Analysis of clinical parameters of different types of α-thalassemia children in Hainan region, China
Ge Gao, Zhengnan Sun, Junhong Chen, Jinyu Kang, Fei Sun, Qi Li, Limei Fu, Yi Gong, Linna Ma, Qiuling Jie, Yanlin Ma

TL;DR
This study examines how different types of α-thalassemia affect children's health in Hainan, China, focusing on blood parameters, growth, and iron levels.
Contribution
The study provides a detailed comparison of clinical parameters across α-thalassemia genotypes in children, highlighting age- and genotype-specific disease manifestations.
Findings
Children with Hb H disease showed the most severe hematological impairments, including growth delays and elevated bilirubin and ferritin.
Non-deletional genotypes (--SEA/αQSα, --SEA/αCSα) had the most pronounced clinical deficits.
Even silent carriers showed developmental delays in later childhood, and older children with Hb H disease exhibited iron overload and organ involvement.
Abstract
Thalassemia, a hereditary hemoglobinopathy characterized by impaired hemoglobin production, results in the premature destruction of erythrocytes and consequent anemia. However, the distinct hematological parameters and phenotypic expressions associated with different α-thalassemia genotypes in the pediatric population remain inadequately characterized. Therefore, this study was designed to perform a comparative analysis of clinical parameters between pediatric patients with