Shiga Toxin Induces Apoptosis via ROS–Caspase Activation in Human Cerebral Endothelial Cell Line hCMEC/D3 and Astrocyte Co-Culture
Mirim Kim, Kyung-Soo Lee, Jun Young Park, Chang-Ung Kim, Yu-Jin Jeong, Moo-Seung Lee

TL;DR
Shiga toxin from E. coli causes brain barrier damage by triggering cell death and inflammation, leading to neurological issues in HUS.
Contribution
This is the first study using hCMEC/D3 cells to model Shiga toxin-induced blood-brain barrier disruption.
Findings
Shiga toxins Stx1a and Stx2a trigger apoptosis and inflammation in brain endothelial cells.
Pharmacologic caspase inhibition prevents toxin-induced barrier damage and cell death.
Reactive oxygen species (ROS) and caspase pathways are central to toxin-induced endothelial injury.
Abstract
Hemolytic uremic syndrome (HUS), a fatal complication of Shiga toxin-producing Escherichia coli (STEC) infection, is classically characterized by acute renal failure, but frequently accompanied by central nervous system (CNS) dysfunction. Because the CNS is normally protected by the blood-brain barrier (BBB), the toxin-mediated BBB injury is considered to be a major cause of neurologic sequelae in STEC infection. Here, we delineate how Shiga toxin type 1a (Stx1a) and Shiga toxin type 2a (Stx2a) compromise BBB-like endothelial barrier integrity with the human brain microvascular endothelial cell line hCMEC/D3 as an in vitro model, complemented by an endothelial–astroglial co-culture system. Stx1a/Stx2a exposure induced the MAPK pathway and ER stress, triggering caspase-mediated apoptosis and pro-inflammatory cytokines expression. Coincidentally, permeability across tight junctions was…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
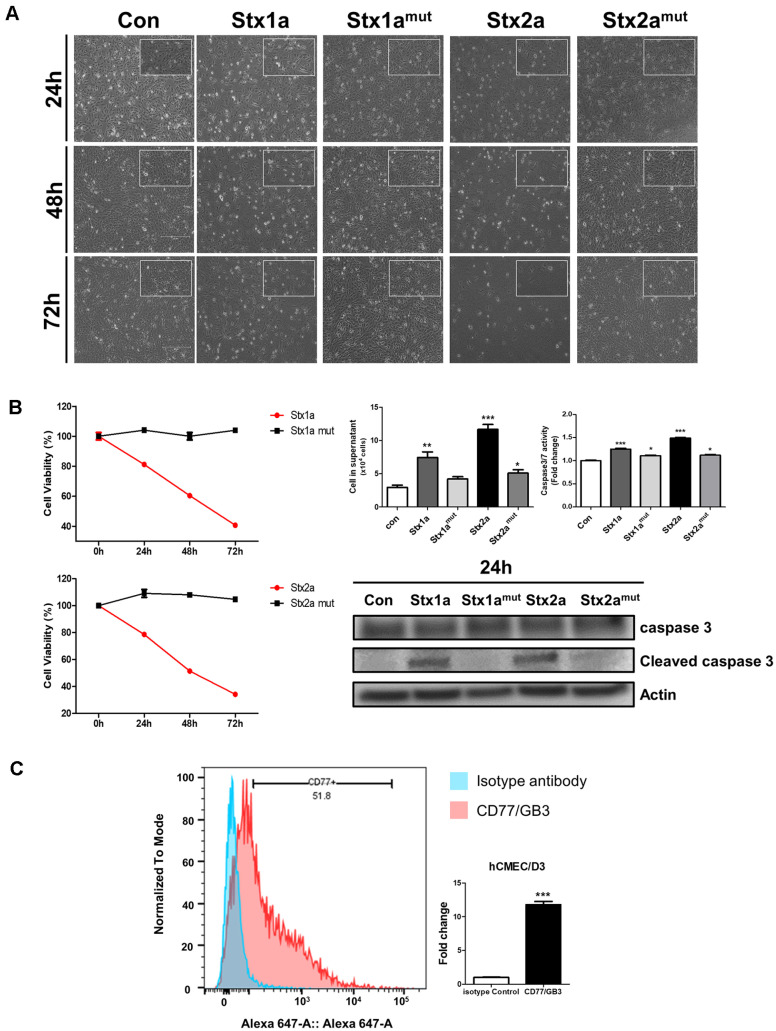 Figure 1
Figure 1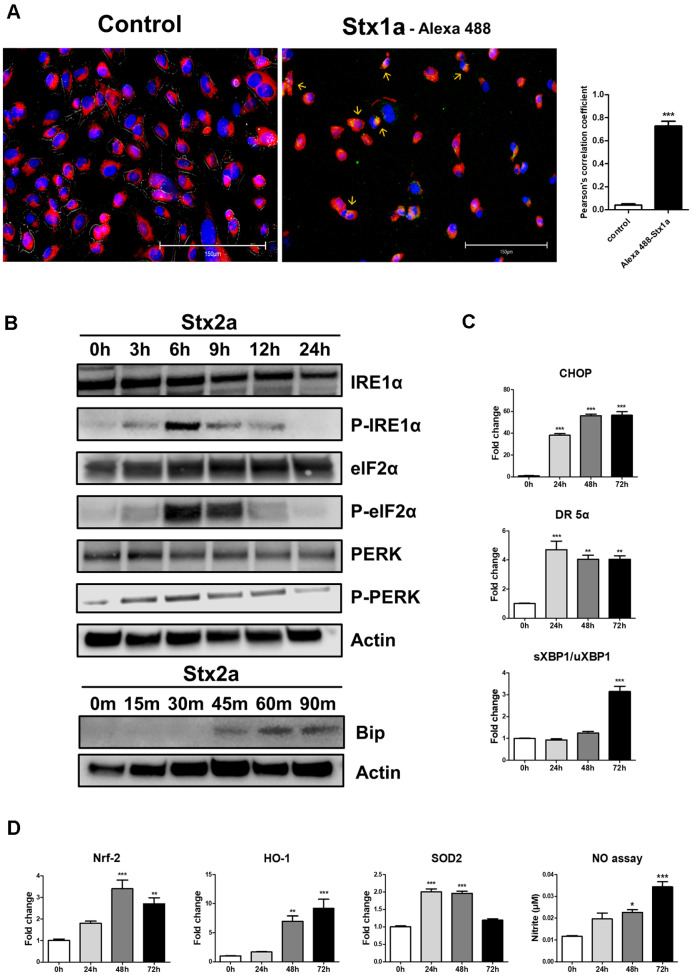 Figure 2
Figure 2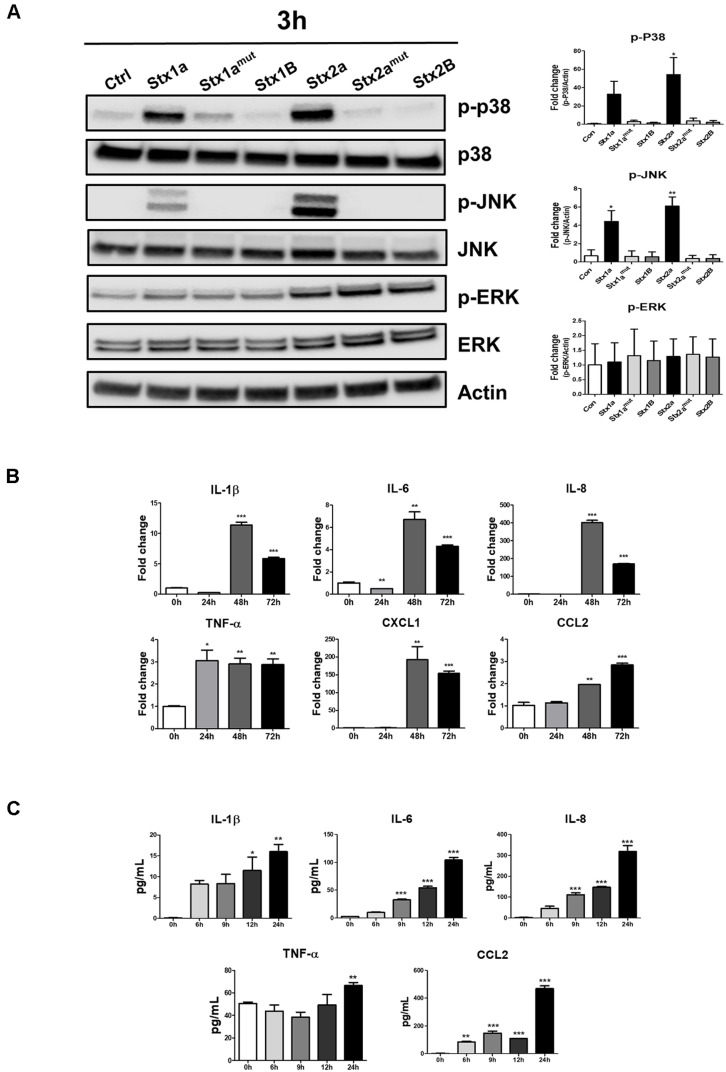 Figure 3
Figure 3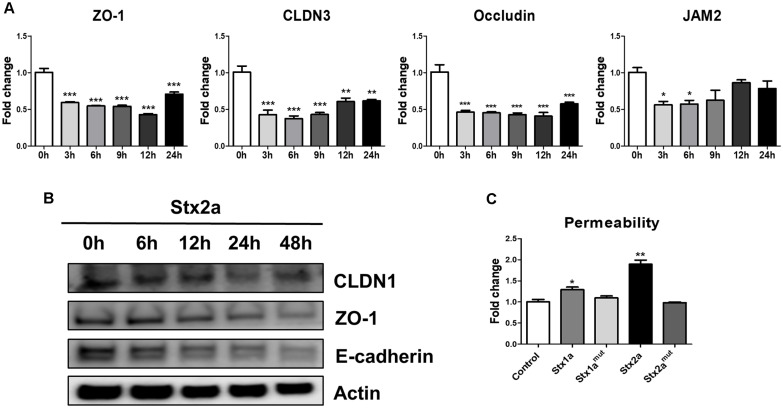 Figure 4
Figure 4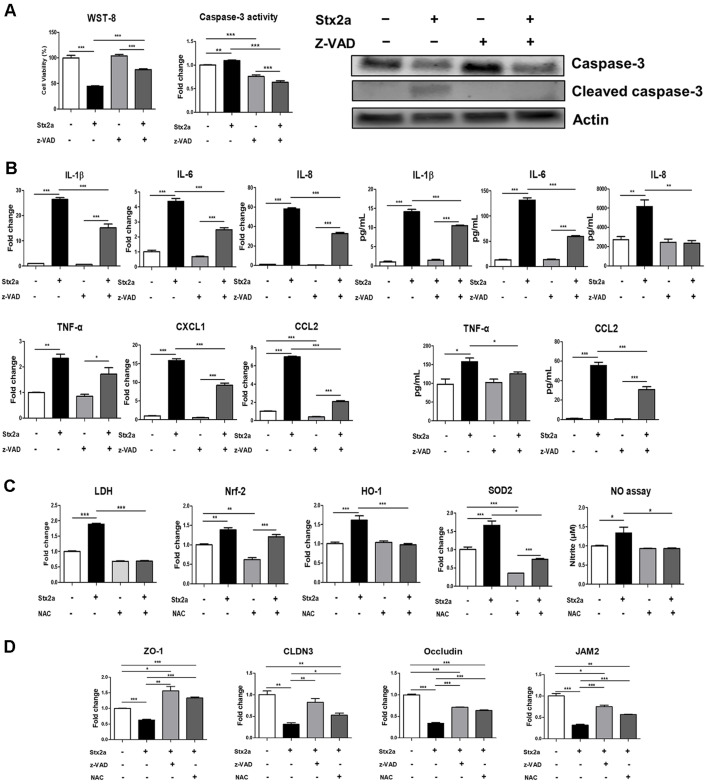 Figure 5
Figure 5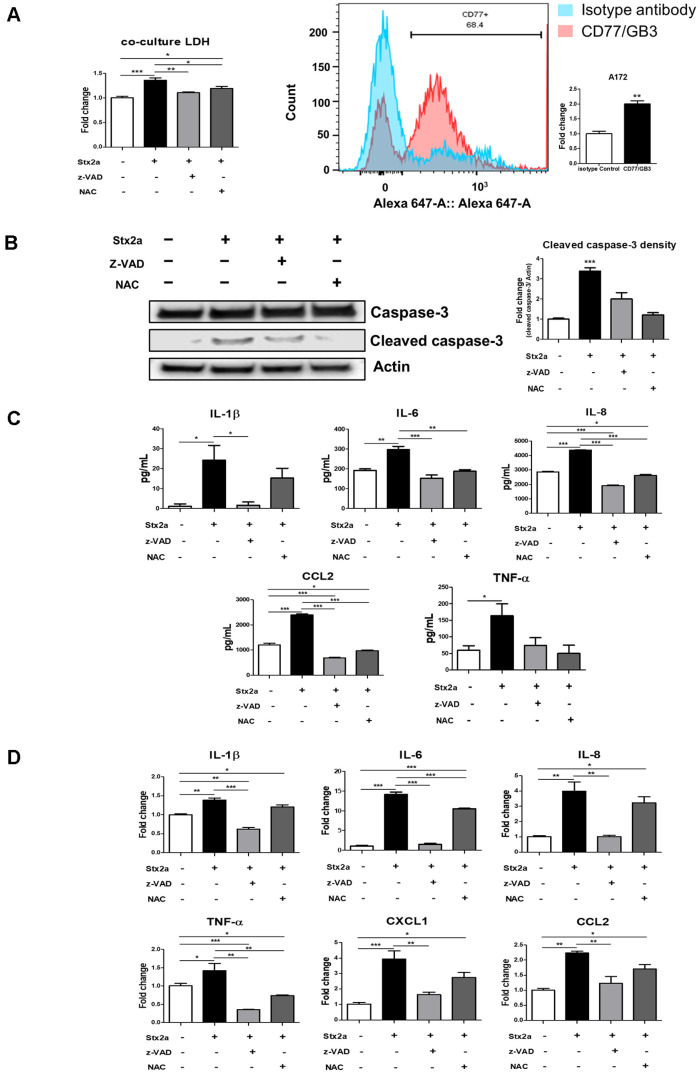 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Barrier Structure and Function Studies · Endoplasmic Reticulum Stress and Disease
Introduction
