Repositioning a Human Kinase Platelet-Derived Growth Factor Receptor Alpha Type II Inhibitor for Malaria and Inhibition of Hemozoin Formation
Mmakwena M. Mmonwa, Oluwatosin Audu, Keletso Maepa, Godwin A. Dziwornu, Preshen Govender, Liso Tshaka, James Burrows, Dale Taylor, Keabetswe Masike, Mathew Njoroge, Kathryn J. Wicht, Lauren B. Coulson, Kelly Chibale

TL;DR
A drug originally designed to inhibit a human kinase was found to also fight malaria by blocking hemozoin formation in parasites.
Contribution
The paper introduces a new application of a human kinase inhibitor for malaria treatment by targeting hemozoin formation.
Findings
Compound GSK190937 showed strong antiplasmodium activity with an IC50 of 0.22 μM.
Compounds 20, 23, and 29 improved metabolic stability and antiplasmodium activity.
The drug series inhibits hemozoin formation, killing late-stage malaria parasites.
Abstract
A type II Platelet-Derived Growth Factor Receptor Alpha (PDGFRA) human kinase inhibitor GSK190937, with antiplasmodium activity against asexual blood stage parasites (PfNF54 IC50 = 0.22 μM) was identified from the Kinase Chemogenomics Set, a collection of narrow-spectrum human kinase inhibitors. Medicinal chemistry progression of the hit focused on improving potency, selectivity, and ADME properties, leading to compounds 20, 23, and 29 with improved microsomal metabolic stability and asexual blood stage antiplasmodium activity. Mechanism of action studies showed that this series inhibits hemozoin formation, killing late-stage trophozoites.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1 2
2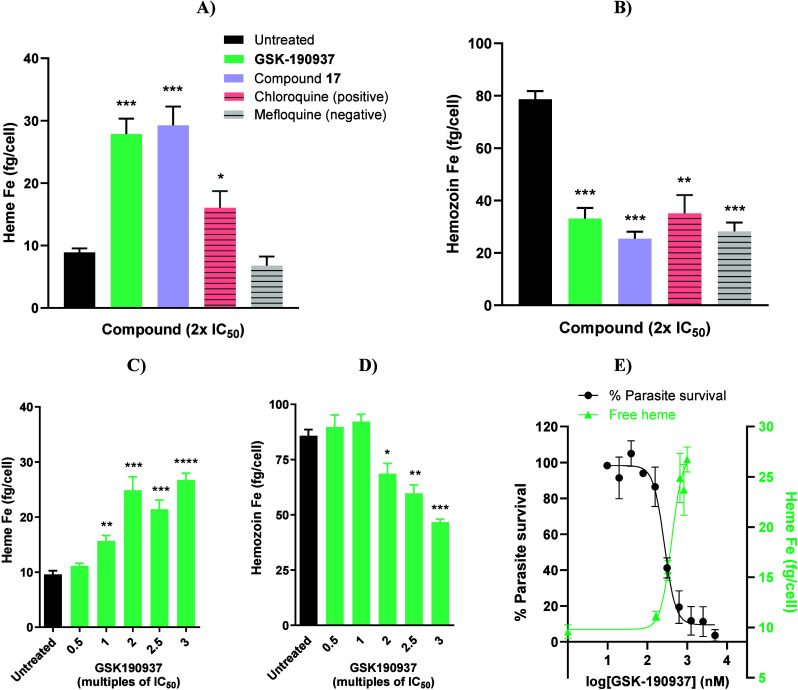 2
2- —National Institute of Allergy and Infectious Diseases10.13039/100000060
- —University of Capetown10.13039/501100001338
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMalaria Research and Control · Platelet Disorders and Treatments · Liver physiology and pathology
Although preventable and curable, malaria, a disease caused by Plasmodium parasites, remains a challenging problem in many parts of the world, particularly in sub-Saharan Africa. According to the World Health Organization (WHO), 263 million cases of malaria and 597 000 deaths were reported globally in 2023.? This is an increase of 14 million cases from 249 million cases in 2022. Of these, 94% of all malaria cases and 95% of deaths in 2023 occurred in the African region. Children under five years of age in the African region accounted for an estimated 76% of all malaria deaths in 2023. The currently approved and recommended antimalarial drugs, particularly artemisinin-based combination therapy (ACT) regimens, are fast becoming less efficacious against the Plasmodium parasite due to the emergence of resistance-conferring mutations in the parasite, resulting in a delayed death phenotype. ?,? While chemoprophylaxis efforts through the administration of the RTS,S/AS01 malaria vaccine to children under the age of 5 in endemic areas is a major advance toward control and eradication of the disease,? this vaccine only provides modest (∼30%) protection against severe malaria. New affordable drugs with a lower propensity for resistance are needed to overcome the rapid rise in parasite resistance toward first line treatments for malaria.
