Quinazolinones as Bioisosteres of Naphthoquinones: A Path to Potent HsDHODH Inhibitors with Optimized Properties
Bruna F. Godoi, Jéssica D. Bueno, Wemenes J. L. Silva, Aline D. da Purificação, Pedro I. P. Leite, Thiago dos Santos, Murillo Freitas, Daniel G. Silva, Tais C. Silva, Josué de Moraes, Caroline S. Freitas, Mayara Mattos, Thiago M. L. Souza, Bianca A. Martin, Renata F. V. Lopez

TL;DR
Researchers developed quinazolinone compounds that inhibit a key enzyme in virus replication, showing promise for antiviral therapies against SARS-CoV-2.
Contribution
Quinazolinones are introduced as bioisosteres of naphthoquinones, offering improved potency and selectivity for HsDHODH inhibition.
Findings
Compound 10c inhibited HsDHODH with an IC50 of 25 μM.
Compound 10e showed high potency (IC50 = 0.59 μM) and antiviral activity (EC50 = 0.15 μM) against SARS-CoV-2.
Quinazolinones demonstrated improved selectivity over naphthoquinone analogs but had solubility challenges.
Abstract
Human dihydroorotate dehydrogenase (HsDHODH) is a key enzyme in pyrimidine biosynthesis and a target for antiviral therapies against RNA viruses like SARS-CoV-2. Building on prior quinone-based inhibitors, we explored quinazolinones as bioisosteric replacements to reduce cytotoxicity and off-target effects. Through structure-based design, we synthesized quinazolinone derivatives aimed at maintaining critical binding interactions. First-generation compounds showed moderate HsDHODH inhibition (up to 60% at 250 μM), with compound 10c having an IC50 of 25 μM. Using computational modeling, we optimized second-generation derivatives, with 10e showing the highest potency (IC50 = 0.59 ± 0.03 μM) and significant antiviral activity against SARS-CoV-2 (EC50 = 0.15 ± 0.03 μM). These compounds demonstrated improved selectivity compared to naphthoquinone analogs, though challenges with aqueous…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
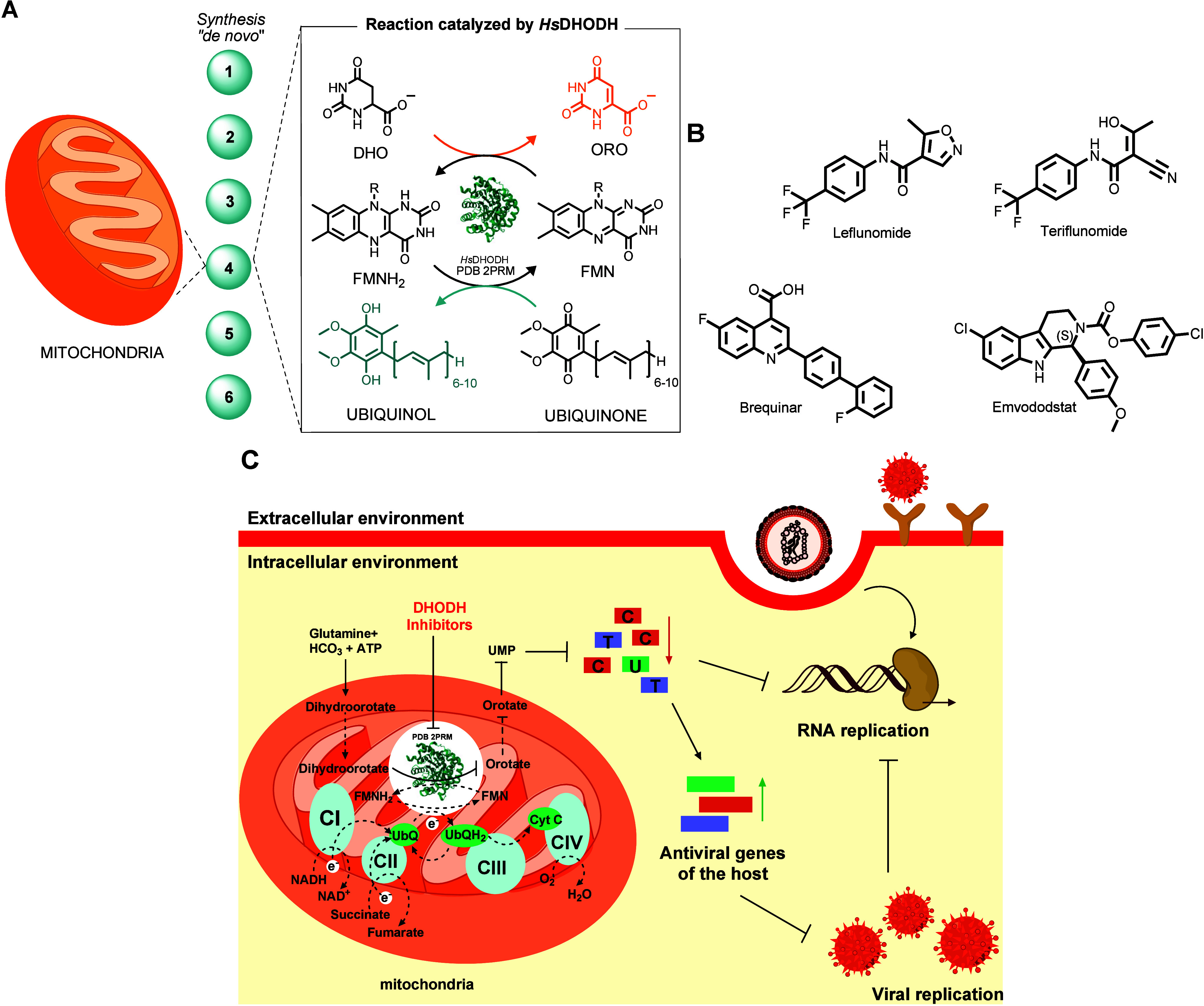 Figure 1