Hemolytic uremic syndrome (HUS) is a fatal, toxin-mediated microangiopathy predominantly manifested as an acute renal failure that is disproportionately prevalent in children and the elderly, typically after the ingestion of contaminated meat, water, or unpasteurized milk [1, 2]. The principal perpetrators responsible for HUS are Shiga toxins (Stxs), AB5 protein exotoxins produced by Shiga toxin–producing Escherichia coli (STEC) and Shigella dysenteriae serotype 1. Globally, foodborne STEC infections have a substantial public health impact, with the World Health Organization estimating over 1 million illnesses and roughly 13,000 disability-adjusted life years in 2020 [3]. Stxs comprise multiple serological variants classified under two main families (Stx1 and Stx2), with clinically significant subtypes such as Stx1a and Stx2a [4]. Structurally, Stxs are assembled as an AB5 holotoxin: a single A subunit noncovalently associated with five identical B subunits forming a pentameric ring [5]. The A subunit possesses N-glycosidase activity that selectively depurinates a single adenine residue (A4324) within 28S rRNA, thereby halting protein synthesis and activating cellular apoptosis [6]. The B pentamer binds the glycosphingolipid globotriaosylceramide (Gb3) present on host-cell membranes, mediating endocytosis and retrograde trafficking to the Golgi and endoplasmic reticulum (ER) while evading lysosomal degradation, ultimately leading to cytotoxicity [7].
Although renal injury is the defining clinical feature of HUS, central nervous system (CNS) involvement including seizures, stroke, encephalopathy/coma, visual loss, and other neurologic symptoms occur in a substantial proportion of patients and is associated with adverse outcomes [8?-10]. In a 33-year pediatric cohort, neurological manifestations ranged from seizures (71%) and altered consciousness (85%) to focal weakness/paralysis (40%) [8]. The CNS is typically shielded by the blood–brain barrier (BBB), a neurovascular unit composed of brain microvascular endothelial cells, pericytes, basement membrane, and astrocytic endfeet, that tightly regulates paracellular and transcellular transport of solutes and cells [11]. The selectivity of BBB is regulated mostly by the junctional complex between endothelial cells—tight junctions (TJs), adherent junctions, and gap junctions—with TJs (e.g., claudins, occludin, and ZO proteins) forming a tightly selective diffusion barrier that maintains endothelial polarity and facilitates intracellular signaling [12, 13]. This stringent barrier is also the principal reason that >98% of small-molecule drugs and nearly all biologics are ineffective in accessing the brain parenchyma [14]. Despite