α-thalassemia and healthy controls, to elucidate genotype-specific disease manifestations, and to inform optimized management strategies. This retrospective cross-sectional study enrolled 160 children with genetically confirmed α-thalassemia and 105 healthy controls in Hainan. Participants were categorized into silent carrier, mild, Hb H disease, and control groups. Comprehensive assessments included hematological…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
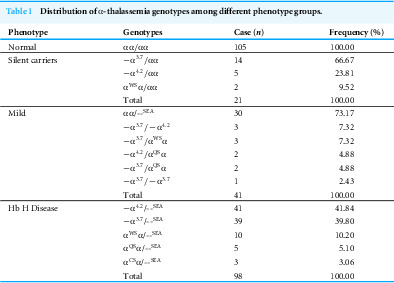 Figure 1
Figure 1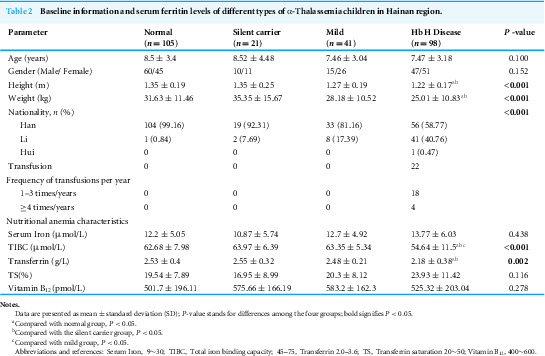 Figure 2
Figure 2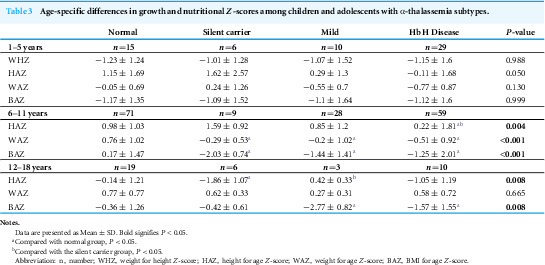 Figure 3
Figure 3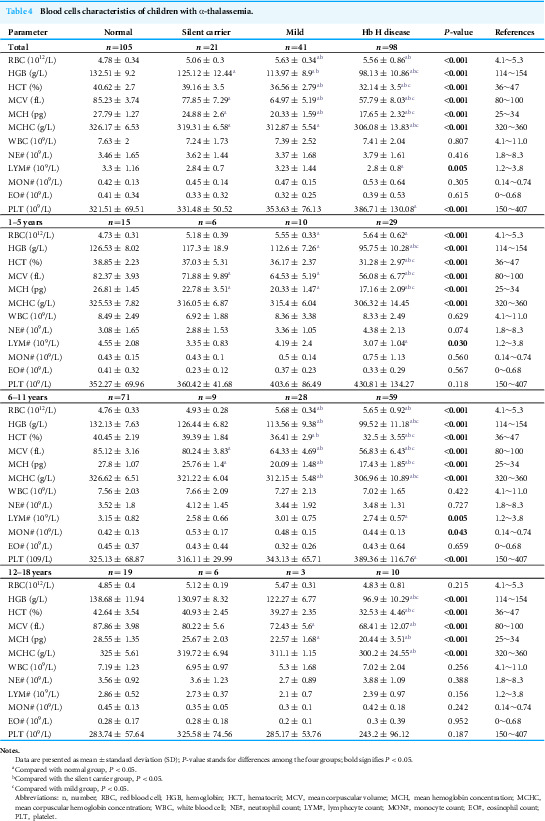 Figure 4
Figure 4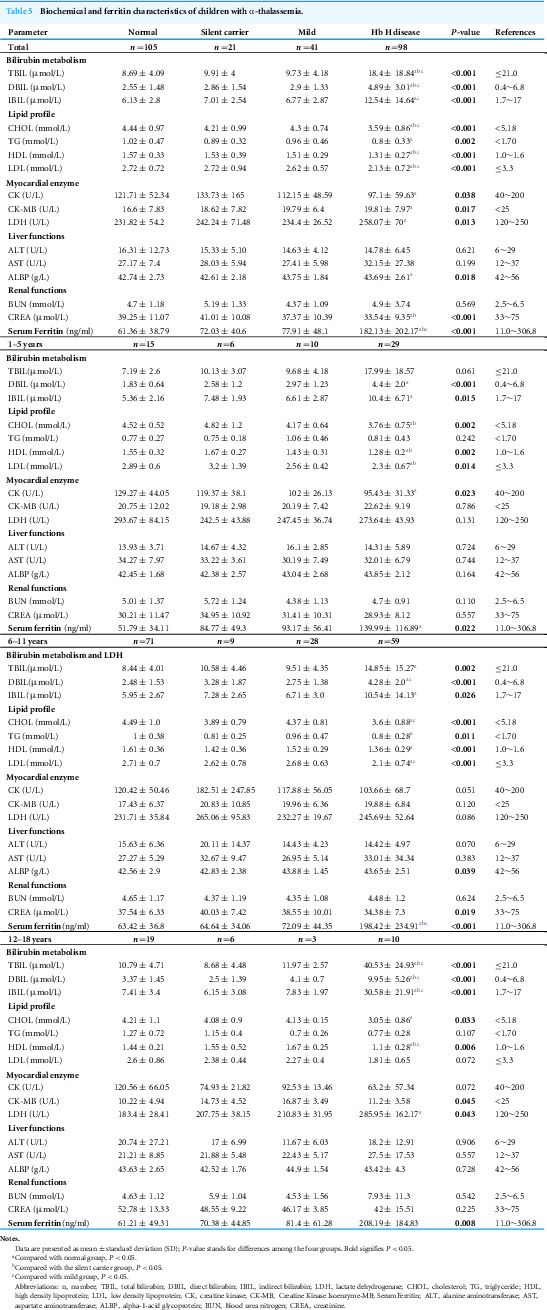 Figure 5
Figure 5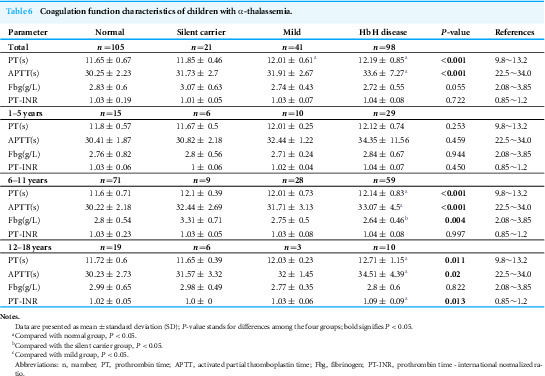 Figure 6
Figure 6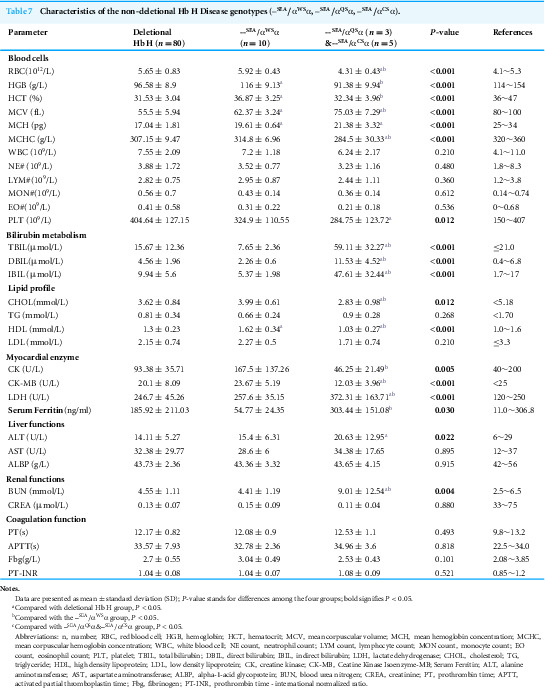 Figure 7
Figure 7- —Hainan Province Science and Technology Project
- —Major Science and Technology Program of Hainan Province
- —National Natural Science Foundation of China
- —Natural Science Foundation of Hainan Province
- —Hainan Province Clinical Medical Center
- —Innovation Platform for Academicians of Hainan Province
- —Hainan Medical University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsHemoglobinopathies and Related Disorders · Iron Metabolism and Disorders · Erythrocyte Function and Pathophysiology
Introduction