Figure 1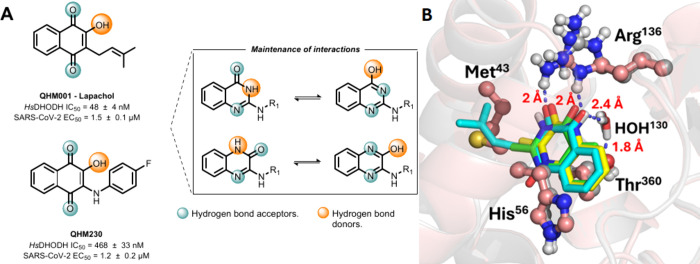 Figure 2
Figure 2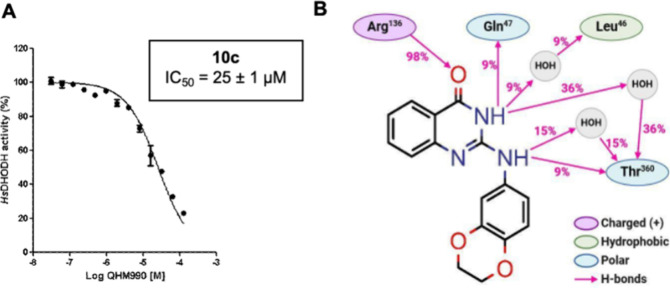 Figure 3
Figure 3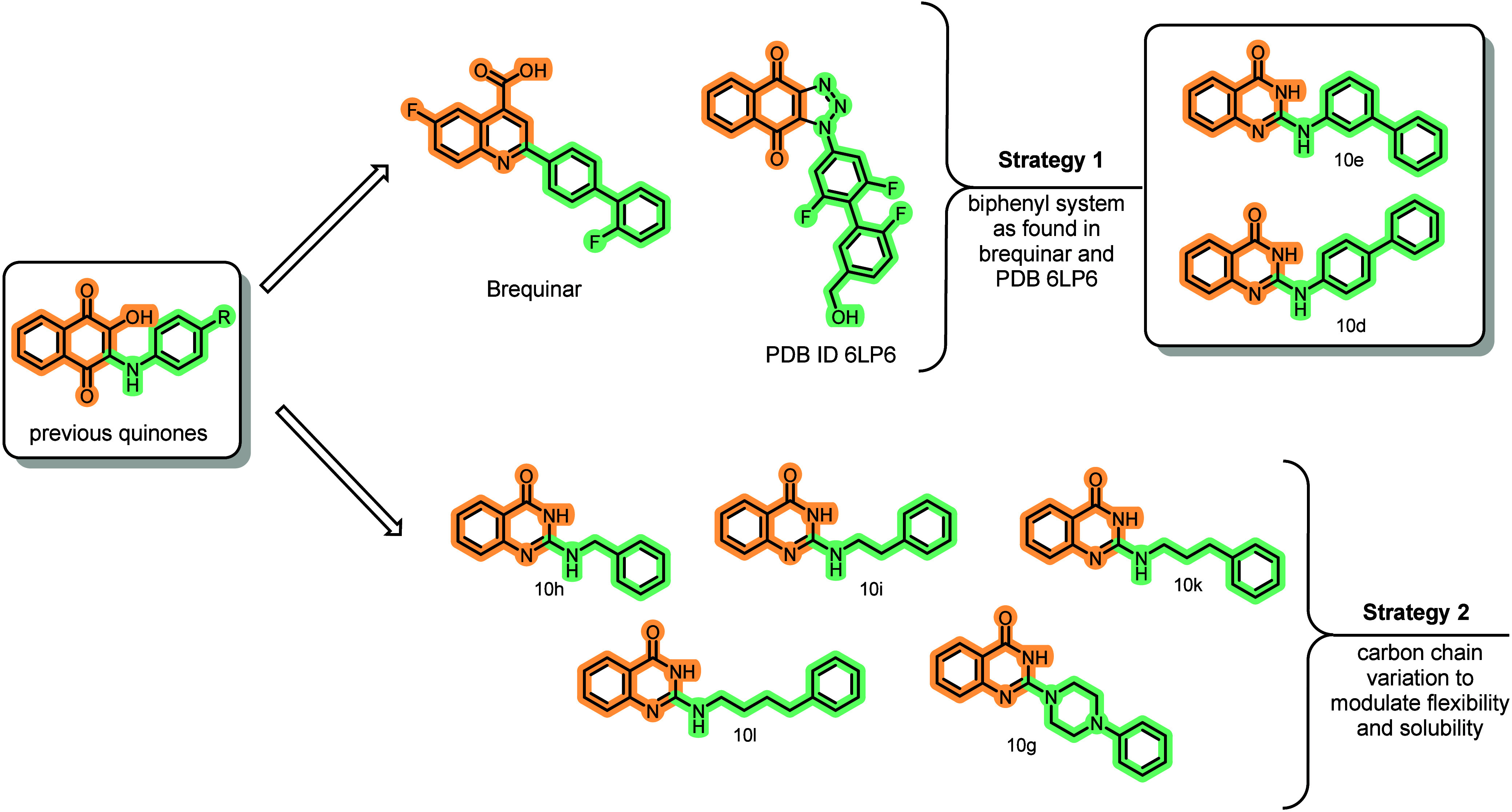 Figure 4
Figure 4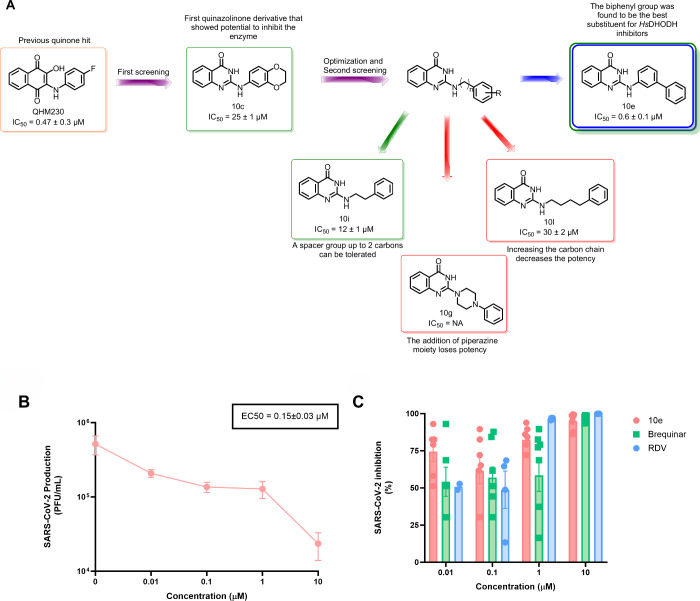 Figure 5
Figure 5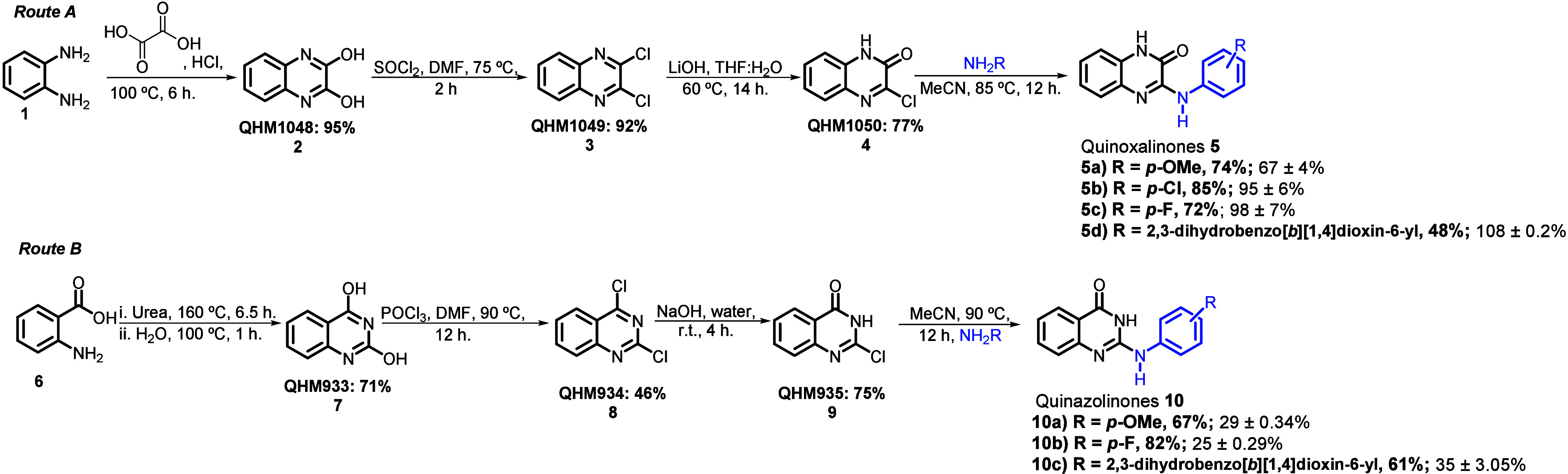 Figure 6
Figure 6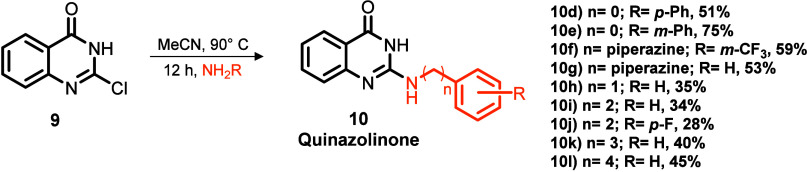 Figure 7
Figure 7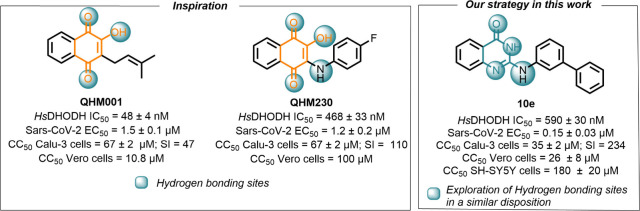 Figure 8
Figure 8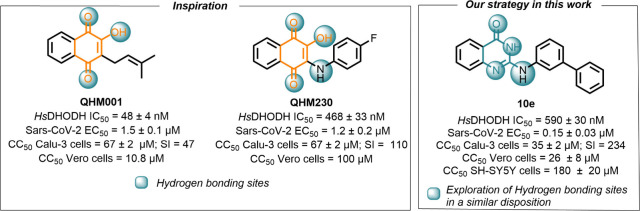 Figure 9
Figure 9- —National Institute on Aging10.13039/100000049
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Funda??o de Amparo ? Pesquisa do Estado de S?o Paulo10.13039/501100001807
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Conselho Nacional de Desenvolvimento Cient?fico e Tecnol?gico10.13039/501100003593
- —Funda??o de Amparo ? Pesquisa do Estado de Goi?s10.13039/501100005285
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsQuinazolinone synthesis and applications · Synthesis and Biological Evaluation · Synthesis and Characterization of Heterocyclic Compounds
The de novo synthesis of pyrimidine nucleotides represents a conserved metabolic pathway across bacteria, protozoans, and animals, providing essential building blocks for DNA and RNA biosynthesis.? Dihydroorotate dehydrogenase (DHODH) catalyzes the fourth and rate-limiting step in this pathway. ?,? Located on the outer surface of the inner mitochondrial membrane, human DHODH (HsDHODH) catalyzes the oxidation of dihydroorotate to orotate through a flavin-dependent redox reaction (FigureA), which is subsequently utilized for the production of uridine monophosphate (UMP) and downstream pyrimidine nucleotides. ?,?
Although they are recognized as validated targets for drug discovery, a limited number of HsDHODH inhibitors have reached clinical trials and advanced to the market. For instance, the prodrug leflunomide, metabolized to teriflunomide, is a potent HsDHODH inhibitor that was approved for the treatment of rheumatoid arthritis (FigureB). ?−? ?