this, clinical observations indicate that Stxs are capable of permeating BBB barriers and causing CNS pathology. Previous studies have shown that Stxs can damage astrocytes and other neural components, yet the endothelial-centric, BBB-resolving processes, injury biomarkers, and paracrine propagation of damage remain incompletely defined [15]. In an effort to fill this gap, we employed the human brain microvascular endothelial cell line hCMEC/D3 as a tractable in-vitro BBB endothelium and questioned cellular and molecular responses to Stx1a and Stx2a. hCMEC/D3 cells express tight junction proteins as well as multiple transporters and efflux pumps and form a low-permeability monolayer, making them a suitable in vitro model for evaluating changes in blood–brain barrier integrity and permeability [16, 17]. We determined that exposure to Stx activates stress-activated MAPK cascades and ER stress, with resultant caspase-dependent apoptosis and a pro-inflammatory transcriptional profile. Coinciding with the disruption of endothelial TJs, with loss of junctional proteins and increased paracellular permeability, such events provide a direct mechanistic link between toxin signaling and failure of the barrier. Functionally, inhibition of caspase activity attenuated cytotoxicity and maintained expression of TJ proteins, implicating the apoptotic pathway as a critical driver of BBB dysfunction. Recognizing that barrier physiology is an emergent property of the neurovascular unit, we extended our analysis to a transwell co-culture of hCMEC/D3 endothelial cells with human astrocytes (A172), better approximating the BBB microenvironment [18, 19]. Notably, astrocytes displayed caspase activation and cytokine induction independent of direct toxin exposure, indicating paracrine endothelial–astroglial signaling as a conduit of injury propagation across the BBB interface. Overall, these studies support a model in which Stx1a/Stx2a compromise BBB integrity via ROS accumulation, MAPK/ER stress signaling, and caspase-executed apoptosis, crosstalk-signaling between endothelial cells and astrocytes may further augment neurovascular injury. By discerning these endothelial-centric mechanisms and their downstream consequences, our work provides mechanistic insight into Stx-mediated BBB dysfunction and a rationale for therapeutic interventions that target oxidative stress, ER stress responses, or apoptotic execution to mitigate CNS complications in HUS.