Thalassemia, a inherited hemoglobinopathy in China, is characterized by impaired hemoglobin synthesis, resulting in the premature destruction of erythrocytes and consequent anemia. The disorder is primarily classified into α-thalassemia and β-thalassemia, which result from mutations in the genes encoding the α-globin and β-globin chains of hemoglobin, respectively (Kattamis, Kwiatkowski & Aydinok, 2022; Piel & Weatherall, 2014). α-thalassemia has a high prevalence in southern provinces in China, especially in areas where malaria was once endemic (Kattamis, Kwiatkowski & Aydinok, 2022). Provinces like Guangdong (9.46%), Guangxi (14.13%) and Fujian have carrier rates as high as 10–15% (Lai et al., 2017), while Hainan island reports carrier rates of 17.74% (Wang et al., 2021). α-Thalassemia is primarily caused by mutations or deletions in the HBA1 and HBA2 genes, which encode the α-globin chains of hemoglobin. Non-deletional mutations often produce more severe phenotypes than deletional forms due to highly unstable hemoglobin variants. Genotype-phenotype correlations are crucial for predicting clinical outcomes (Hamid et al., 2022). These genetic alterations lead to a deficient synthesis of α-globin chains, creating an imbalance in the α- to β-globin chain ratio. This imbalance promotes the formation of unstable β-globin tetramers (Hb H) and other abnormal hemoglobin species, which ultimately leads to premature erythrocyte destruction and chronic hemolytic anemia.