Recent investigations have explored HsDHODH as a target to discover drug candidates for COVID-19. ?,?,? This metabolic pathway has gained significant attention as a promising target for antiviral therapeutics, as viruses rely extensively on host pyrimidine biosynthesis to support their replication cycles. This host-directed approach offers potential advantages for developing broad-spectrum antivirals with reduced susceptibility to viral resistance mechanisms (FigureC). ?−? ? Brequinar, originally developed as an anticancer agent,? exhibits highly specific and potent inhibitory activity against HsDHODH and shows low micromolar anti-SARS-Cov-2 activity (FigureB). ?,?
Emvododstat (PTC299) emerged during the COVID-19 pandemic as a promising drug candidate targeting HsDHODH, demonstrating dual therapeutic mechanisms through direct inhibition of pyrimidine biosynthesis and suppression of pro-inflammatory cytokine production (FigureB).? Beyond SARS-CoV-2, emvododstat exhibits broad-spectrum antiviral activity against other RNA viruses, including Ebola virus and Hepatitis C virus.? Although Phase 2/3 clinical trials for hospitalized COVID-19 patients initially showed encouraging results, these studies were ultimately terminated before completion.? The clinical trial failure of HsDHODH inhibitor monotherapy for SARS-CoV-2 is not discouraging the attempts to find clinical candidates targeting this enzyme, as combination therapy with antivirals is a promising approach to fight infections with RNA viruses. ?,? These preliminary data nonetheless contributed valuable insights regarding the therapeutic potential of inhibiting HsDHODH as a host-directed antiviral drug discovery approach.
In our previous studies, we identified a series of quinone-based compounds as potent inhibitors of HsDHODH (as Lapachol and QHM230, FigureA), exhibiting nanomolar IC_50_ values and demonstrating in vitro activity against SARS-CoV-2 in the micromolar range.? These findings highlight the therapeutic potential of HsDHODH inhibition as a host-directed antiviral strategy. Molecular docking analyses revealed that these quinoidal inhibitors establish binding interactions similar to Brequinar at the enzyme’s ubiquinone binding site. Despite these promising results, the naphthoquinone series displayed significant limitations including high cytotoxicity, poor aqueous solubility, and inherent scaffold liabilities related to promiscuous reactivity and metal chelation. ?−? ?
To address these liabilities while maintaining potent inhibitory activity, we hypothesized that replacing the hydroxynaphthoquinone core with isosteric quinazolinone or quinoxalinone scaffolds could enhance drug-like properties while preserving critical binding interactions. In this work, through structure-based drug design (SBDD), we identified and validated quinazolinones as promising bioisosteres of the naphthoquinone core. Herein, we report the design, synthesis, molecular interactions, and biological evaluation of novel HsDHODH inhibitors that demonstrate anti-SARS-CoV-2 activity with improved physicochemical and toxicological profiles. Our optimization strategy focused on enhancing both polar and hydrophobic interactions within the enzyme’s binding site. In the first screening, we selected matched pairs (such as fluorine and chlorine in the para position, the most potent compounds in the previous paper) in order to compare the inhibitory activity of quinones, quinazolinones, and quinoxalinones. Moreover, since electron-donating groups were not assessed in the past paper, we also added p-methoxy and 2,3-dihydrobenzo[b][1,4]dioxin-6-yl groups to better understand the SAR.
We established two linear synthetic routes to obtain the designed quinoxalinones (5a–d) and quinazolinones (10a–c) as depicted in Scheme. For quinoxalinones (Route A), we conjugated oxalic acid with 1,2-diaminobenzene under acidic conditions to afford 1,4-dihydroquinoxaline-2,3-dione (2) in 95% yield,? which was further chlorinated via thionyl chloride to yield 2,3-dichloroquinoxaline (3).? Selective hydrolysis of 3 using lithium hydroxide yielded 3-chloroquinoxalin-2(1H)-one (4), which was then reacted with anilines to provide the desired set of 3-amino-substituted quinoxalinones with yields ranging from 48 to 85%. ?,? For 2-amino-substituted quinazolinones (Route B), we reacted 2-aminobenzoic acid and urea according to Gong and co-workers’ procedure? to produce quinazoline-2,4-diol (7, 71%) on a gram scale. After dichlorination with phosphoryl chloride, selective hydrolysis with sodium hydroxide, and nucleophilic substitution with the respective anilines, we obtained 2-amino-quinazolin-4-(3H)-ones with yields ranging from 61 to 82%. ?−? ? ?
