A Novel Class of Orally Bioavailable Phenylglycine–Benzoxaborole Conjugates with Antimalarial Activity and Potentially Novel Mechanism of Action
Mokhitli Morake, Dale Taylor, Dina Coertzen, Mathew Njoroge, Liezl Krugmann, Meta Leshabane, Shanté da Rocha, Tarrick Qahash, Gareth Girling, Rachael Coyle, Marcus C. S. Lee, Sergio Wittlin, Manuel Llinás, Lyn-Marie Birkholtz, Gregory S. Basarab, Kelly Chibale

TL;DR
Researchers discovered a new class of malaria-fighting compounds with promising activity and a potentially new mechanism of action.
Contribution
A novel class of phenylglycine–benzoxaborole conjugates with antimalarial activity and high selectivity was developed.
Findings
Compounds showed potent in vitro activity against both drug-sensitive and drug-resistant malaria strains.
Selected compounds demonstrated high solubility and metabolic stability in human and animal liver microsomes.
Oral dosing of two compounds in a mouse malaria model showed encouraging in vivo efficacy.
Abstract
A new class of benzoxaboroles with a phenylglycine appendage was found to display in vitro blood stage activity against the human malaria parasite Plasmodium falciparum (Pf). Structure–activity relationship studies of the starting hit compound 3 resulted in compounds active against PfNF54 drug-sensitive and PfK1 drug-resistant strains with an in vitro antiplasmodium IC50 < 0.4 μM, selectivity over mammalian cell-lines (selectivity index > 47) and high aqueous solubility (160 to >200 μM). Selected compounds showed good in vitro metabolic stability when incubated with human, rat, and mouse liver microsomes and showed no cross-resistance against barcoded mutant lines. Two frontrunner compounds, 6 and 7, were dosed orally at 50 mg·kg–1 using a standard quadrupole dosing regimen in a P. berghei mouse infection model and showed encouraging in vivo efficacy. This work identifies a promising…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
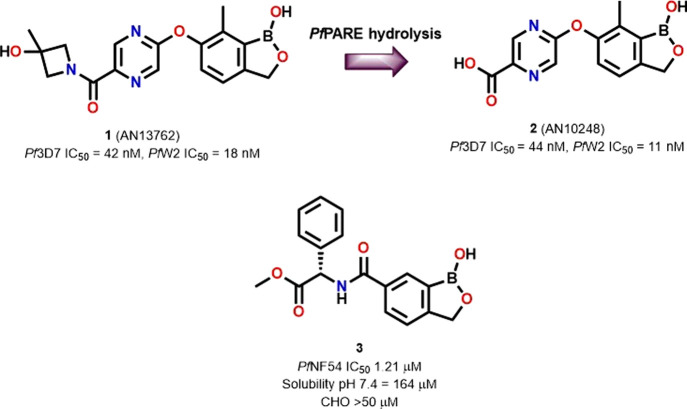 Figure 1
Figure 1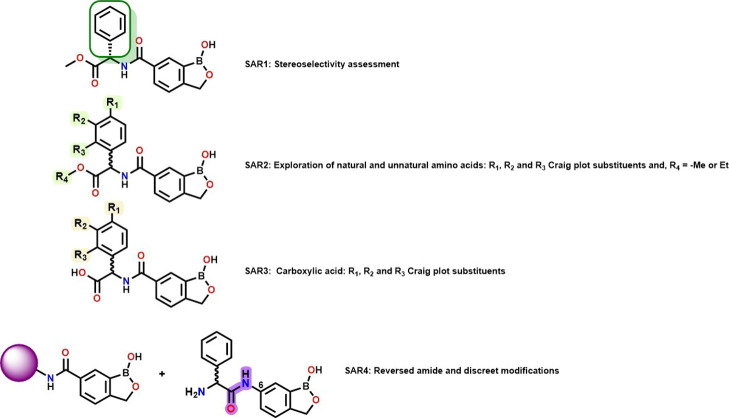 Figure 2