The clinical severity of α-thalassemia exhibits a broad spectrum, ranging from silent carriers to severe. 1–2 gene defects lead to silent (−α/αα, α^ND^α/αα, αα^ND^/αα) or mild (--/αα, −α/−α, --α/α^ND^α, α^ND^α/α^ND^α) forms, and patients have mild anemia or no clinical symptoms (Musallam et al., 2024). The most severe form, resulting from the deletion of all four α-globin genes (Hb Bart’s hydrops fetalis), is typically fatal during the perinatal period (Horvei, MacKenzie & Kharbanda, 2021). However, a significant source of clinical complexity is hemoglobin H disease (Hb H disease), which is caused by defects in three α-globin genes (--/−α, --/α^ND^α) (Fucharoen & Viprakasit, 2009). In pediatric patients, it can lead to growth retardation, developmental delays, and a reduced quality of life. A critical complication is iron overload, which may result in irreversible organ damage (Tenuta et al., 2024). Clinical management is complicated by phenotypic heterogeneity and diagnostic complexity. Early genetic screening, individualized treatment, and lifelong monitoring of iron burden are essential.
Previous studies have predominantly focused on the molecular genetics and hematological features of α-thalassemia (Hamid et al., 2022), and most analyses have been conducted in adult populations, relatively limited attention has been paid to the broader systemic manifestations of Hb H disease in children. This gap in the literature is particularly evident for parameters such as hepatic and renal function, coagulation profiles, iron overload, and growth development. Moreover, the influence of different genotypes on these systemic parameters remains incompletely understood in pediatric populations.
To address this knowledge gap, clinical parameters were collected from 160 pediatric patients with α-thalassemia and 105 healthy controls in Hainan Island. A comprehensive analysis was conducted to compare clinical indicators across various α-thalassemia genotypes and against healthy controls, with a particular focus on Hb H disease. A detailed assessment of these clinical features is essential for developing tailored prevention and management strategies. The findings from this study are anticipated to enhance the current understanding of Hb H disease phenotypes and inform the development of optimized management strategies for affected children.
Patients and Methods
Recruitment and inclusion of research subjects
All participants were recruited at the First Affiliated Hospital of Hainan Medical University between May 2022 and April 2024. Genetic diagnosis for thalassemia genotypes was performed using standardized molecular techniques (BGI Genomics, Shenzhen, China) to screen for 508 known pathogenic variants in the α-globin (HBA1/HBA2) and β-globin (HBB) genes. Based on the genotyping results, 160 children with a confirmed α-thalassemia diagnosis and 105 age- and gender-matched healthy controls, who tested negative for both α- and β-thalassemia mutations, were included in the study. Participants were selected from a pediatric population (age ≤ 18 years). Exclusion criteria comprised comorbidities such as β-thalassemia, active infections, iron deficiency, other hematological or metabolic disorders, and any chronic conditions that could confound the results, including hepatic, cardiac, or renal diseases, as well as malignancies. The study protocol received approval from the Institutional Review Board (IRB) of the Ethics Committee of The First Affiliated Hospital of Hainan Medical University (Approval No.: 2022 [Scientific Research L] No. [60]). Written informed consent was obtained from all participants or their legal guardians after a comprehensive explanation of the study’s objectives and procedures. All study procedures were conducted in accordance with the principles outlined in the Declaration of Helsinki. Based on the molecular diagnosis of α-globin genotypes, participants were categorized into four groups: (1) silent carrier group (one mutated or deleted α-globin gene): −α/αα, α^ND^α/αα, αα^ND^/αα; (2) mild group (two mutated or deleted α-globin genes): --/αα, −α/−α, −α/α^ND^α, α^ND^α/α^ND^α; (3) Hb H disease group (three mutated or deleted α-globin genes): --/−α, --/α^ND^α; (4) control group (normal α-globin genotype, no mutations or deletions): αα/αα.