Compounds 5a–d and 10a–c were evaluated for HsDHODH inhibition in a single-dose assay at 250 μM, with enzymatic inhibition [±SD (%)] reported without preincubation. Notably, while quinazolinones demonstrated significant inhibitory activity under the assay conditions, the quinoxalinone scaffold failed to inhibit the enzyme. The differences in inhibition profiles can be rationalized by computational analysis revealing suboptimal hydrogen bonding distances between quinoxalinones and the conserved water molecule (3.1 Å, Figure S3) compared to the more favorable interactions observed with Lapachol (2.4 Å) and quinazolinones (2.3 Å, Figure S3). The altered positioning and orientation of the quinoxalinone carbonyl hydrogen bond acceptor, compared to the optimal geometry observed in lapachol (FigureB), appears to be a critical factor preventing effective enzyme inhibition, thus failing to mimic the essential interaction pattern established by our previous hits.? The quinazolinone derivatives (10a–c), however, exhibited promising inhibitory activity, with up to 75% inhibition of HsDHODH in a single-dose assay. Compound 10c demonstrated dose-dependent inhibition with an IC_50_ value of 25 ± 1 μM. Despite showing higher percent inhibition at the screening conditions, compounds 10a and 10b exhibited nonsigmoidal dose–response relationships that precluded reliable IC_50_ determination.
FigureA presents the dose–response curve for compound 10c, calculated IC_50_ for HsDHODH inhibition, and a 2D interaction analysis derived from a 50 ns molecular dynamics (MD) simulation, illustrating the relative frequency (%) of molecular interactions between compound 10c and HsDHODH. Throughout the MD simulation, 10c maintained stable binding conformation, as evidenced by a root-mean-square deviation (RMSD) of less than 2 Å (Figure S1A). The analysis reveals a highly conserved hydrogen bond (98% occupancy) between the carbonyl group in 10c and the side chain of Arg^136^ in HsDHODH (FigureB). Additionally, transient hydrogen bonds (9% occupancy) were observed between the two N–H groups of 10c and residues Gln^47^ and Thr^360^. Water-mediated interactions were also identified, bridging the N–H moieties of 10c with residues Leu^46^ and Thr^360^. Notably, this hydrogen-bonding network between 10c and residues Arg^136^, Thr^360^, and Gln^47^ corresponds to interactions previously reported for naphthoquinoidal inhibitors,? suggesting a conserved binding mechanism despite the scaffold modification.
Interestingly, during MD analysis, while the benzodihydrodioxine moiety exhibited no observable interactions within the HsDHODH enzyme active site, the 2-aminoquinazolinone core showed multiple interactions (FigureB). These observations support our hypothesis that the quinazolinone scaffold effectively mimics the essential binding features of quinones, as demonstrated for lapachol (FigureB, PDB: 9CCC). This structural mimicry, coupled with no benzodihydrodioxine-mediated interactions, guided the computational design of a second-generation quinazolinone series to extend binding interactions toward unexplored hydrophobic regions of the enzyme binding pocket. This rational new design focused on enhancing HsDHODH inhibitory potency through strategic optimization of key pharmacophoric elements while improving drug-like properties, particularly aqueous solubility, and preserving structural features critical for target engagement.
To accomplish this goal, we employed computational analysis of 10c’s binding mode and developed compounds using two distinct optimization strategies (Figure):
The first approach was based on structural insights from the potent naphthotriazoledione inhibitor (IC_50_ = 1.2 nM), cocrystallized with HsDHODH (PDB ID: 6LP6),? which exhibits interaction patterns analogous to our previously characterized quinone-based inhibitors. The naphthotriazoledione scaffold incorporates a biphenyl ring system that effectively occupies subsite 1, a nomenclature adopted by Baumgartner et al. in 2006? (Figure). Notably, this biphenyl motif is also present in Brequinar,? further validating the significance of this pharmacophoric element in establishing favorable hydrophobic interactions within this region of the binding pocket (Strategy 1 in Figure).
The second approach evaluated the importance of an aliphatic linker between the amine −NH and the phenyl substituent to understand whether the added flexibility could improve potency by enabling different interaction patterns (Strategy 2 in Figure). Moreover, increasing the sp^3^ fraction (Fsp^3^) of designed compounds could modulate druglike properties, including solubility.? Furthermore, the strategy also contemplates the ability of the series to occupy the hydrophobic subpocket of HsDHODH.