Figure 2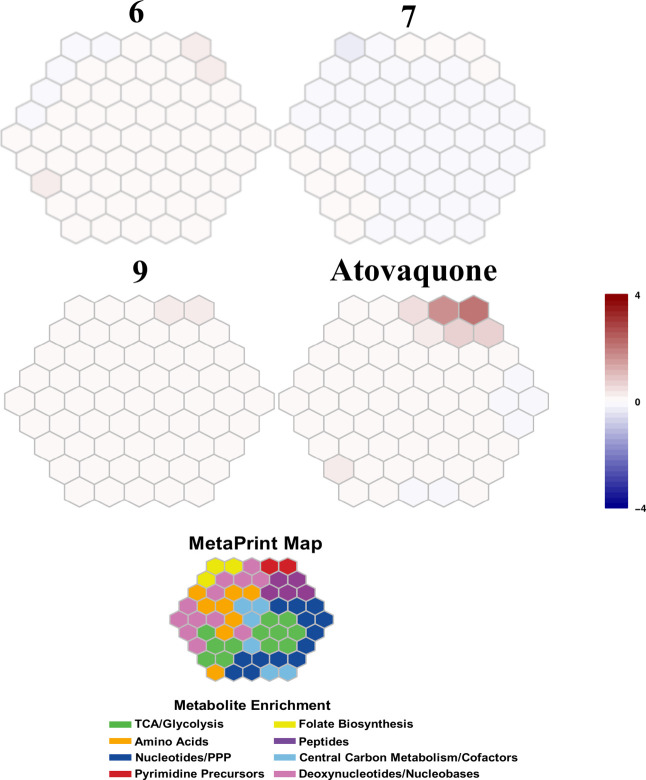 Figure 3
Figure 3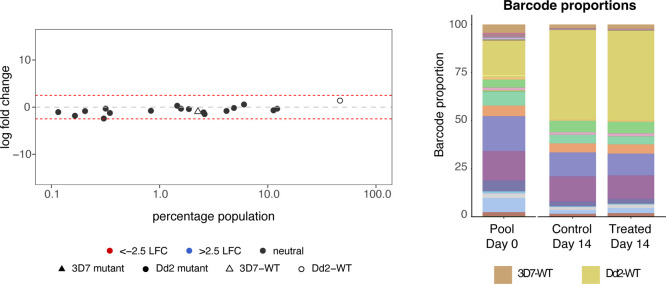 Figure 4
Figure 4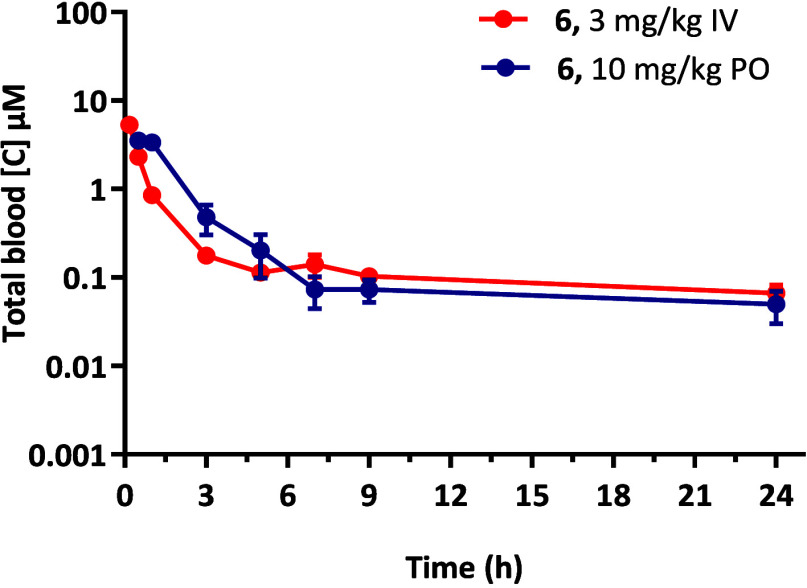 Figure 5
Figure 5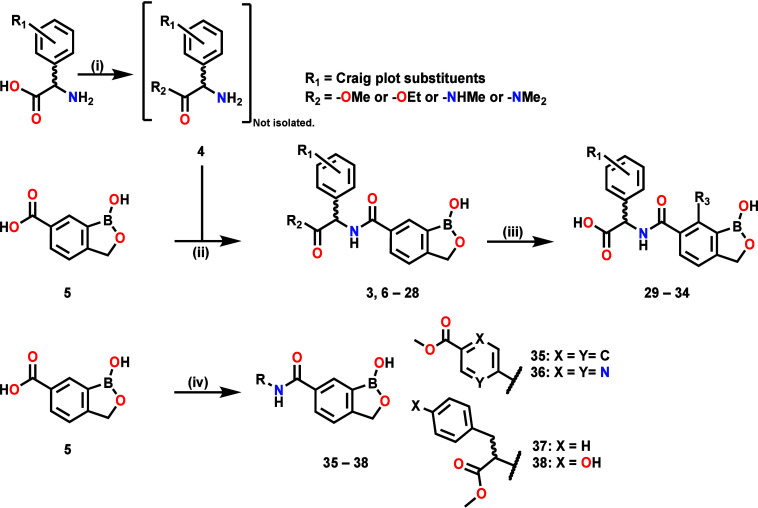 Figure 6
Figure 6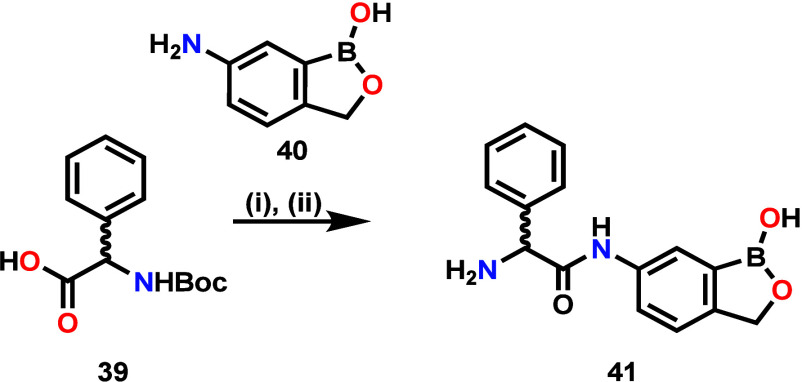 Figure 7
Figure 7 Figure 8
Figure 8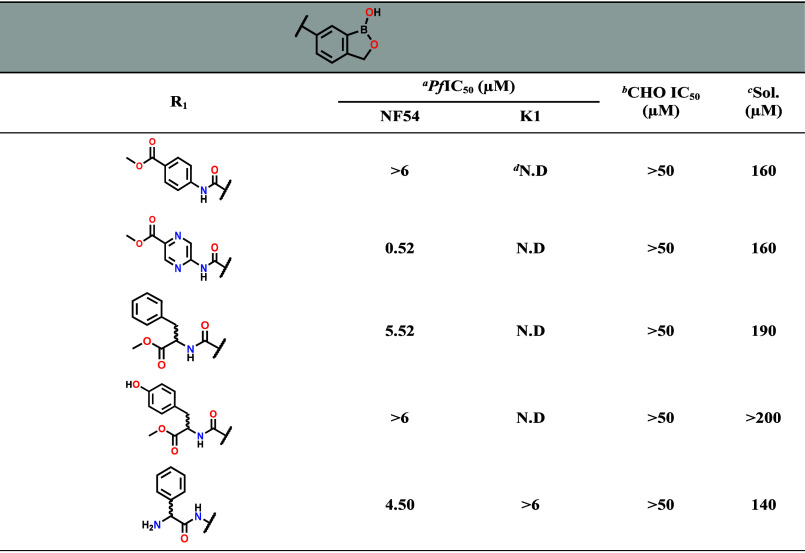 Figure 9
Figure 9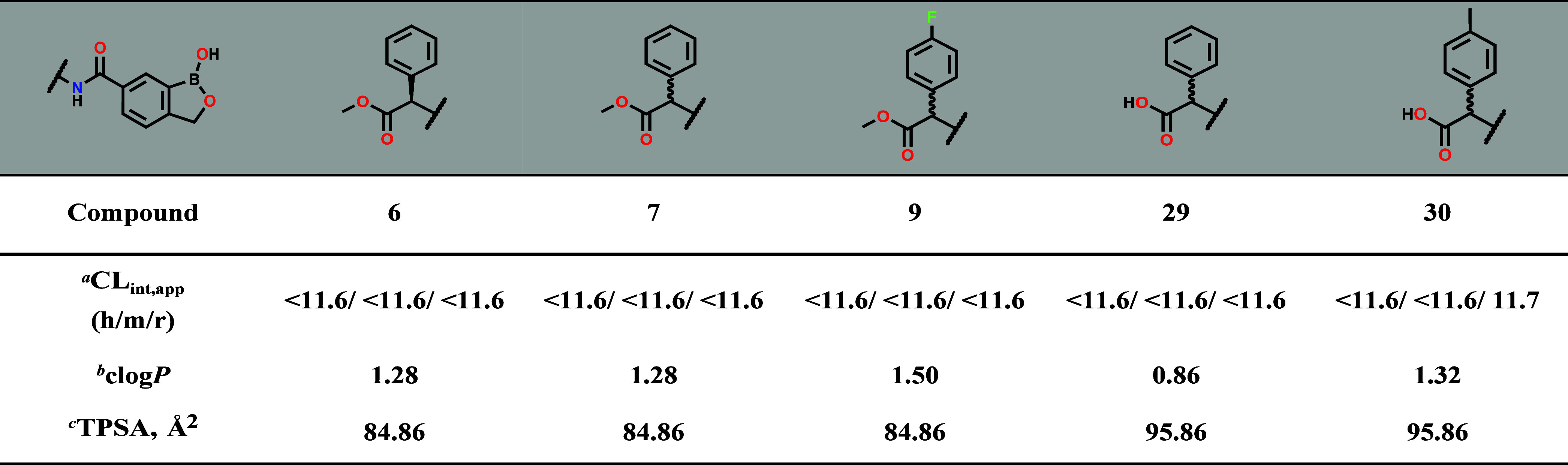 Figure 10
Figure 10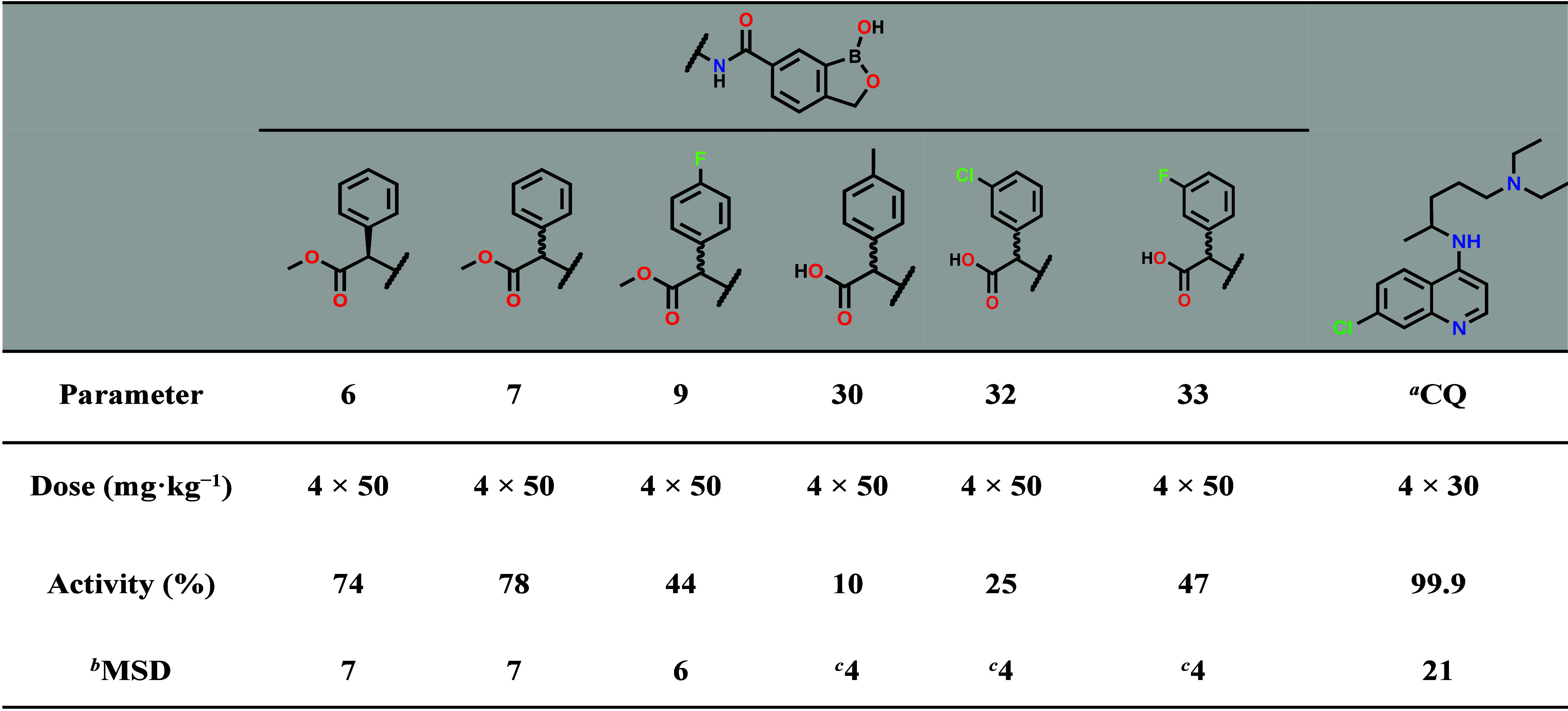 Figure 11
Figure 11 Figure 12
Figure 12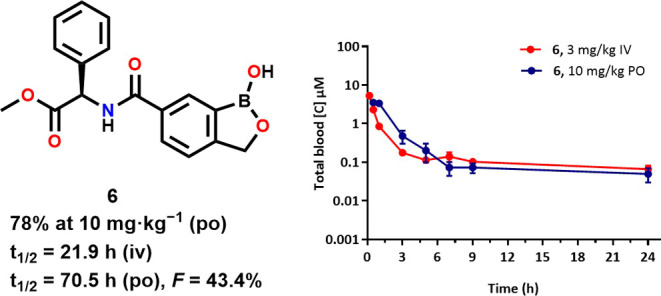 Figure 13
Figure 13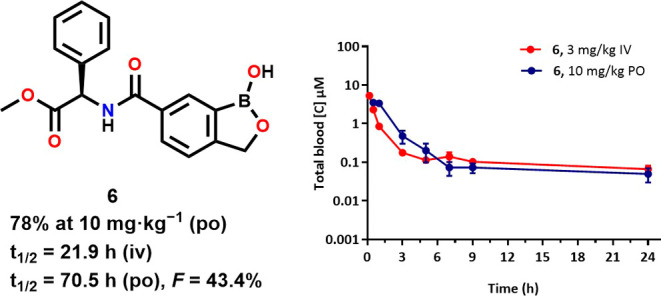 Figure 14
Figure 14- —Bill and Melinda Gates Foundation10.13039/100000865
- —Bill and Melinda Gates Foundation10.13039/100000865
- —Wellcome10.13039/100004440
- —Medicines for Malaria VentureNA
- —Huck Institutes? Metabolomics Core FacilityNA
- —South African National Research Foundation (NRF)NA
- —South African National Research Foundation (NRF)NA
- —South African National Research Foundation (NRF)NA
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOrganoboron and organosilicon chemistry · Synthesis and bioactivity of alkaloids · Quinazolinone synthesis and applications
Malaria continues to be a major global health problem with a disproportionate distribution in the impoverished regions of the world. Most cases, particularly in Africa, are due to Plasmodium falciparum (Pf) infections. The World Health Organization (WHO) estimated morbidity and mortality due to malaria in 2023 to be 263 million and 597 000, respectively. These are marked increases compared to pre-COVID-19 pandemic numbers in 2019 where 233 million cases and 576 000 deaths were estimated by the WHO. This is in contrast with the steady decline in malaria incidences reported between 2000 and 2014, although they had already started to plateau since 2015.? Several factors have influenced the reversal of the gains previously made to combat malaria. These include a rise in insecticide and drug resistant vectors and parasites, respectively.?