Sample collection and laboratory values analysis
Peripheral venous blood samples (2–5 mL) were collected from each participant into EDTA-anticoagulated tubes. Genomic DNA was extracted using a commercial kit (QIAamp DNA Blood Mini Kit; Qiagen) according to the manufacturer’s protocol. Next-generation sequencing (NGS) was performed to screen for 508 known pathogenic variants in the HBA1 and HBA2 genes (BGI Genomics, Shenzhen, China). All participants underwent a comprehensive panel of laboratory tests, including complete blood count (CBC), biochemical analysis, coagulation profiling, and serum ferritin measurement. All data and samples were anonymized and maintained with strict confidentiality. Complete blood count (CBC) analysis was conducted using an automated hematology analyzer (Sysmex XN-3000). Coagulation profiles were assessed using an automated coagulation analyzer (Sysmex CS-2500). Biochemical parameters, including liver and renal function, were measured using an automated biochemistry analyzer (Mindray BS-2800M). Serum ferritin levels were quantified by chemiluminescence immunoassay. Samples exceeding the upper detection limit (>1,500 ng/mL) were appropriately diluted and re-analyzed to obtain accurate quantitative values.
Z-score calculation
Growth status was assessed using age- and sex-specific Z-scores, which were calculated in accordance with the World Health Organization (WHO) Growth Standards (2006 for children aged 0–5 years and 2007 for children and adolescents aged 5–19 years). The Z-scores for weight-for-age (WAZ), height-for-age (HAZ), weight-for-height (WHZ), and body mass index-for-age (BAZ) were computed utilizing the WHO Anthro (version 3.2.2) and Anthro Plus (version 1.0.4) software.
Statistical methods
All statistical analyses were performed using IBM SPSS Statistics version 25.0 (IBM Corp., Armonk, NY, USA). Continuous variables are presented as mean ± standard deviation (SD) for the clinical parameters and hemoglobin profiles of each thalassemia genotype. The normality of data distribution was assessed using the Shapiro–Wilk test. For comparisons between two groups, the independent samples t-test was used for normally distributed data, and the Mann–Whitney U test was employed for non-normally distributed data. For comparisons across more than two groups, one-way analysis of variance (ANOVA) was used for parametric data, with post-hoc multiple comparisons conducted using the Bonferroni correction. A two-tailed p-value of less than 0.05 was considered statistically significant.
Results
Basic information of patients
In total, 265 children were enrolled for this study, comprising 160 with α-thalassemia and 105 healthy controls. Based on their α-thalassemia genotypes, the participants were classified into four groups: silent carrier, mild, Hb H disease, and normal. The distribution of α-thalassemia genotypes across these groups is presented in Table 1. The frequencies of deletional and non-deletional mutations in the Hb H disease group were 81.63% and 18.37%, respectively. The height and weight of patients in the Hb H disease group were significantly lower than those of the normal and silent carrier groups, but not substantially different from the mild group. The proportion of Li ethnic children in the α-thalassemia groups was significantly higher than that in the normal group, increasing with the severity of α-globin gene defects. Among the α-thalassemia children, 22 (13.75%) received blood transfusions, all of whom were from the Hb H disease group. Of these, 18 patients received only occasional blood transfusions (Table 2).
Table 1: Distribution of α-thalassemia genotypes among different phenotype groups.
Table 2: Baseline information and serum ferritin levels of different types of α-Thalassemia children in Hainan region.
Age-specific impairments in growth and development among children and adolescents with α-Thalassemia
To further investigate the effects of different types of α-thalassemia on the growth and development of children across various age groups, we categorized the enrolled children into three groups: early childhood (0–5 years), middle childhood (6–11 years), and adolescence (12–18 years). We assessed the changes in growth and development using age- and sex-specific Z-scores. The results indicate that as age increases, the differences in growth and development among the groups become more pronounced. In the 0–5 years group, there were no significant differences in Weight-for-Height Z-scores (WHZ), Height-for-Age Z-scores (HAZ), Weight-for-Age Z-scores (WAZ), and Body Mass Index Z-scores (BAZ) between the different α-thalassemia types and the normal group. However, with advancing age, significant differences emerged in growth and development among the groups. In the 6–11 years age group, WAZ (P < 0.001) and BAZ (P < 0.001) for the Hb H disease and mild groups were significantly lower than those of the normal control group, indicating delayed growth in weight and BMI. Even in the 12–18 years group, BAZ remained significantly lower (P = 0.008), suggesting that BMI development continued to be affected during adolescence. Additionally, in the 6–11 years age group, silent carriers exhibited significantly lower WAZ (P < 0.001) and BAZ (P < 0.001) compared to the normal group. By the age of 12–18 years, their HAZ also showed significant delays (P = 0.008). These data demonstrate age-specific effects of α-thalassemia on growth, even among silent carriers (Table 3).