The new designed quinazolinones (10d–l) were obtained according to the same synthetic route, and the isolated yields varied from 28 to 75% (Table). Table summarizes the enzymatic activity, IC_50_ of the most potent compounds, cytotoxicity in Vero and SH-SY5Y cells, and water solubility. Compound 10c, the most potent inhibitor from our initial screening, demonstrates substantially lower potency compared to our second-generation compounds, validating our strategic scaffold optimization approach. The enzymatic inhibition data confirm the efficacy of our computational design strategy, with compound 10e emerging as the most potent inhibitor of this series, exhibiting 96% inhibition of HsDHODH and an IC_50_ of 0.59 ± 0.03 μM at 250 μM (Table). The 2D interaction analysis derived from a 50 ns MD simulation of compound 10e reveals critical binding interactions (Figure 3C). Root mean square deviation (RMSD) analysis demonstrates that 10e maintains a stable binding conformation throughout the simulation, with minimal positional fluctuations (<1.5 Å) (Figure S2C). In addition to preserving the key interactions with Arg^136^ and the water-mediated hydrogen bond with Thr^360^ observed in 10c, compound 10e establishes an additional hydrogen bond between the quinazolinone carbonyl group and Gln^47^, which contributes significantly to binding stability. Furthermore, a novel water-mediated hydrogen bond forms between Ala^55^ and the N–H from the substituted aniline, providing additional anchoring points within the binding pocket.
More significantly, the enhanced potency of the m-substituted 10e can be attributed to two key π–π stacking interactions formed between Phe^62^, Tyr^38^, and the terminal aromatic ring of the biphenyl substituent, interactions that are coherent with those observed in the cocrystal structure of the naphthotriazoledione inhibitor (PDB: 6LP6).? The structural features of Brequinar and the tricyclic derivative? further corroborate the critical importance of this biphenyl system in establishing optimal contacts with key amino acid residues. The computational prediction of diminished interactions with residues Ala^55^ and Tyr^38^ can rationalize the substantially reduced potency observed with the p-substituted analog.
To analyze the influence of aliphatic spacer groups between the −NH substitution of quinazolinone ring and the phenyl on inhibitory potency, we systematically varied the carbon chain length in a homologous series (10h–l). Comparison of compound 10b (p-fluor-aniline, lack of dose–response) with compound 10j (p-fluor-phenethyl-amine), which incorporates an ethylene linker, revealed the latter as a moderate inhibitor, with an IC_50_ of 29 ± 2 μM for 10j. Interestingly, these results did not align precisely with computational predictions which indicated that a larger spacer group would increase potency. Our experimental results suggest that an ethylene linker represents the optimal substitution pattern. Extension of this linker to three (10k) or four (10l) methylene units resulted in progressively diminished potency, despite MD analysis predicting a π-stacking interaction between compound 10l and Phe^62^ (Figure S2G,H).
Compounds incorporating the piperazine moiety (10f and 10g) exhibited favorable computational docking scores (Table S1) but demonstrated negligible inhibitory activity against HsDHODH. This discrepancy suggests that the absence of a −N-H hydrogen bond donor at the C-2 position of the quinazolinone scaffold is critical for activity. Furthermore, the nonaromatic heterocycle is more rigid that the linear ethylene linker. The loss of this key hydrogen bond donor eliminates essential interactions with active site residues, particularly Ala^55^ and Gln^47^. This structure–activity observation underscores the importance of maintaining specific hydrogen bonding capabilities within this region of the inhibitor scaffold for effective target engagement (FigureA).
In addition to the enhanced potency observed within our second-generation quinazolinone series, a notable advantage emerged in their improved cytotoxicity profiles compared to our naphthoquinone inhibitors (like Lapachol).? Although compounds 10d and 10e exhibited moderate cytotoxicity in Vero cell lines, they demonstrated substantially reduced toxicity toward human SH-SY5Y neuroblastoma cells, confirming that the quinazolinone scaffold provides optimized toxicological properties for HsDHODH inhibition (Table). However, regarding aqueous solubility, the quinazolinone series did not demonstrate significant improvement compared to the naphthoquinone precursors. Consequently, enhancement of water solubility remains a critical parameter requiring further optimization in subsequent medicinal chemistry efforts.
Following the identification of 10e as a potent inhibitor of HsDHODH, we evaluated its potential as an antiviral candidate by assessing its cytotoxicity and antiviral activity against SARS-CoV-2 and determining the selectivity index (SI). As shown in Table, compound 10e demonstrates potent anti-SARS-CoV-2 activity (EC_50_ = 0.15 ± 0.03 μM) in infected Calu-3 cells, with SI equal to 234, indicating a favorable safety profile. Notably, while 10e exhibited comparable HsDHODH inhibitory potency to our previously inhibitors,? it demonstrated a 10-fold enhancement in antiviral activity compared to lapachol (EC_50_ = 1.5 ± 0.1 μM)the most potent antiviral quinone in our studiescoupled with significantly reduced cytotoxicity.