Human kinase inhibitors with potent antiplasmodium activity have been identified as promising starting points for malaria drug discovery and have led to the identification of several promising Plasmodium kinase targets. ?−? ?
GSK190937 (Figure), a human kinase Platelet-Derived Growth Factor Receptor Alpha (PDGFRA) type II inhibitor, was identified from a phenotypic screen of the Kinase Chemogenomics Set (KCGS)? against asexual blood stage Plasmodium falciparum (Pf) NF54 parasites. GSK190937 displayed an IC_50_ of 0.22 μM against PfNF54 parasites and no significant cross resistance with the multidrug resistant PfK1 strain, albeit with low solubility and low microsomal metabolic stability.
Here we describe the medicinal chemistry exploration of GSK190937 with the goal of optimizing potency, selectivity, and ADME properties. This exploration focused on three regions of the molecule, the left-hand side (LHS), the right-hand side (RHS) and the core (Figure). We demonstrate that this series inhibits β-hematin formation in a cell-free assay with representative compounds inhibiting hemozoin formation within the parasite, leading to a dose-dependent accumulation of toxic heme.
Synthesis
We first investigated the structure–activity relationship (SAR) on the LHS of the molecule. Hydrolysis of ethyl 5-bromonicotinate with aqueous NaOH in methanol afforded 5-bromonicotinic acid 1, which was subjected to amide coupling with 2-(2,4-dichlorophenyl)ethan-1-amine to deliver 5-bromonicotinamide intermediate 2. The vinyl group was introduced via a palladium-catalyzed Stille cross-coupling reaction of the latter with tributyl(vinyl)stannane in toluene to afford 5-vinylnicotinamide 3. A palladium-catalyzed Heck cross-coupling reaction of intermediate 3 with various aryl bromides afforded target compounds 4–13 (Scheme). In addition, compounds 22 and 23, as well as 26–29 were prepared following Scheme using relevant bromo aryl-esters in place of the ethyl 5-bromonicotinate (see Supplementary Information).
Synthesis of Compounds 4–13
Compounds for RHS SAR exploration were synthesized, as depicted in Scheme. A Stille cross-coupling reaction of ethyl 5-bromonicotinate with butyl(vinyl)stannane in toluene afforded 5-vinylnicotinamide 14, which was subjected to a Heck cross-coupling reaction with 3-bromo-5-methylpyridin-2-amine to generate intermediate 15. This intermediate was hydrolyzed with aqueous NaOH in methanol to deliver the carboxylic derivative 16, which was subjected to amide coupling with various amines to afford target compounds 17–21 and 24.
Synthesis of Compounds 17–21, 24
Antiplasmodium Activity and Cytotoxicity
The resynthesized hit GSK190937 and derivatives were evaluated for in vitro asexual blood stage (ABS) activity against drug sensitive PfNF54 parasites and cytotoxicity against Chinese hamster ovarian (CHO) cells. Compounds with PfNF54 IC_50_ ≤ 1 μM were further tested against multidrug-resistant PfK1 and/or PfDd2 strains (Tables and ?).