Table 3: Age-specific differences in growth and nutritional Z-scores among children and adolescents with α-thalassemia subtypes.
The erythrocyte system, the platelet system varied greatly between α-thalassemia groups
To understand the anemia conditions of participants with different types of α-thalassemia, we analyzed the routine blood indicators of the enrolled participants. Compared to the normal group, the levels of erythrocyte system indicators, including hemoglobin (HGB), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC), decreased in all three α-thalassemia groups, with the most significant decrease observed in the Hb H group (P < 0.001), falling below clinical normal reference ranges. In contrast to the normal and silent carrier groups, the red blood cell (RBC) counts were higher in the mild and Hb H disease groups, exceeding clinical normal reference ranges. The platelet (PLT) values in the Hb H disease group were significantly higher than those in the normal group; however, this difference lacks clinical significance (Table 4). Given that many clinical hemoglobin parameters vary significantly with age and sex, we further conducted an age-stratified analysis. The results indicated that, regardless of age group, the differences in MCV, MCH, and MCHC were consistent with the overall findings. Nonetheless, the RBC, HGB, lymphocyte (LYM) count, and PLT levels of children under 11 years old aligned with the overall trend. For children over 12 years old, the HGB levels exhibited differences only in the Hb H disease group, with no significant differences observed in RBC, LYM count, and PLT levels (Table 4).
Table 4: Blood cells characteristics of children with α-thalassemia.
Differences in biochemical indicators in patients with different α-thalassemia genotypes
The assessment of bilirubin metabolism indicators revealed that, compared to the normal group and other thalassemia groups, the mean values of total bilirubin (TBIL), direct bilirubin (DBIL), and indirect bilirubin (IBIL) were significantly elevated in the Hb H group (P < 0.05). Furthermore, the evaluation of blood lipid indicators indicated that patients with Hb H disease exhibited significantly lower levels of cholesterol (CHOL), high-density lipoprotein (HDL), and low-density lipoprotein (LDL) compared to the normal group and other thalassemia groups. Notably, triglyceride (TG) levels were only reduced in the Hb H group when contrasted with the normal group. Additionally, myocardial enzyme indicators, including creatine kinase (CK), CK-MB, and lactate dehydrogenase (LDH), as well as liver function indicators such as alkaline phosphatase (ALBP) and renal function indicators like creatinine (CREA), demonstrated significant differences in the Hb H group compared to the normal group and other thalassemia groups. However, all these differential indicators remained within the clinical reference ranges and lacked clinical significance. A further age-stratified analysis indicated that for children over 12 years old, the levels of TBIL, DBIL, and IBIL were significantly increased in the Hb H group (P < 0.05) and exceeded the clinical normal reference ranges. Moreover, serum ferritin levels were significantly higher in the Hb H disease group than in the other groups (P < 0.001), with levels gradually increasing with age (Table 5).
Table 5: Biochemical and ferritin characteristics of children with α-thalassemia.