FigureB and C illustrates the comparative inhibition of SARS-CoV-2 production (PFU/mL) by compounds 10e, Brequinar (EC_50_ = 0.8 ± 0.2 μM), and Remdesivir (EC_50_ = 0.03 ± 0.0015 μM), the latter two used as positive controls. The results highlight the exceptional antiviral potential of quinazolinone derivative 10e, demonstrating approximately 5-fold greater potency in suppressing viral replication compared to Brequinar under identical assay conditions and concentrations. This enhanced antiviral efficacy, combined with favorable selectivity indexes, positions this scaffold as promising candidates for further optimization as SARS-CoV-2 inhibitors targeting host pyrimidine biosynthesis.
In conclusion, this study demonstrates the successful design, synthesis, and anti-SARS-CoV-2 evaluation of novel quinazolinone derivatives as potent inhibitors HsDHODH. Employing bioisosteric replacement of the naphthoquinone scaffold, we developed quinazolinones that effectively mimic critical binding interactions within the ubiquinone binding site of the studied enzyme while addressing cytotoxicity and limitations of promiscuous properties inherent to quinone-based inhibitors. Through structure-based design and SAR analysis, we identified 10e as the most promising compound, exhibiting high HsDHODH inhibition (IC_50_ = 0.59 ± 0.03 μM) and antiviral activity against SARS-CoV-2 (EC_50_ = 0.15 ± 0.03 μM) with a favorable SI (234). MD simulations revealed key determinants of binding affinity, including conserved hydrogen bonding networks with Arg^136^, Thr^360^, and Gln^47^, as well as critical π–π stacking interactions with Tyr^38^ and Phe^62^ facilitated by the optimized biphenyl substituent. While our quinazolinone derivatives demonstrate significantly improved potency and reduced cytotoxicity compared to their naphthoquinone counterparts, aqueous solubility remains suboptimal and represents a primary focus for subsequent optimization efforts. Overall, these findings establish quinazolinones as a promising core for HsDHODH inhibition and advance the development of anti-SARS-CoV-2 candidates targeting host pyrimidine biosynthesis.
Safety Statement
No unexpected or unusually high safety hazards were encountered.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Boschi D.Pippione A. C.Sainas S.Lolli M. L.Dihydroorotate dehydrogenase inhibitors in anti-infective drug research Eur. J. Med. Chem.201918311168110.1016/j.ejmech.2019.11168131557612 · doi ↗ · pubmed ↗
- 2Reis R. A. G.Calil F. A.Feliciano P. R.Pinheiro M. P.Nonato M. C.The dihydroorotate dehydrogenases: Past and present Arch. Biochem. Biophys.201763217519110.1016/j.abb.2017.06.01928666740 · doi ↗ · pubmed ↗
- 3Lipowska J.Pyrimidine biosynthesis in pathogens - Structures and analysis of dihydroorotases from Yersinia pestis and Vibrio cholerae Int. J. Biol. Macromol.20191361176118710.1016/j.ijbiomac.2019.05.14931207330 PMC 6686667 · doi ↗ · pubmed ↗
- 4Loffler M.Jockel J.Schuster G.Dihydroorotat-ubiquinone oxidoreductase links mitochondria in the biosynthesis of pyrimidine nucleotides Mol. Cell. Biochem.199717412510.1023/A:10068591154509309676 · doi ↗ · pubmed ↗
- 5Cherwinski H. M.The immunosuppressant leflunomide inhibits lymphocyte proliferation by inhibiting pyrimidine biosynthesis Journal of Pharmacology and Experimental Therapeutics 199527246010.1016/S 0022-3565(25)24292-67529314 · doi ↗ · pubmed ↗
- 6Kaur H.Efficacy and safety of dihydroorotate dehydrogenase (DHODH) inhibitors “leflunomide” and “teriflunomide” in Covid-19: A narrative review Eur. J. Pharmacol.202190617423310.1016/j.ejphar.2021.17423334111397 PMC 8180448 · doi ↗ · pubmed ↗
- 7Miller A. E.An Updated Review of teriflunomide’s Use in Multiple Sclerosis Neurodegener Dis Manag 20211138740910.2217/nmt-2021-001434486382 · doi ↗ · pubmed ↗
- 8Coelho A. R.Oliveira P. J.Dihydroorotate dehydrogenase inhibitors in SARS-Co V-2 infection European Journal of Clinical Investigation 202050 e 1336610.1111/eci.1336632735689 PMC 7435507 · doi ↗ · pubmed ↗