1: In Vitro Asexual Blood Stage Activity Against P. falciparum NF54, Dd2 and K1 Strains, Chinese Hamster Ovarian (CHO) Cytotoxicity, Kinetic Aqueous Solubility, and Inhibition of β-Hematin Formation (βH) (LHS SAR)
2: In Vitro Asexual Blood Stage P. falciparum NF54/Dd2/K1 Parasite Activity, Chinese Hamster Ovarian (CHO) Cytotoxicity, Kinetic Aqueous Solubility, and Inhibition of β-Hematin Formation (βH) IC50 Values (Core Change SAR)
LHS SAR
Resynthesized GSK190937 displayed ABS activity comparable to that of the original hit from the KCGS library (PfNF54 IC_50_ = 0.22 μM) with a selectivity index (SI, CHO IC_50_/PfNF54 IC_50_) of 113 relative to CHO cells. No significant cross-resistance was observed against the PfK1 and PfDd2 strains (IC_50_ = 0.59 and 0.25 μM, respectively). Removal of pyridine nitrogen or amino substituent in compounds 5 and 6, respectively, led to significantly reduced PfNF54 activity (IC_50_ = 2.60 and 1.18 μM, respectively). When the pyridine methyl substituent was removed in compound 7, the PfNF54 activity was retained (IC_50_ = 0.34 μM). While compound 7 showed marginal cross-resistance against the PfK1 strain (IC_50_ = 0.96 μM), significant cross-resistance was observed against the PfDd2 strain (IC_50_ = 5.47 μM). Over 2-fold loss in PfNF54 activity (IC_50_ = 0.51 and 0.48 μM) was observed when the amino substituent was moved to position 5 or 6 of the pyridine ring as in compounds 8 and 9, respectively. These compounds showed selectivity indices comparable to those of the parent compound (SI = 98 and 104). Cross-resistance was also observed for compounds 8 and 9 with the former displaying an IC_50_ of 2.21 μM against PfDd2, while the latter showed significant cross resistance against PfK1 strain (IC_50_ > 6 μM). Interestingly, PfNF54 activity (IC_50_ = 0.14 μM) was retained when the amino substituent was moved to position 4 of the pyridine ring in compound 12. Reduced PfNF54 activity (IC_50_ = 2.63 μM) was observed when both the methyl and amino substituents were omitted from pyridine in compound 10. On the other hand, there was an ∼20-fold loss in PfNF54 activity (IC_50_ = 4.28 μM) for compound 11 where the 2-amino-5-methylpyridin-3-yl was replaced with a phenyl group. Lastly, replacing the 2-amino-5-methylpyridin-3-yl with 4-aminopyrimidin-5-yl in compound 13 resulted in a PfNF54 IC_50_ of 0.48 μM and a SI of 104.
RHS SAR
Comparable PfNF54 activity to the hit compound was observed when either of the chlorine atoms or both were removed in compounds 17, 18, and 19 [IC_50_ (SI) = 0.27 μM (53), 0.26 μM (100) and 0.32 μM (56), respectively] with activity of <1 μM against both PfK1 and PfDd2 strains. Equipotent activity was also observed when the carbon chain linker was reduced or removed, as in compounds 20 and 22, respectively (PfNF54/K1/Dd2 IC_50_ = 0.16/0.25/0.19 and 0.20/ND/0.35 μM, respectively). Compound 20 showed improved selectivity (SI = 304) compared to 4 (SI = 113). Shifting the chlorine atoms in compound 20 to positions 3 and 5 as in compound 21 led to comparable PfNF54 and PfK1 activities (PfNF54/K1 IC_50_ = 0.09/0.46 μM) to the hit. Compound 21 also showed high selectivity relative to that of CHO cells (SI = 400). Significant loss of PfNF54 activity was observed when the dichlorophenyl group was replaced with basic groups (4-pyridine and saturated 4-piperidine) as in compounds 24 and 25. Compound 24 displayed weak PfNF54 activity (IC_50_ = 2.06 μM), while there was a complete loss of activity for 25 at the highest concentration tested (IC_50_ > 6 μM).
Core Change and Amide Flip SAR
Scaffold hopping led to reduced PfNF54 activity as well as cross resistance against PfK1 and/or PfDd2 strains relative to resynthesized 4 (Table). Moving the amide and vinyl substituents to positions 2 and 6 of the pyridine core, respectively, as in compound 26, resulted in a 4-fold loss of PfNF54 activity as well as cross resistance against the PfDd2 strain (PfNF54/Dd2 IC_50_ = 0.78/3.19 μM). Similarly, compound 27 where the pyridine core was replaced with a phenyl group gave PfNF54 IC_50_ = 0.62 μM and showed some cross resistance against the PfK1 strain (IC_50_ = 1.65 μM). Cross-resistance was also observed when a thiophene core was used in place of the pyridine core in compound 28 ( PfNF54/K1 IC_50_ = 0.59 μM/2.36 μM). Flipping the amide group to produce compound 29 resulted in retention of PfNF54 activity (IC_50_ = 0.20 μM) albeit this compound was 4-fold less active against PfDd2 (IC_50_ = 0.75 μM).