Materials and Methods
Toxins
Stx2a purified from Escherichia coli was purchased from List Biological Laboratories, Inc. (USA). Stx1-B subunit was purchased from Sigma (Merck, Germany). Stx2B was purchased from Creative Diagnostics (USA). Stx1a and Stx2a mutant containing the triple mutations Y77S/E167Q/R170L in the enzymatic active site of the A subunit were a generous gift from Professor Vernon L. Tesh, Texas A&M University, USA.Stx1a was purified in the laboratory of Professor Vernon L. Tesh from recombinant Stx1a-expressing E. coli DH5α (pCKS112) using sequential ion exchange and immunoaffinity chromatography. Purity was confirmed by SDS-PAGE, silver staining, and Western blot analysis. Endotoxin contaminant levels were reduced to <0.1 ng/ml using an ActiClean Etox column (Sterogene Bioseparations, USA). Residual endotoxin contamination was assessed using the Limulus Amoebocyte lysate assay (Associates of Cape Cod, USA). A purified Stx1a mutant containing double mutations (E167Q and R170L) in the A subunit was a generous gift from Dr. Shinji Yamasaki, Osaka Prefecture University, Japan.
Cell Culture
The human brain endothelial cell line hCMEC/D3 (Merck) was cultured in Endothelial Cell Growth Medium (PromoCell, Germany) supplemented with 1% Antibiotic-Antimycotic (Thermo Fisher Scientific, USA), and human astrocyte cell line A172 was cultured in RPMI 1640 medium (Corning, Thermo Fisher Scientific) supplemented with 10% FBS and 1% Antibiotic-Antimycotic (Thermo Fisher Scientific) at 37°C in a humidified incubator containing 5% CO_2_. hCMEC/D3 and A172 cells were seeded at 5.0 × 10^5^ cells/well into 6-well plates, washed once with sterile Dulbecco's phosphate-buffered saline (DPBS) (Sigma-Aldrich, USA), and treated with 10 ng/ml Stx2a in Endothelial Cell Growth Medium or RPMI containing 0.5% FBS without supplements for the indicated time periods.
For the co-culture of hCMEC/D3 and A172 cells, hCMEC/D3 cells were seeded at 3.0 × 10^5^ cells/well in insert of a trans-well (Corning, Thermo Fisher Scientific, USA), and A172 cells were seeded at 5.0 × 10^5^ cells/well in the lower well. After washing once with sterile Dulbecco’s phosphate-buffered saline (DPBS) (Sigma-Aldrich), cells were treated with 10 ng/ml Stx2a for various times in Endothelial Cell Growth Medium and RPMI containing 0.5% FBS and without supplements in all experiments.
Cytotoxicity Assay
Cell supernatants were used to determine cytotoxicity by measuring lactate dehydrogenase (LDH) release. Experiments were performed using the Pierce LDH Cytotoxicity Assay Kit according to the manufacturer's instructions. Absorbance was measured at 490 nm and 680 nm using a SpectraMAX 190 Microplate Reader (Molecular Devices, USA). The background concentration was removed by subtracting the 680 nm absorbance of each sample from the 490 nm absorbance. Cell death was assessed using the Muse Annexin V & Dead Cell Kit (Cytek Biosciences, USA) according to the manufacturer's protocol. The WST-8 dye-based assay was performed using the WST-8 Cell Counting Kit (QM1000) (Biomax, Republic of Korea) according to the manufacturer's protocol. Briefly, one-tenth of the cultured hCMEC/D3 cell suspension treated with Stx2 was incubated with WST-8 reagent at RT for an appropriate amount of time. The incubated culture medium was then transferred to a 96-well plate in triplicate, and the absorbance was measured at 450 nm using a SpectraMAX 190 microplate reader (Molecular Devices, USA).
Flow Cytometry Analysis to Confirm Gb3 (CD77) Expression
hCMEC/D3 cells were seeded in 6-well plates at 5.0 × 10^5^ cells/well and cultured in Endothelial Cell Growth Medium (PromoCell) containing 1% Antibiotic-Antimycotic (Thermo Fisher Scientific). Cells were maintained at 37°C in a humidified incubator containing 5% CO_2_. After 24 h, hCMEC/D3 cells were washed three times with cold PBS, hCMEC/D3 cells were detached with trypsin-EDTA (Gibco, Thermo Fisher Scientific) and collected by centrifugation at 780 × g for 5 min. Cells were then stained with Alexa Fluor 647-conjugated anti-human Gb3/CD77 monoclonal antibody (BD Biosciences, USA) or Alexa Fluor 647-conjugated mouse IgM (isotype control) for 30 min at 4°C in the dark and analyzed by flow cytometry using a BD FACS Canto ІІ cell analyzer (BD Bioscience) [10].
Caspase-3/7 Activity Analysis
hCMEC/D3 cells were seeded in 6-well plates at 5.0 × 10^5^ cells/well and cultured in Endothelial Cell Growth Medium (PromoCell) containing 1% Antibiotic-Antimycotic (Thermo Fisher Scientific). Cells were maintained at 37°C in a humidified incubator containing 5% CO_2_. Cells were pretreated with z-VAD (Selleckchem, USA) for 1h before exposure to Stx2a (10 ng/ml). After 24 h, the culture medium was removed, and 100 μl of the supernatant was returned to the wells. Then, the cells were incubated with 100 μl of Caspase-Glo 3/7 reagent (Promega, USA) containing substrate for 1–3 h. Next, the reaction mixture was transferred in triplicate to a 96-well Flat Clear bottom white microplate (Corning, Thermo Fisher Scientific) and luminescence was measured using a VICTOR Nivo Microplate Reader (PerkinElmer, USA).
