.J. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic Biol Med. 2017;107:35-52. doi 10.1016/j.freeradbiomed.2016.11.030
Fleming A.M., Burrows C.J. Chemistry of ROS-mediated oxidation to the guanine base in DNA and its biological consequences. Int J Radiat Biol. 2022;98(3):452-460. doi 10.1080/09553002.2021. 2003464
Genereux J.C., Barton J.K. Mechanisms for DNA charge transport. Chem Rev. 2010;110(3):1642-1662. doi 10.1021/cr900228f
Giese B. Long-distance electron transfer through DNA. Annu Rev Biochem. 2002;71:51-70. doi 10.1146/annurev.biochem.71.083101. 134037
Halliwell B., Gutteridge J.M.C. Free Radicals in Biology and Medicine. Oxford Univ. Press, 2015
Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci USA. 1992;89(22):10915-10919. doi 10.1073/pnas.89.22.10915
Higa M., Fujita M., Yoshida K. DNA replication origins and fork progression at mammalian telomeres. Genes. 2017;8(4):112. doi 10.3390/genes8040112
Ijdo J.W., Baldini A., Ward D.C., Reeders S.T., Wells R.A. Origin of human chromosome 2: an ancestral telomere-telomere fusion. Proc Natl Acad Sci USA. 1991;88(20):9051-9055. doi 10.1073/pnas.88. 20.9051
Kino K., Kawada T., Hirao-Suzuki M., Morikawa M., Miyazawa H. Products of oxidative guanine damage form base pairs with guanine. Int J Mol Sci. 2020;21(20):7645. doi 10.3390/ijms21207645
Koh G., Degasperi A., Zou X., Momen S., Nik-Zainal S. Mutational signatures: emerging concepts, caveats and clinical applications. Nat Rev Cancer. 2021;21(10):619-637. doi 10.1038/s41568-021- 00377-7
Kouchakdjian M., Bodepudi V., Shibutani S., Eisenberg M., Johnson F., Grollman A.P., Patel D.J. NMR structural studies of the ionizing radiation adduct 7-hydro-8-oxodeoxyguanosine (8-oxo-7H-dG) opposite deoxyadenosine in a DNA duplex. 8-Oxo-7H-dG(syn)·dA(anti) alignment at lesion site. Biochemistry. 1991;30(5):1403-1412. doi 10.1021/bi00219a034
Kucab J.E., Zou X., Morganella S., Joel M., Nanda A.S., Nagy E., Gomez C., Degasperi A., Harris R., Jackson S.P., Arlt V.M., Phillips D.H., Nik-Zainal S. A compendium of mutational signatures of environmental agents. Cell. 2019;177(4):821-836.e816. doi 10.1016/j.cell.2019.03.001
Kunkel T.A., Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497-529. doi 10.1146/annurev.biochem.69.1.497
Kurbanyan K., Nguyen K.L., To P., Rivas E.V., Lueras A.M.K., Kosinski C., Steryo M., González A., Mah D.A., Stemp E.D.A. DNA-protein cross-linking via guanine oxidation: dependence upon protein and photosensitizer. Biochemistry. 2003;42(34):10269-10281. doi 10.1021/bi020713p
Liao X., Zhu W., Zhou J., Li H., Xu X., Zhang B., Gao X. Repetitive DNA sequence detection and its role in the human genome. Commun Biol. 2023;6:954. doi 10.1038/s42003-023-05322-y
Lipscomb L.A., Peek M.E., Morningstar M.L., Verghis S.M., Miller E.M., Rich A., Essigmann J.M., Williams L.D. X-ray structure of a DNA decamer containing 7,8-dihydro-8-oxoguanine. Proc Natl Acad Sci USA. 1995;92(3):719-723. doi 10.1073/pnas.92. 3.719
Liu B., Xue Q., Tang Y., Cao J., Guengerich F.P., Zhang H. Mechanisms of mutagenesis: DNA replication in the presence of DNA damage. Mutat Res. 2016;768:53-67. doi 10.1016/j.mrrev.2016.03.006
Livingstone C.D., Barton G.J. Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Comput Appl Biosci. 1993;9(6):745-756. doi 10.1093/bioinformatics/ 9.6.745
Maga G., Villani G., Crespan E., Wimmer U., Ferrari E., Bertocci B., Hübscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447(7144):606-608. doi 10.1038/nature05843
McAuley-Hecht K.E., Leonard G.A., Gibson N.J., Thomson J.B., Watson W.P., Hunter W.N., Brown T. Crystal structure of a DNA duplex containing 8-hydroxydeoxyguanine-adenine base pairs. Biochemistry. 1994;33(34):10266-10270. doi 10.1021/bi00200a006
McMurray C.T. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11(11):786-799. doi 10.1038/nrg2828