Mode of Action Studies
As already mentioned, GSK190937 (4) is a PDGFRA type II inhibitor.? PDGFRA is part of the receptor tyrosine kinase family, for which there are no Plasmodium orthologs.? However, antiplasmodium compounds containing a nicotinamide group have been reported to inhibit hemozoin formation in the parasite via π–π interactions with heme/hemozoin.? Notably, the synthesized compounds 4–26 herein contain a nicotinamide core. In addition, the pyridine core in compounds 4–26 has the potential to contribute to π-π interactions with heme, while the amide H could contribute to H-bonding, disrupting hemozoin crystal packing.? Based on these hypotheses, selected compounds were tested for their ability to inhibit β-hematin (synthetic hemozoin) formation in a cell free assay (Tables and ?). All tested compounds, except for 25, showed β-hematin inhibition activity within 5-fold of the control, chloroquine, with IC_50_ < 100 μM. Compounds 21, 22, 23, and 29 showed the most potent inhibition of cell free β-hematin formation, with IC_50_ values at least 2-fold less than chloroquine (IC_50_ < 10 μM).
In the cellular context, antiplasmodium activity mediated by disruption of the hemoglobin degradation pathway is largely dependent on the extent to which compounds accumulate in the acidic digestive vacuole via heme binding and pH trapping. ?,? To confirm that inhibition of hemozoin is contributing to antiplasmodium activity in the cellular context, intracellular heme fractionation assays were carried out. ?,?
In the cell fractionation assay, a compound that inhibits the formation of intracellular hemozoin in the digestive vacuole of the parasite causes an increase in the absolute amount (fg/cell) of free heme and a decrease in the amount of hemozoin relative to the untreated control. Single-point cell fractionation was performed with compounds 4(GSK190937) and 17 at 2× their IC_50_ values using chloroquine and mefloquine as positive and negative controls, respectively (FigureA). Both tested compounds showed significantly increased levels of absolute free heme at 2 × IC_50_, even more so than the classical hemozoin formation inhibitor chloroquine, relative to the untreated control parasites, with an equally significant decrease in the hemozoin levels (FigureB). These high levels may be indicative of the compounds killing the parasites later in the trophozoite stage compared to chloroquine, allowing more time for the buildup of heme. Furthermore, compound 4 was tested using this assay at increasing concentrations from 0.5× to 3× IC_50_, resulting in a dose-dependent increase in free heme and decrease in hemozoin (FigureC and ?D). Additionally, an inverse correlation was observed between parasite survival and the amount of free heme, where the curves intersected close to the IC_50_ value of compound 4. This strongly suggests that elevated levels of cytotoxic heme cause inhibition of parasite growth and, therefore, that inhibition of hemozoin formation is the primary mode of action for this series.
*Cell fractionation results showing A) the increase in free heme levels (fg of heme-derived Fe per cell) and B) hemozoin levels (fg of hemozoin-derived Fe per cell) at 2× IC50 (single point) for GSK190937 (4) and compound 17 relative to controls, chloroquine and mefloquine. Bars represent mean ± SEM from two independent experiments where n = 4. C) The dose–response increase in free heme or D) dose–response decrease in hemozoin for GSK190937 (4) relative to the untreated control (black). Bars represent mean ± SEM from two independent experiments where n = 4. Significance was determined using unpaired t-tests where comparisons are shown relative to the untreated control; *p < 0.05; **p < 0.01; ***p < 0.001; ***p < 0.0001. E) The inverse correlation between parasite survival and levels of free heme for GSK190937 (4). The curves intersect close to the IC50 value, suggesting that increased heme levels are driving parasite cell death. Values represent mean ± SEM from n = 4.
Aqueous Solubility and Microsomal Metabolic Stability
A good oral drug candidate should meet acceptable levels of solubility and microsomal stability, among other parameters.? With this in mind, emphasis was also put on improving these parameters for the prepared derivatives (Tables and ?). Using the kinetic solubility assay at pH 6.5, improved solubility relative to the hit compound (solubility <5 μM) was observed for compounds 8, 12, 13, 18, 19, 24, and 25. Shifting the LHS amino substituent to position 6 or 4 of the pyridine in compounds 8 and 12 improved the solubility to 50 and 75 μM, respectively, while the amino pyrimidinyl group in compound 13 had a solubility of 55 μM. On the other hand, the removal of 2-chloro from the RHS phenyl group resulted in a small improvement in solubility to 15 μM, while removing both chlorine atoms in compound 19 resulted in a solubility of 40 μM. Replacing the dichlorophenyl group on the RHS with hydrophilic groups, pyridine and piperidine groups, in compounds 24 and 25 significantly improved the solubility to 105 and 190 μM, respectively, albeit reduced PfNF54 and cell-free β-hematin formation inhibition activity.
Lastly, due to low microsomal metabolic stability of the hit compound 4, we tested selected compounds for metabolic stability using mouse, rat and human liver microsomes (Table). Despite most of these compounds showing low microsomal stability, compounds 20 and 23 with a reduced or removed RHS alkyl chain linker, respectively, as well as compound 29 with a flipped amide group showed improved microsomal stability across all three species.