Western Blot Analysis and Antibodies
All cells were harvested at 1h intervals and lysed with CETi lysis buffer (TransLab, Republic of Korea). Protein concentration was measured using the Pierce BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein samples (20–40 μg/lane) were dispensed onto Bolt Bis-Tris Plus 4–12% gradient gels (Thermo Fisher Scientific) and transferred to polyvinylidene fluoride (PVDF) membranes via electrophoresis at 180 V. Membranes were blocked with PVDF Blocking Reagent (TOYOBO, Japan) for 1 h, followed by three 5-min washes with TBST (20 mM Tris [pH 7.6], 137 mM NaCl, 0.1% Tween 20). The PVDF membranes were then incubated overnight at 4°C with the appropriate primary antibody. After incubation, the PVDF membranes were washed three times with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 90 min at room temperature (21–23°C) in the dark. Band detection was performed using the Immobilon Forte Western HRP substrate (Merck) and Odyssey Scanner (LI-COR, USA). The antibodies used were as follows:
Primary antibodies: PERK, Phospho-PERK, IRE1α, eIF2α, Phospho-eIF2α, Bip, Cleaved Caspase-3, Caspase-3, p-P38, P38, p-JNK, JNK, P-ERK, ERK, Claudin-1, ZO-1, E-cadherin (Cell Signaling Technology, USA), phospho-IRE1α (Abcam, UK). HRP-conjugated human β-actin-specific monoclonal antibody (Cell Signaling Technology). Secondary antibody: HRP-conjugated anti-rabbit IgG (Cell Signaling Technology)
Intracellular Trafficking Assay
Cell culture slides (SPL Life Sciences, Republic of Korea) were coated with collagen type 1, rat tail (Merck), and hCMEC/D3 cells were seeded at 1.0 × 10^5^ cells/well. Alexa Fluor 488-conjugated Stx1a (100 ng/ml) was treated for 1 h and 30 min. Cells were washed with DPBS (Sigma-Aldrich), and the cells were fixed by treating with 1 ml of 4% formaldehyde (Biosolution, Republic of Korea) for 2 min. Next, wash the formaldehyde with DPBS and add ER-Tracker Red (BODIPY TR Glibenclamide) (Invitrogen, Thermo Fisher Scientific) and NucBlue Fixed Cell ReadyProbes Reagent (DAPI) (Invitrogen, Thermo Fisher Scientific, USA) and incubate for 20 min at 37°C in a humidified 5% CO_2_ environment. Afterwards, the cells were washed, covered with cover glass, and mounted with Mountant with NucBlue (Invitrogen, Thermo Fisher Scientific). Using a fluorescence microscope EVOS M5000 (Thermo Fisher Scientific) with fluorescence signals detected in red, green, and blue emission channels.
Reverse Transcription - Quantitative PCR
Total RNA was extracted using NucleoSpin RNA Plus (Macherey-Nagel, Germany). cDNA synthesis and PCR amplification using reverse transcriptase were performed using the NanoHelix RT-qPCR kit (NanoHelix, Republic of Korea) according to the manufacturer's protocol. The cycling conditions for real-time PCR were as follows: 40 min at 50°C, followed by 15 min of PCR enzyme activation at 95°C. This was followed by 40 cycles of 20 s at 95°C, 30 sec at each primer annealing temperature, and 1 min at 72°C. Fluorescence data were collected using SYBR Green with a qTOWER³ (Analytik Jena, Germany). All mRNA expression data were normalized to GAPDH expression. Primer sequences are listed below (Table 1).
NO Assay
Collect the supernatant from hCMEC/D3 cells treated with Stx2a (10 ng/ml) and NAC (MedChemExpress, USA). Use the nitrite standard solution included in the Griess Reagent Kit (Invitrogen, ThermoFisher Scientific) to create a standard curve. Supernatants were transferred in triplicate. Mix N-(1-naphthyl) ethylenediamine dihydrochloride (Component A) and Sulfanilic acid (Component B) in a 1:1 ratio and add to each well of a 96-well plate. After 30 min at room temperature, measure the absorbance at 548 nm using a SpectraMax190 microplate reader.
ELISA
The concentrations of human IL-1β, IL-6, IL-8, CCL-2, and TNF-α, which are secreted cytokines and chemokines in the culture medium, were measured using ELISA kits (Invitrogen, ThermoFisher Scientific) according to the manufacturer's instructions. Cell supernatants were collected from hCMEC/D3 or A172 cells in a co-culture model treated with Stx2a (10 ng/ml). Briefly, 50 μl of each capture antibody solution was coated on 96-well half microplates and incubated overnight at 4°C. The next day, wash the wells once with PBS-T (10 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, and 0.05% Tween 20, [pH 7.4]) and add 100 μl of 5X ELISA buffer diluted to 1X to each well and block for 1 h at room temperature. After 1 h, wash the wells three times, add 50 μl of each sample, and incubate for 2 h at room temperature. Wash the wells three times as described above, add 50 μl of each detection antibody, and incubate for 1 h at room temperature. Next, wash the wells three times, add 50 μl of streptavidin-HRP, and incubate for 30 min at room temperature, protected from light. Finally, wash the wells five times, add 50 μl of substrate solution, and incubate for 15 min, away from light. When a color change occurs, stop the reaction by adding 25 μl of 2 M H_2_SO_4_, and measure the absorbance at 450 nm using a SpectraMax190 microplate reader (Molecular Devices, USA).
Cell-to-Cell Permeability Analysis
hCMEC/D3 cells were seeded at 3.0 × 10^5^ cells/well in the insert chambers of trans-well plates. Cells were treated with Stx1a (100 ng/ml), Stx1a^mut^ (100 ng/ml), Stx2a (10 ng/ml), or Stx2a^mut^ (10 ng/ml) for 24 h. Fluorescein-conjugated ovalbumin (Invitrogen, ThermoFisher Scientific), 45 kDa, was added to the insert wells containing toxin-treated hCMEC/D3 cells at the concentration recommended by the manufacturer. After 1 h, the supernatant was collected from the bottom well of the trans-well. The supernatant was transferred to a 96-well plate in triplicate (100 μl per well), and fluorescence was measured using a VICTOR Nivo Microplate Reader (PerkinElmer). The measured fluorescence values were used to evaluate cell permeability in response to each toxin.
Quantitative Analysis
Image quantification was performed using ImageJ/Fiji. The Pearson's correlation coefficients of multiple image sets were calculated using the 'Colocalization' plugin in ImageJ/Fiji. The values ranged between 0 and 1; a value of 1 indicated complete co-localization, while a value of 0 indicated no co-localization.
Statistical Analysis
Data were expressed as the means ± standard error of the mean (SEM). Group differences were evaluated by one-way ANOVA followed by Tukey post test, using GraphPad Prism version 5.00. (GraphPad Software, USA). A p-value < 0.05 was considered statistically significant. (* = p < 0.05; ** = p < 0.01; *** = p < 0.001).