Miller H., Grollman A.P. Kinetics of DNA polymerase I (Klenow fragment exo–) activity on damaged DNA templates: effect of proximal and distal template damage on DNA synthesis. Biochemistry. 1997; 36(49):15336-15342. doi 10.1021/bi971927n
Mirkin S.M. Expandable DNA repeats and human disease. Nature. 2007;447(7147):932-940. doi 10.1038/nature05977
Moriya M. Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G·C→T·A transversions in simian kidney cells. Proc Natl Acad Sci USA. 1993;90(3):1122-1126. doi 10.1073/pnas.90.3.1122
Nurk S., Koren S., Rhie A., Rautiainen M., Bzikadze A.V., Mikheenko A., Vollger M.R., … Zook J.M., Schatz M.C., Eichler E.E., Miga K.H., Phillippy A.M. The complete sequence of a human genome. Science. 2022;376(6588):44-53. doi 10.1126/science. abj6987
Okonechnikov K., Golosova O., Fursov M.; UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012; 28(8):1166-1167. doi 10.1093/bioinformatics/bts091
O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., … Tatusova T., DiCuccio M., Kitts P., Murphy T.D., Pruitt K.D. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733-D745. doi 10.1093/nar/ gkv1189
Opresko P.L., Sanford S.L., De Rosa M. Oxidative stress and DNA damage at telomeres. Cold Spring Harb Perspect Biol. 2025;17(6): a041707. doi 10.1101/cshperspect.a041707
Pfeiffer V., Lingner J. Replication of telomeres and the regulation of telomerase. Cold Spring Harb Perspect Biol. 2013;5(5):a010405. doi 10.1101/cshperspect.a010405
Pilati C., Shinde J., Alexandrov L.B., Assié G., André T., Hélias-Rodzewicz Z., Ducoudray R., Le Corre D., Zucman-Rossi J., Emile J.-F., Bertherat J., Letouzé E., Laurent-Puig P. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J Pathol. 2017;242(1):10-15. doi 10.1002/ path.4880
Prorok P., Grin I.R., Matkarimov B.T., Ishchenko A.A., Laval J., Zharkov D.O., Saparbaev M. Evolutionary origins of DNA repair pathways: role of oxygen catastrophe in the emergence of DNA glycosylases. Cells. 2021;10(7):1591. doi 10.3390/cells10071591
Richard G.-F., Kerrest A., Dujon B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol Mol Biol Rev. 2008;72(4):686-727. doi 10.1128/MMBR.00011-08
Saito I., Nakamura T., Nakatani K., Yoshioka Y., Yamaguchi K., Sugiyama H. Mapping of the hot spots for DNA damage by one-electron oxidation: efficacy of GG doublets and GGG triplets as a trap in long-range hole migration. J Am Chem Soc. 1998;120(48):12686- 12687. doi 10.1021/ja981888i
Shibutani S., Takeshita M., Grollman A.P. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349(6308):431-434. doi 10.1038/349431a0
Sugiyama H., Saito I. Theoretical studies of GG-specific photocleavage of DNA via electron transfer: significant lowering of ionization potential and 5′-localization of HOMO of stacked GG bases in B- form DNA. J Am Chem Soc. 1996;118(30):7063-7068. doi 10.1021/ja 9609821
Taylor W.R. The classification of amino acid conservation. J Theor Biol. 1986;119(2):205-218. doi 10.1016/S0022-5193(86)80075-3
Tubbs A., Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644-656. doi 10.1016/j.cell.2017.01.002
Viel A., Bruselles A., Meccia E., Fornasarig M., Quaia M., Canzonieri V., Policicchio E., … Maestro R., Giannini G., Tartaglia M., Alexandrov L.B., Bignami M. A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine. 2017;20:39-49. doi 10.1016/ j.ebiom.2017.04.022
Wood M.L., Esteve A., Morningstar M.L., Kuziemko G.M., Essigmann J.M. Genetic effects of oxidative DNA damage: comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro- 8-oxoadenine in Escherichia coli. Nucleic Acids Res. 1992;20(22): 6023-6032. doi 10.1093/nar/20.22.6023
Yudkina A.V., Shilkin E.S., Endutkin A.V., Makarova A.V., Zharkov D.O. Reading and misreading 8-oxoguanine, a paradigmatic ambiguous nucleobase. Crystals. 2019;9(5):269. doi 10.3390/cryst 9050269