The level of PT and APTT significantly increased in the Hb H group
Next, we analyzed the coagulation indices of the enrolled participants. Our findings revealed that, compared to the normal group, the levels of prothrombin time (PT) and activated partial thromboplastin time (APTT) were significantly elevated in the Hb H group, while the PT level also increased in the mild group, indicating a prolonged clotting time. However, all these differential indicators remained within the clinical reference ranges and lacked clinical significance. Further age-stratified analysis indicated that, for children over 12 years old, the APTT levels were significantly increased in the Hb H group (P < 0.05) and exceeded the clinical normal reference ranges (Table 6). Additionally, we further stratified the patients by age and gender for comparison. Nonetheless, the sample size of each group was relatively small, and the data should be interpreted with caution (Tables S1–S3). In the future, we plan to increase the sample size for more robust comparative analysis.
Table 6: Coagulation function characteristics of children with α-thalassemia.
Hematologic and biochemical characteristics among deletional and non-deletional Hb H genotypes
To investigate the clinical manifestations of both non-deletional and deletional Hb H disease, we classified patients with Hb H disease into these two categories and examined differences in blood cell counts, biochemical parameters, coagulation parameters, ferritin levels, and other relevant indicators. Our analysis revealed no significant differences in most of the indicators between non-deletional Hb H disease and deletional Hb H disease across the three age groups (Tables S4–S6). This finding is inconsistent with previous studies, which indicated that non-deletional Hb H disease patients exhibit more severe clinical manifestations than patients with deletional Hb H disease. We further divided non-deletional Hb H disease into --^SEA^/α^WS^α and --^SEA^/α^QS^α&--^SEA^/α^CS^α subgroups for analysis and found that --^SEA^/α^QS^α and --^SEA^/α^CS^α exhibits more severe manifestations compared to deletional Hb H disease and --^SEA^/α^WS^α. Patients with --^SEA^/α^QS^α and --^SEA^/α^CS^α genotypes showed significantly reduced red cell indices (HGB, HCT, MCV, MCH, MCHC) and elevated bilirubin (TBIL, IBIL, DBIL), which exhibited the most severe hematological impairment and hemolytic markers. --^SEA^/α^QS^α and --^SEA^/α^CS^α also had significantly lower RBC counts, PLT, lipid levels (CHOL, HDL), and myocardial enzymes (CK, CK-MB), alongside higher LDH, ALT, and BUN, indicating pronounced hemolysis, possible liver involvement, and altered metabolic and renal function compared to other genotypes. Although ferritin levels in all three groups did not reach iron overload levels, levels in the --^SEA^/α^QS^α and --^SEA^/α^CS^α groups were significantly higher than those in the --^SEA^/α^WS^α and deletion groups (Table 7). We did not further subdivide the various genotypes of Hb H disease due to the small number of patients in each group, which limited our ability to conduct more detailed analyses. We will continue to collect samples for future research.
Table 7: Characteristics of the non-deletional Hb H Disease genotypes (–SEA/αWSα, –SEA/αQSα, –SEA/αCSα).
Discussion
Hb H disease is characterized by a significant deficiency of α-globin, leading to the excessive formation of β-globin chains that result in the production of unstable hemoglobin, known as Hb H. This condition can present with varying degrees of anemia, ranging from mild to severe, and exhibits considerable clinical heterogeneity (Lal et al., 2011). The variability in clinical presentation complicates treatment strategies. Typically, patients experiencing severe anemia receive blood transfusions as symptomatic treatment; however, early monitoring and prevention of disease progression remain challenging. In this study, we employed a cross-sectional approach to stratify patients based on age, sex, and genotype. We systematically analyzed multiple dimensions of disease impact to investigate the clinical heterogeneity of pediatric α-thalassemia, focusing particularly on growth characteristics, hematological and biochemical profiles, and genotype-phenotype correlations across different subtypes. Our findings indicate a strong relationship between the clinical phenotype of α-thalassemia, particularly Hb H disease, and both the patient’s age and genotype. Furthermore, some non-deletional Hb H disease phenotypes exhibit greater severity than their deletional counterparts.