The WHO has recently recommended the RTS,S/AS01 and R21/Matrix M malaria vaccines for use in moderate to high transmission endemic areas, and this has led to a decline in childhood mortality due to malaria.? While RTS,S/AS01 achieves only 36% efficacy in infants, R21/Matrix M has ≥75% efficacy, and it is by far the most effective vaccine for seasonal malaria administration. ?,? These are still being explored for adult populations and are thus currently limited for full scale use in endemic areas.? Other prevention strategies such as the use of insecticidal bed nets and indoor residual spraying are limited by Anopheles resistance to pyrethroids and are thus not effective in eliminating malaria.? Therefore, the mainstay of malaria control relies on chemotherapy, and currently, numerous drugs are used for chemoprophylaxis and treatment of malaria. Pf has developed resistance to almost all of these drug classes, with the artemisinins remaining the most effective first-line drugs in clinical use for the treatment of the disease. They are used in combination with other drug classes in Artemisinin-based combination therapy (ACT) regimens.? Recent reports indicating partial artemisinin resistance, as shown by decreased cure rates and high recrudescence following the use of ACT regimens, are alarming. While these were largely noted in Southeast Asia, parasites with novel resistance markers or similar in the PfKelch13 gene have recently been reported in Africa. ?,? This signifies that the parasite has developed tolerance to artemisinins and thus a threat for imminent resistance, compounded by the bleak prospects of novel antimalarials for clinical use in the near future.
Novel antimalarial drugs and chemotypes are urgently needed to combat malaria and overcome current drug resistance challenges. In recent years, the benzoxaborole scaffold has shown promise for drug discovery in various disease areas ranging from parasitic to bacterial infections.? Recent reports of this class with activity against parasites such as Toxoplasma gondii, Cryptosporidium, Leishmaniasis and Pf are promising for their exploration as a novel chemotype. ?,?,? Although the exact target has not been fully deconvoluted, these compounds have been proposed to inhibit the Leucyl-RNA synthetase (LeuRS) by arresting editing by LeuRS or targeting the pre-mRNA processing Cleavage and Polyadenylation Specificity Factor 3 (CPSF3). A number of other proteins including those involved in ubiquitination such as SUMO-activating enzyme subunit 2 and ubiquitin-activating enzyme 1 have also been implicated as targets for the benzoxaboroles. ?−? ?,?,?
Recently a preclinical candidate, benzoxaborole 1 (AN13762), was reported with in vitro antiplasmodium activity against Pf3D7 (IC_50_ = 42 nM) and PfW2 (IC_50_ = 18 nM) (Figure). Biochemical and genetic studies of the mechanism of action of this compound revealed that in addition to PfCPSF3, mutations were observed in prodrug activation and resistance esterase (PfPARE) suggesting that the amide of 1 is cleaved to give its carboxylic acid derivative 2 (AN10248, Figure). However, the development of AN13762 was reported to have been halted due to toxicity in animals.? Metabolite 2 was roughly equipotent to parent compound 1 against the parasite. The ease of amide hydrolysis to carboxylic acid by the parasite is limiting due to the facile resistance resultant from the PfPARE mutation. AN10248 itself would likely show reduced permeability (and hence bioavailability) in vivo and would be susceptible to metabolic processes commonly associated with carboxylic acids.? Hence, there is a need for more stable benzoxaboroles if this promising class is to yield future antimalarials.
Our group recently reported a crystallographic study of boron-containing compounds that inhibited the bacterial Penicillin Binding Protein.? Inspired by the precedence of antimalarial benzoxaboroles mentioned above, we cross-screened these reported compounds for in vitro antiplasmodium activity against the drug-sensitive PfNF54 strain and identified hit compound 3 (Figure) with an IC_50_ = 1.21 μM and no cytotoxicity against the Chinese Hamster Ovarian cell line (CHO) at the highest concentration tested (IC_50_ > 50 μM). Compound 3 had a high aqueous solubility of 164 μM at pH 7.4 in PBS. Based on these data, we conducted a Formal Hit Assessment (FHA) campaign and explored structure–activity relationship (SAR) studies or variations of the amino acid group appendage of 3 to identify compounds with improved antiplasmodium activity.