Result
hCMEC/D3 Cells Undergo Cytotoxicity and Apoptosis Exposure to Stx1a and Stx2a
To determine the effect of Stxs on hCMEC/D3 cells, we treated monolayers with Stx2a (10 ng/ml), catalytically inactive Stx2a^mut^ (10 ng/ml), Stx1a (100 ng/ml), and Stx1a^mut^ (100 ng/ml) at different time points [10, 20] and examined the cellular morphological characteristics of hCMEC/D3 cells (Fig. 1A). The concentrations of Stx1a and Stx2a were selected to induce comparable levels of cellular responses in hCMEC/D3 cells, reflecting previous studies demonstrating that Stx2a exhibits significantly higher biological activity and toxicity than Stx1a [10, 20]. hCMEC/D3 cells showed a typical spread morphology with extended processes in the control group, whereas cells treated with Stx1a and Stx2a displayed time-dependent cytopathic morphological changes, including intercellular gap widening and loss of cellular extensions. In contrast, cells treated with the Stx2a^mut^ and Stx1a^mut^ lacking the N-glycosidase activity of the A subunit exhibited morphological characteristics similar to those of the control cells. A cytotoxic assay (WST) on hCMEC/D3 cells exhibited a significant reduction in Stx1a and Stx2a-treated cells (Fig. 1B). The expression of Gb3, an essential receptor for Stx binding and endocytosis, was measured by fluorescence-activated cell sorting (FACS). Cultured hCMEC/D3 cells showed Gb3 expression (Fig. 1C, Fig. S1). Moreover, to assess apoptotic execution in Stxs-treated hCMEC/D3 cells expressing Gb3, we measured caspase-3/7 activity and the expression of immunoblotted cleaved caspase-3, a key protein mediating apoptosis (Fig. 1B). We confirmed that cleaved caspase-3 expression was significantly increased in cells treated with Stx1a and Stx2a, and that caspase-3/7 activity was also increased. These results indicate that hCMEC/D3 cells are sensitive to Stxs-induced cytotoxicity, supporting caspase-mediated apoptosis as a principal mode of Stx cytotoxicity in hCMEC/D3.
Intracellular Translocation of Stxs Induces ER Stress and ROS Production in hCMEC/D3 Cells
To visualize Stxs internalization in hCMEC/D3 cells [7], we examined intracellular trafficking using immunofluorescence microscopy. To identify the cellular pathway, hCMEC/D3 cells were treated with Alexa Fluor 488-conjugated Stx1a. Cell translocation was observed after 2 h (Fig. 2A). Fluorescent signals were observed to coalesce around the nucleus, indicating intracellular translocation of the Stxs. Notably, Alexa Fluor 488-conjugated Stx1a was co-localized with an ER-specific fluorescent marker, resulting in a yellow fluorescence signal in the image, indicating retrograde translocation of the Stxs to the ER. We then examined ER stress and increased ROS in hCMEC/D3 cells exposed to Stxs. Inositol-requiring enzyme 1α (IRE1α) and eukaryotic translation initiation factor-2α (eIF2α) were confirmed to be phosphorylated to the highest level at 6 h, while protein kinase RNA-like endoplasmic reticulum kinase (PERK) was activated at 3 h and binding immunoglobulin protein (BiP) at 45 min, respectively (Fig. 2B). In addition, C/EBP homologous protein (CHOP) and death receptor 5 (DR5), ER stress-related factors that induce apoptosis, were significantly increased after Stx2a treatment compared to the control (Fig. 2C). Concordantly, oxidative stress-responsive proteins were upregulated, including Nuclear factor erythroid 2-related factor 2 (Nrf-2), Heme Oxygenase-1 (HO-1), Superoxide dismutase 2 (SOD-2), and nitrosative signals (NO_2_), indicating a Stx2a-dependent elevation in ROS. These results suggest that Stx2a is associated with the induction of ER stress and the rise of ROS synthesis in hCMEC/D3 cells.
Stxs Activate MAPK Signaling and Upregulate Inflammatory Cytokines in hCMEC/D3 Cells
ER stress response activated by Stxs induces the MAPK signaling pathway [21, 22]. Therefore, we examined whether Stxs activate MAPK signaling in hCMEC/D3 cells. When the cells were treated with Stx1a and Stx2a, robust phosphorylation of p38 and c-Jun N-terminal kinase (JNK) was observed at 90 to 180 min (Fig. S2), whereas no phosphorylation was observed when treated with Stx1a^mut^, Stx2a^mut^, and the B subunit. In contrast, extracellular signal–regulated kinase (ERK) was phosphorylated in the B subunit and Stx1a^mut^, Stx2a^mut^ (Fig. 3A), suggesting that the apoptosis-linked MAPK activity in this context is dominated by p38/JNK rather than ERK. To confirm that Stx2a induces the expression of inflammatory cytokines in hCMEC/D3 cells, we measured the mRNA and protein levels of representative inflammatory cytokines, such as IL-1β, IL-6, TNF-α, and IL-8. The expression of inflammatory cytokines significantly increased at all time points compared to the control group (Fig. 3B and 3C). Therefore, the exposure to Stx activates a MAPK pathway centered on p38/JNK, which aligns with the induction of pro-inflammatory cytokines in hCMEC/D3 cells.
Stxs Impair the Integrity of Tight Junctions and Increase Endothelial Permeability
TJs between brain endothelial cells assemble into intercellular complexes that strictly limit the transport and diffusion of specific substances across the BBB, thereby serving as its essential barrier [13, 23?-25]. Representative TJ markers include Zonula Occludens-1 (ZO-1), Claudin3 (CLDN3), Occludin, and Junctional Adhesion Molecule 2 (JAM2). In this study, we confirmed that the mRNA levels of TJs were significantly reduced beginning at 3 h after Stxs treatment (Fig. 4A). In particular, the downward trends of ZO-1, CLDN3, and occludin spanned 9–12 h, that of JAM2 progressed until ~9 h with subsequent restoration in part. Followed at the protein level, CLDN1, E-cadherin (a component of the adherens junction), and ZO-1, known as markers of endothelial cells, declined over time gradually decreased over time (Fig. 4B). Functionally, permeability in hCMEC/D3 monolayers treated for 24 h in Stx1a or Stx2a was elevated to ovalbumin, whereas no increase in permeability was discernible in control or in enzymatically inactive mutants Stx1a^mut^, Stx2a^mut^ (Fig. 4C). Accordingly, Stx exposure reduces TJ protein expression and increases paracellular flux in human cerebral endothelium.