3: In Vitro Microsomal Stability for Selected Compounds
GSK190937 (4), identified from the Kinase Chemogenomics Set, showed antiplasmodium activity against both sensitive PfNF54 and multidrug resistant PfK1 strains. The medicinal chemistry efforts to address the low aqueous solubility and microsomal metabolic stability led to identification of compounds 20, 23, and 29 with equipotent antiplasmodium activities and improved microsomal stability. Reduced or lost antiplasmodium activity was observed with solubility improvement. Mode of action studies revealed that this series inhibits hemozoin formation with significantly elevated levels of free heme at concentrations ≥ IC_50_. We conducted a formal hit assessment to evaluate if the series has a tractable SAR and early ADME studies toward identifying issues to be addressed in a future lead optimization campaign, including addressing solubility, which remains an issue for the series. Work to further improve the antiplasmodium activity and physicochemical properties toward a pharmacology proof-of-concept in a malaria mouse infection model is ongoing in our laboratories.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1WHO . World Malaria Report; WHO, 2024.
- 2Blasco B.Leroy D.Fidock D. A.Antimalarial drug resistance: linking Plasmodium falciparum parasite biology to the clinic Nat. Med.20172391792810.1038/nm.438128777791 PMC 5747363 · doi ↗ · pubmed ↗
- 3Madhav H.Hoda N.An insight into the recent development of the clinical candidates for the treatment of malaria and their target proteins Eur. J. Med. Chem.202121011295511299410.1016/j.ejmech.2020.11295533131885 · doi ↗ · pubmed ↗
- 4Laurens M. B.RTS,S/AS 01 vaccine (Mosquirix): an overview Human Vaccines & Immunotherapeutics 20201648048910.1080/21645515.2019.166941531545128 PMC 7227679 · doi ↗ · pubmed ↗
- 5Njoroge M.Njuguna N. M.Mutai P.Ongarora D. S. B.Smith P. W.Chibale K.Recent approaches to chemical discovery and development against malaria and the neglected tropical diseases Human African Trypanosomiasis and schistosomiasis Chem. Rev.2014114111381116310.1021/cr 500098 f 25014712 · doi ↗ · pubmed ↗
- 6Wang L.Bohmer M. J.Wang J.Nardella F.Calla J.De Souza M. L.Schindler K. A.Montejo L.Mittal N.Rocamora F.Treat M.Charlton J. T.Tumwebaze P. K.Rosenthal P. J.Cooper R. A.Chakrabarti R.Winzeler E. A.Chakrabarti D.Gray N. S.Discovery of potent antimalarial type II kinase inhibitors with selectivity over human kinases J. Med. Chem.2024671460148010.1021/acs.jmedchem.3c 0204638214254 PMC 10950204 · doi ↗ · pubmed ↗
- 7Arendse L. B.Murithi J. M.Qahash T.Pasaje C. F. A.Godoy L. C.Dey S.Gibhard L.Ghidelli-Disse S.Drewes G.Bantscheff M.Lafuente-Monasterio M. J.Fienberg S.Wambua L.Gachuhi S.Coertzen D.van der Watt M.Reader J.Aswat A. S.Erlank E.Venter N.Mittal N.Luth M. R.Ottilie S.Winzeler E. A.Koekemoer L. L.Birkholtz L.-M.Niles J. C.Llinás M.Fidock D. A.Chibale K.The anticancer human m TOR inhibitor sapanisertib potently inhibits multiple Plasmodium kinases and life cycle stages Sci. Transl. Med.202214 eabo 721910.1126/scitranslmed.abo 721936260 · doi ↗ · pubmed ↗
- 8Wells C. I.Al-Ali H.Andrews D. M.Asquith C. R. M.Axtman A. D.Dikic I.Ebner D.Ettmayer P.Fischer C.Frederiksen M.Futrell R.Gray N. S.Hatch S. B.Knapp S.Lücking U.Michaelides M.Mills C. E.Müller S.Owen D.Picado A.Saikatendu K. S.Schröder M.Stolz A.Tellechea M.Turunen B. J.Vilar S.Wang J.Zuercher W. J.Willson T. M.Drewry D. H.The kinase chemogenomic set (KCGS): An open science resource for kinase vulnerability identification Int. J. Mol. Sci.20212256658310.3390/ijms 2202056633429995 PMC 7826789 · doi ↗ · pubmed ↗