Four SAR exploration strategies were undertaken, maintaining the benzoxaborole core unchanged as in 3 (SAR1-SAR4, Figure). Starting with commercially sourced carboxylic acid intermediate 5 (Scheme), SAR1 focused on the assessment of the stereospecificity of the putative target by synthesizing the opposite enantiomer 6 of 3 and the corresponding racemic mixture 7. SAR2 explored different substituents on the phenylglycine group including methyl and ethyl esters via compounds 8–26 (Table). The selection of substituents on the phenylglycine were guided by the Craig plot with groups selected from different quadrants based on their hydrophobicity and electronic nature.? This SAR also incorporated derivatives with the methyl ester of 3 replaced with methylamine and dimethylamine carboxamides affording 27 and 28, respectively. SAR3 focused on the carboxylic acid matched pairs 29–34 of selected methyl and ethyl esters while SAR4 incorporated several discrete modifications that included aromatic carboxamide substituents 35–36, phenylalanine (37) and tyrosine (38) replacements for the phenylglycine and, a reversed amide 41 (Table).
Synthesis of compounds covered in SAR1–4 (Scheme) involved (i) Fischer–Speier esterification of relevant commercially sourced amino acids in methanol or ethanol to give relevant intermediates followed by (ii) and (iv) amide coupling (SAR1, 2, and 4) and (iii) ester hydrolysis (SAR3). The reversed amide compound 41 of SAR4 was synthesized (Scheme) via amide coupling of Boc-protected phenylglycine 39 with commercially available 6-amino-benzoxaborole 40 followed by Boc-deprotection under acidic conditions.
All the compounds were profiled for in vitro blood stage antiplasmodium activity against drug-sensitive PfNF54 and multidrug-resistant PfK1 parasites using either the 72 h metabolic pLDH or 72 h proliferative SYBR Green I assays, both of which are known to produce correlative IC_50_ values.? Mammalian cell cytotoxicity was determined using the CHO cell line (Table). Hit validation was conducted with resynthesized compound 3, and its antiplasmodium activity against PfNF54 was confirmed with an IC_50_ of 1.06 μM, comparable to 1.21 μM obtained in the initial screen. We then synthesized both opposite enantiomer 6 starting with the R-enantiomer phenylglycine and racemic mixture 7. The latter was made to assess if the racemate retains sufficient antiplasmodium activity so that the other compounds could be synthesized in racemic form, thereby streamlining synthetic efforts. The PfNF54 IC_50_ values for 6 and the racemic mixture 7 were 0.64 and 0.85 μM, respectively (Table). These data show that the opposite enantiomer 6 is 2-fold more active compared to 3 and, that the racemate 7 retains antiplasmodium activity comparable to that of 6. Given that the racemic mixture had viable blood stage antiplasmodium activity, it became expedient to synthesize all subsequent compounds as racemic mixtures (Scheme and ?).
In SAR2, the most active compounds in the series were para-fluoro 9 (PfNF54 IC_50_ = 0.72 μM), and methyl amide 27 (PfNF54 IC_50_ = 0.56 μM) with blood stage antiplasmodium activities comparable to that of 7 (PfNF54 IC_50_ = 0.85 μM), within a 2-fold variation inherent in the assay. The regioisomer of 9, meta-fluoro 10 had slightly lower blood stage antiplasmodium activity against PfNF54 (IC_50_ = 1.39 μM). The dimethyl amide 28 lost blood stage antiplasmodium activity (PfNF54 IC_50_ > 6 μM) at the highest concentration tested. Unlike 6 that showed lower blood stage activity against the drug-resistant PfK1 strain (PfK1 IC_50_ = 3.88 μM), 27 showed equipotent blood stage activity against the PfK1 strain with an IC_50_ = 0.60 μM. Compound 27 also maintained favorable solubility (150 μM) and showed no cytotoxicity at the highest concentration tested (CHO IC_50_ > 50 μM) (Table). Other substituents on the phenylglycine in the series led to lower blood stage activity relative to 7, i.e. PfNF54 IC_50_ > 1 μM. The ethyl ester derivatives 20–26 all had lower blood stage antiplasmodium activity with PfNF54 IC_50_ > 1 μM compared to 7 irrespective of the substituent on the phenylglycine. When phenylalanine in 37 and tyrosine in 38 were introduced to explore effects of the spacer and addition of the polar group (OH), respectively, the former was 10-fold less active (PfNF54 IC_50_ = 5.52 μM) while the latter was not active at the highest concentration tested (PfNF54 IC_50_ > 6 μM) (Table). Derivatives with the methyl 4-aminobenzoate appendage 35 and aminopyrazine-2-carboxylate appendage 36 had PfNF54 IC_50_ values of >6 and 0.52 μM, respectively. The high blood stage antiplasmodium activity for 36 is of interest in that it contains the pyrazine-2-carboxy moiety present in the preclinical candidate 1 (Figure) suggesting that this group may be contributing to the slight increase in potency.