Caspase-Mediated Apoptosis and ROS Propel Stxs-Induced Cell Damage and Tight Junction Disruption
To identify whether apoptosis and oxidative stress engage in Stx damage, we pretreated hCMEC/D3 cells with z-VAD (pan-caspase inhibitor) or N-acetylcysteine (NAC; ROS scavenger) and tested viability, cytokine expression, and junctional protein expression. Pretreatment with z-VAD substantially improved viability in comparison with Stx2a alone, reducing viability (Fig. 5A). Our dependence on caspases was defined by cleaved caspase-3 immunoblotting: there was evident cleaved caspase-3 in Stx2a-treated cultures, no evident signal in control or control+z-VAD cultures, and no evident signal in Stx2a+z-VAD cultures (Fig. 5A). To assess inflammatory outputs, we tested levels of IL-1β, IL-6, IL-8, TNF-α, CXCL1, and CCL2 at mRNA/protein levels; all were upregulated with Stx2a alone and downregulated by z-VAD (Fig. 5B). To define the role of the oxidative stress, NAC pretreatment reduced LDH release in comparison with Stx2a alone (Fig. 5C). Stx2a robustly upregulated markers of oxidative stress (Nrf-2, HO-1, SOD-2) as well as nitrosative signals (NO2), and NAC reduced these readouts, confirming toxin-induced ROS/RNS accumulation. Finally, in order to correlate these pathways with loss of barrier, we examined the expression of ZO-1, CLDNs, occludin, and JAM2 after pretreatment with z-VAD or NAC. Stx2a reduced TJ proteins by itself, while pretreatment with z-VAD or NAC reversed their expression partially, confirming that caspase-dependent apoptosis and ROS signaling engage to stimulate.
Stxs Induce Caspase-Dependent Apoptosis and Inflammatory Responses in an hCMEC/D3-A172 Co-Culture Model of the BBB
A transwell co-culture in vitro model was established using the human brain endothelial cell line, hCMEC/D3, and the human astrocyte cell line, A172, to more closely resemble an efficient and functional microenvironment of the BBB [18, 19]. The setup allows the indirect or paracrine effects of molecules, including toxins and inflammatory mediators, to be transmitted between the two cell types [26]. After exposure of hCMEC/D3 in the insert to Stx, A172 cells in the basal compartment showed impaired viability relative to untreated controls (Fig. 6A, left panel). Flow cytometry shows a clear rightward fluorescence shift for CD77/Gb3 (red) relative to the isotype control (blue), with the gate indicating ~69% CD77^+^ cells. The accompanying bar graph quantifies a significant increase in signal (fold-change), confirming surface Gb3 expression on A172 astrocytes (Fig. 6A, right panel). Although the A172 cells were not exposed directly to Stx, the presence of Gb3 accompanies their inherent susceptibility as well as provides a mechanism for the paracrine injury evident in the co-culture. In parallel, cleavage of caspase-3 was readily apparent in A172 after challenge of the overlying endothelium by Stx, yet was attenuated by z-VAD or NAC, reflecting paracrine, caspase-dependent apoptosis with a facet of ROS (Fig. 6B). In concordance, A172 showed higher levels of mRNA/protein expression of the inflammatory/apoptotic mediators IL-1β, IL-6, IL-8, TNF-α, and CCL2 in the Stx2a-only condition, yet pretreatment with z-VAD or NAC diminished these reactions to the cytokines (Fig. 6C and 6D). These observations suggest that Stx-injured endothelium delivers ROS- and caspase-linked inflammatory/apoptotic signals to the astrocytes, consistent with endothelial-to-astroglial crosstalk as an explanation for neurovascular damage mediated by the toxin.
Discussion
While acute renal failure is the signature of Stx-mediated HUS, the pathobiology at the blood–brain barrier (BBB) remains relatively undefined. Here, we delineated how human brain microvascular endothelial cells (hCMEC/D3) respond to Shiga toxins and demonstrated that TJ architecture, a determining factor in BBB function, is disrupted following exposure to the toxin. Importantly, we report that Stx1a and Stx2a induce apoptosis in hCMEC/D3 cells, confirming endothelial susceptibility to Stx cytotoxicity and revealing time-dependent morphological deterioration consistent with prior reports [27, 28]. Cytotoxicity was verified by phase-contrast microscopy, WST assays, and caspase-3/7 activity, and mechanistically attributed to toxin internalization, ER-stress and MAPK activation, and the release of pro-inflammatory cytokines [29?-31]. Congruent with the paradigm that Gb3 expression confers Stx susceptibility [2, 32], hCMEC/D3 cells exhibited highly extensive availability of cell surface receptor, as indicated by FACS analysis of A4GALT (Gb3 synthase) and Gb3. Fluorescent recombinant Stx were efficiently internalized and retrogradely trafficked to the ER within 60 min, in favor of canonical Gb3-dependent uptake and retrograde routing (Fig. 2). Once delivered to the ER, Stxs activated the unfolded protein response (UPR), with induction of BiP, CHOP, XBP1, PERK, and IRE1α, and concomitantly increased intracellular ROS (Fig. 2). Because UPR and oxidative stress often intersect with stress-activated kinase cascades, we interrogated the MAPK axis (Fig. 3): Stx exposure robustly phosphorylated p38 and JNK, whereas an enzymatically inactive Stx mutant (Stx1a^mut^, Stx2a^mut^) and isolated B-subunit did not. Conversely, ERK phosphorylation was observed with the B-subunit and with Stx mutant (Stx1a^mut^, Stx2a^mut^), suggesting that the apoptosis- and inflammation-linked MAPK signal in this model follows p38/JNK rather than ERK [10]. Together, these data support a model in which Stx activation induces ER stress and ROS accumulation, driving activation of p38/JNK, cytokine induction, caspase activation, and apoptosis in brain endothelial cells. Functionally, these events compromise BBB structure and function. TJs, comprised of claudins, occludin, junctional adhesion molecules, and scaffold proteins such as ZO-1, govern paracellular barrier selectivity and endothelial polarity [33, 34]. In our model, CLDN3, occludin, JAM2, and ZO-1 were significantly reduced following Stx exposure (Fig. 4), in line with TJ destabilization and barrier failure. CLDN3 is a reported BBB-endothelial constituent and is stably expressed in hCMEC/D3 [25], with ZO-1 scaffolds