SAR3 with carboxylic acid matched pairs showed the highest in vitro blood stage antiplasmodium activity compared to other series with activity ranging between IC_50_ = 0.12–3.25 μM against PfNF54 (Table). Compound 30 with the para-methyl substituent showed the highest activity (PfNF54 IC_50_ = 0.12 μM) notably more active than 6 and 7. Against the drug-resistant strains PfK1 and PfDd2, 30 was slightly less active with IC_50_ values 0.45 and 0.97 μM, respectively. In this series, compounds 32 with meta-chloro and 33 with meta-fluoro also had the highest combination of blood stage antiplasmodium activity against both PfNF54 (0.39 and 0.34 μM, respectively) and PfK1 (0.39 and 0.48 μM, respectively). When these compounds were evaluated for their transmission blocking activity against immature (iGc > 90% stage II–III) and late stage (lGc > 90% IV–V) gametocytes using a luciferase assay platform, 32 was inactive; however, 33 showed moderate activities against both stages (IC_50_ = 3.95 μM and 4.46 μM against iGc and lGc, respectively). Cytotoxicity evaluation for all the compounds across series in the study shows that regardless of the different antiplasmodium potencies, all the compounds profiled for cytotoxicity in the CHO cell line had an IC_50_ > 50 μM as well as favorable solubility, 80 to
200 μM (Table and ?). The reversed amide 41 (Table) led to decreased blood stage activity (PfNF54 IC_50_ = 4.5 μM) but maintained the favorable CHO cytotoxicity (IC_50_ > 50 μM) and solubility (140 μM). Compounds 6 and 30 were evaluated for their hemolytic potential, and no hemolysis was noted across the tested concentration range, indicating that the observed activity is not attributable to host cell damage.
The metabolic stability of selected compounds was then assessed via incubation with human, mouse, and rat liver microsomes for 0.5 h. Compounds 3, 6, 9, 29, and 30 were metabolically stable across the species with apparent intrinsic clearances (CLint, app) < 11.6 μL/min/mg, predicting low hepatic metabolic clearance in vivo (Table).
Toward shedding light on the potential novelty of the mechanism of action (MoA) and/or putative targets of these compounds, selected compounds were subjected to targeted hydrophilic metabolomics and barcoded mutant cross-resistant studies. ?,? The former was aimed at identifying the pathways in which these benzoxaboroles are acting and the latter to show cross resistance (or lack thereof) with known targets of this class. An assessment of the metabolic fingerprints (metaprints) ?,? of compounds 6 and 9 showed a weak increase in pyrimidine biosynthesis precursors (Figure). Additionally, compound 6 resulted in a slight increase in hemoglobin-derived peptides and folate intermediates were also observed for compound 7. However, overall, the metaprint of the compounds studied results in ambiguous profiles under the conditions tested. As previous studies have implicated CPSF3, which is involved in pre-mRNA processing, as a target of benzoxaboroles, it is likely that the pathways perturbed by these compounds do not result in easily defined metabolite changes. Thus, these ambiguous profiles may result from undetectable cellular perturbations as has been previously observed for some compound classes such as trioxolanes and translation inhibitors which give weak signals and, cluster in unclassified pathways.?
Compound 7 was further assessed for cross resistance against a pool of parasite barcoded drug-resistant mutants (Table S2). This compound showed no significant changes in the count of barcode proportions in treated (3 × IC_50_) and untreated parasites in both Pf3D7 and Dd2 backgrounds (Figure). The data suggest that the compound has no cross-resistance with any of the mutants present in the parasite pool and implies that it is likely acting through a novel MoA. The pool included parasites with the CPSF3-Y408S mutation, previously reported in resistant parasite lines raised against AN3661, and compound 7 was not cross-resistant to these parasite lines.? This further suggested that this new class of benzoxaboroles is acting through a novel MoA, dissimilar to other benzoxaborole classes, and that it has no cross-resistance to the current antimalarials.
As the ester groups are metabolically labile being prone to hydrolysis, and that PfPARE has been reported to hydrolyze pepstatin-based antimalarials, future work should include selection of resistant mutants in vitro to determine if this class of benzoxaboroles also shows PfPARE resistant markers.? It is indeed possible that the observed superior activity of the methyl esters compared to the ethyl esters (Table) may be due to the former being hydrolyzed faster by PfPARE or other esterases.
Given the promising in vitro metabolic stability and blood stage antiplasmodium data, compounds 6, 7, 9, 30, 32, and 33 were evaluated for in vivo efficacy in the P. berghei mouse infection model. When dosed orally in a 50 mg·kg^–1^ standard quadrupole dosing regimen, compounds 6 and 7 showed 74 and 78% reduction in parasitaemia, respectively, with mean survival days of 7 days (Table). Pharmacokinetic evaluation of 6 in healthy Balb/c mice shows that the compound has a low clearance (20 mL·min^–1^·kg^–1^) and a high volume of distribution (V d = 34.4 L·kg^–1^), which results in a long half-life (Table and Figure). This, combined with good oral exposure (bioavailability = 43%) would have supported the observation of good efficacy for compound 6. However, when the dose of the compound was increased to 50 mg/kg, the increase in exposure was less than dose proportional, suggesting saturation of absorption. The lower-than-expected exposures would not maintain coverage above the IC_50_ for the 24 h period between doses, which explains why 6 was not more efficacious in vivo.