multiple TJ/adherens components to regulate cell–cell adhesion [35]. In keeping with the structural loss, we observed elevated paracellular permeability, including with Stx vs Stx1a^mut^, Stx2a^mut^ treatment (Fig. 4C) [36]. Notably, co-treatment with z-VAD (pan-caspase inhibitor) lowered cytotoxicity, preserved TJ proteins, and dampened cytokine production, indicating that caspase-dependent apoptosis is a principal executioner of the observed loss of barrier. Similarly, N-acetylcysteine (NAC) pretreatment mitigated ROS and reduced subsequent apoptotic signaling, supporting a ROS→apoptosis axis in the toxin-injured endothelium [37]. Collectively, these data indicate that Stx-induced inflammatory and apoptotic programs are major drivers of the observed permeability increase. Noting that BBB integrity emerges from neurovascular unit crosstalk, we then employed an in vitro transwell co-culture model of hCMEC/D3 endothelium in the presence of human astrocytes (A172) [18, 19]. Strikingly, astrocytes not directly exposed to Stx nonetheless exhibited cytotoxicity, cytokine induction, and increased cleaved caspase-3, indicating that paracrine endothelial–astroglial signaling can propagate injury across the barrier interface (Fig. 6). Although A172 cells were not directly exposed to Stx, the presence of Gb3 supports their intrinsic susceptibility and provides a mechanistic basis for the paracrine injury observed in the co-culture (i.e., once toxin or toxin-induced mediators traverse the endothelial layer, astrocytes may possess the receptor to engage downstream responses). As shown in Fig. 6D, pharmacologic rescue at the endothelial compartment with pan-caspase inhibition (z-VAD) or ROS scavenging (NAC) partially preserved astrocyte viability and reduced caspase activation and cytokine release, suggesting that endothelial caspase activation and oxidative stress are essential upstream initiators of astroglial damage caused by Stx2a. These observations align with a model in which endothelial injury serves as a nexus that amplifies inflammatory/apoptotic cues to neighboring glia, presumably through the action of soluble factors and damage-associated signals, thereby potentially further expanding the impact of neurovascular damage [8, 38].
In conclusion, our findings demonstrate that hCMEC/D3 endothelium is highly susceptible to Stx1a/Stx2a, undergoing time-dependent apoptosis via ER-stress and stress-kinase pathways, with resultant secondary ROS accumulation and pro-inflammatory signaling. This apoptotic program reduces TJ components (CLDN3, ZO-1, occludin, JAM2) and increases paracellular permeability. Pharmacologic caspase blockade (z-VAD) and ROS scavenging (NAC) maintain viability, lessen cytokine production, and stabilize TJ expression, indicating that apoptosis in endothelial cells may promote BBB compromise. We note the following limitations: the in vivo BBB consists of endothelium, pericytes, and astrocytes, and our co-culture lacked pericytes; additionally, in vivo shear stress and full immune components were not modeled [12, 39]. Nonetheless, by integrating receptor-dependent toxin entry, retrograde ER trafficking, UPR/ROS–MAPK signaling, and apoptotic TJ loss with endothelial–astroglial crosstalk, our data provide a coherent mechanistic scheme linking circulating Stx to cerebrovascular injury and neurologic manifestations, in particular stroke, seizures, and encephalopathy, in atypical HUS [8, 40, 41]. Our findings suggest a pathophysiological sequence in which Stx is transported via the bloodstream to the cerebral microvessels, binds Gb3 on brain endothelium, undergoes retrograde entry to the ER, triggers ROS-coupled apoptotic and inflammatory pathways, and structurally disrupts the BBB, thereby facilitating CNS complications. Further studies with the inclusion of pericytes, immune components, and physiological flow, alongside targeted modulation of ER-stress/ROS–caspase cascades, will be helpful in refining therapeutic strategies for preventing Stx-mediated neurovascular injury.
Supplemental Materials
Supplementary data for this paper are available on-line only at http://jmb.or.kr.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coliand haemolytic uraemic syndrome. Lancet 365: 1073-1086. https://doi.org/10.1016/s 0140-6736(05)71144-2. 10.1016/S 0140-6736(05)71144-2 15781103 · doi ↗ · pubmed ↗
- 2Chan YS, Ng TB. 2016. Shiga toxins: from structure and mechanism to applications. Appl. Microbiol Biotechnol. 100: 1597-1610. https://doi.org/10.1007/s 00253-015-7236-3. 10.1007/s 00253-015-7236-3 26685676 · doi ↗ · pubmed ↗
- 3Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, et al. 2015. World health organization global estimates and regional comparisons of the burden of foodborne disease in 2010. P Lo S Med. 12: e 1001923. https://doi.org/10.1371/journal.pmed.1001923. 10.1371/journal.pmed.1001923 26633896 PMC 4668832 · doi ↗ · pubmed ↗
- 4Liu Y, Tian S, Thaker H, Dong M. 2021. Shiga toxins: an update on host factors and biomedical applications. Toxins (Basel) 13: 222. https://doi.org/10.3390/toxins 13030222. 10.3390/toxins 13030222 33803852 PMC 8003205 · doi ↗ · pubmed ↗
- 5Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, et al. 2004. Structure of Shiga toxin type 2 (Stx 2) from Escherichia coli O 157:H 7. J Biol Chem. 279: 27511-27517. https://doi.org/10.1074/jbc.m 401939200. 10.1074/jbc.M 401939200 15075327 · doi ↗ · pubmed ↗
- 6Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. 1988. Site of action of a Vero toxin (VT 2) from Escherichia coli O 157:H 7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 171: 45-50. https://doi.org/10.1111/j.1432-1033.1988.tb 13756.x. 10.1111/j.1432-1033.1988.tb 13756.x 3276522 · doi ↗ · pubmed ↗
- 7Sandvig K, Garred O, Prydz K, Kozlov JV, Hansen SH, van Deurs B. 1992. Retrograde transport of endocytosed Shiga toxin to the endoplasmic reticulum. Nature 358: 510-512. https://doi.org/10.1038/358510 a 0. 10.1038/358510 a 0 1641040 · doi ↗ · pubmed ↗
- 8Trachtman H, Austin C, Lewinski M, Stahl RA. 2012. Renal and neurological involvement in typical Shiga toxin-associated HUS. Nat. Rev. Nephrol. 8: 658-669. https://doi.org/10.1038/nrneph.2012.196. 10.1038/nrneph.2012.196 22986362 · doi ↗ · pubmed ↗