Compounds 9, 30, 32, and 33 showed lower efficacy in vivo, 10–47% reduction in parasitaemia (Table). The suboptimal efficacy of the carboxylic acid derivatives compared to the ester compounds contrasts with in vitro blood stage antiplasmodium data, which shows better activity of the former compared to the latter. This may be due to low permeability of the carboxylic acid derivatives on oral dosing resulting from low diffusion across intestinal epithelial cells and thus, lower drug exposure.?
Benzoxaboroles are emerging as a promising scaffold for development of next generation antimalarials with advantages including activity likely against novel target(s), drug-like properties, and selectivity for the parasite over mammalian cells. The FHA campaign herein enclosed reports on the promising hit compounds with high cross species in vitro microsomal metabolic stability, and SAR exploration identified submicromolar hit compounds with potential for further optimization into lead compounds. Compound 30 showed the highest blood-stage antiplasmodium activity (PfNF54 IC_50_ = 0.12 μM) in vitro. Other carboxylic acid derivatives, 32 and 33 also showed the high blood-stage antiplasmodium activity in in vitro assays adding to the report of the carboxylic acid derivative 2 having activity against Pf similar to that of amide 1 (Figure). That ester derivatives had efficacy in vivo may be due to the proclivity of the esters to undergo metabolic hydrolysis to active carboxylic acids, suggesting that these compounds likely act as prodrugs in vivo. Lack of cross-resistance and ambiguous metabolomics profile of selected benzoxaboroles suggest a novel MoA for this class. Further biochemical studies are required around the esters and amides of the current series to ascertain PfPARE’s involvement for resistance development and, for prodrug convertase activity as well as the relevance to antiplasmodium activity of the corresponding acids. Given inconclusive knowledge of the exact target of the benzoxaboroles and observed polypharmacology of this compound class,? additional studies will be essential to identify target/s for these compounds. The pharmacokinetic data of compound 6 with respect to moderate oral bioavailability and long half-life are promising for lead optimization for an oral drug. This FHA campaign has identified a new class of benzoxaborole compounds with in vivo efficacy, which motivates further optimization for potential hit-to-lead transition.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization . World malaria report 2024. https://www.who.int/publications/i/item/9789240104440 (accessed December 10, 2024).
- 2Gao L.Shi Q.Liu Z.Li Z.Dong X.Impact of the COVID-19 Pandemic on Malaria Control in Africa: A Preliminary Analysis Trop. Med. Infect. Dis.2023816710.3390/tropicalmed 801006736668974 PMC 9863638 · doi ↗ · pubmed ↗
- 3Datoo M. S.Dicko A.Tinto H.Ouédraogo J.-B.Hamaluba M.Olotu A.Beaumont E.Lopez F. R.Natama H. M.Weston S.Safety and efficacy of malaria vaccine candidate R 21/Matrix-M in African children: a multicentre, double-blind, randomised, phase 3 trial Lancet 20244031042653354410.1016/S 0140-6736(23)02511-438310910 PMC 7618965 · doi ↗ · pubmed ↗
- 4Rts S.Efficacy and safety of RTS, S/AS 01 malaria vaccine with or without a booster dose in infants and children in Africa: final results of a phase 3, individually randomised, controlled trial Lancet 20153869988314510.1016/S 0140-6736(15)60721-825913272 PMC 5626001 · doi ↗ · pubmed ↗
- 5Stanisic D. I.Good M. F.Malaria Vaccines: Progress to Date Bio Drugs 202337673775610.1007/s 40259-023-00623-437728713 PMC 10581939 · doi ↗ · pubmed ↗
- 6Ingham V. A.Grigoraki L.Ranson H.Pyrethroid resistance mechanisms in the major malaria vector species complex Entomol. Gen.202343351552610.1127/entomologia/2023/1880 · doi ↗
- 7van der Pluijm R. W.Amaratunga C.Dhorda M.Dondorp A. M.Triple artemisinin-based combination therapies for malaria–a new paradigm?Trends Parasitol.2021371152410.1016/j.pt.2020.09.01133060063 · doi ↗ · pubmed ↗
- 8Begolo D.Vincent I. M.Giordani F.Pöhner I.Witty M. J.Rowan T. G.Bengaly Z.Gillingwater K.Freund Y.Wade R. C.The trypanocidal benzoxaborole AN 7973 inhibits trypanosome m RNA processing P Lo S Pathog.2018149 e 100731510.1371/journal.ppat.100731530252911 PMC 6173450 · doi ↗ · pubmed ↗