In our study, α-thalassemia was categorized into three groups: silent carriers, mild carriers, and Hb H disease. Analysis of the four groups with different genotypes and blood parameters revealed statistically significant differences in Hb, RBC count, MCV, MCH, and MCHC. The Hb levels in the Hb H group were notably lower than those in the other three groups and fell below the clinical reference values, indicating clinical diagnostic significance. Hb levels are essential for diagnosing anemia. Chronic anemia can lead to insufficient systemic oxygenation and adversely affect various physiological functions, including growth and development. In our study, the height and weight of children with thalassemia were significantly lower than those without the condition, with growth indicators in the Hb H group substantially lagging behind those in the silent carrier and mild groups. This result was further confirmed by the Z-scores for weight-for-age (WAZ), height-for-age (HAZ), and BMI-for-age (BAZ) (De Onis et al., 2007). Meanwhile, we also found that in patients with more severe types of non-deficient Hb H disease, the degree of anemia was more severe, and their height and weight also decreased accordingly, reflecting the systemic burden of chronic anemia. These findings align with previous research linking α-thalassemia, particularly Hb H disease, to growth delays due to chronic hemolytic anemia and associated metabolic disturbances (Delvecchio & Cavallo, 2010; Higgs, Engel & Stamatoyannopoulos, 2012). The severity of growth impairment correlated with the severity of the thalassemia type (Zhu et al., 2021), and this influence gradually intensifies with age. Notably, subtle growth disturbances were also observed in silent carriers during middle childhood and adolescence, suggesting that even milder genotypes may contribute to developmental challenges. Ineffective erythropoiesis likely exacerbates these effects by promoting red cell destruction and increasing bone marrow stress, which may alter cytokine profiles and hormonal regulation (Lal et al., 2024). Chronic anemia significantly affects children’s growth and development of α-thalassemia, especially hindering physical maturation and adolescent development (Harteveld et al., 2022). These findings underscore the critical need for genotype-specific monitoring and early intervention to optimize developmental outcomes in affected children. We advocate for an integrated, multidisciplinary management approach that includes routine hematologic monitoring, regular growth assessments and targeted nutritional support to improve long-term outcomes for pediatric patients.
Our study demonstrated that the TBIL and DBIL levels in the silent carrier, mild, and Hb H disease groups were all significantly higher than those in the normal group. Notably, the Hb H group exhibited a significantly greater increase compared to the silent carrier and mild Hb H disease groups. These differences are clinically significant among children over 12 years of age. Previous studies have reported similar patterns, indicating that the production of elongated or unstable α-globin chains leads to precipitation within erythroid precursors, thereby exacerbating hemolysis and bone marrow stress (Lal et al., 2024). Hemolysis can result in an increase in bilirubin decomposition, subsequently elevating bilirubin-related indicators. Concurrently, heightened hemolysis can lead to the accumulation of ferritin, which aligns with our observed results. As patients with Hb H age, their ferritin levels tend to rise, suggesting that the impact of hemolysis intensifies with age. This finding underscores the importance of early recognition and intervention for emerging issues in these patients.
Evidence suggests that β-thalassemia induces a hypercoagulable state, attributed to factors such as the exposure of phosphatidylserine on red blood cells, the increased fragility of these cells, and the absence of the spleen. Laboratory tests confirm elevated coagulation markers in thalassemia, particularly among patients who have undergone splenectomy and are not regularly transfused (Taher et al., 2008). Although research on α-thalassemia is limited, our findings indicate that PT and APTT in the three thalassemia groups were significantly higher than in the normal group. This suggests that as the severity of the disease increases, the risk of thrombosis also rises due to abnormal coagulation in patients. Notably, the difference in APTT was clinically significant only in patients aged 12 years, indicating that this effect may also be cumulative with age. Therefore, the coagulation function of patients with α-thalassemia, particularly those with Hb H disease, should be monitored concurrently before the age of 12 to detect abnormalities early and prevent thrombosis.
The values of PLT, platelet large cell ratio (PLC-R), mean platelet volume (MPV), and plateletcrit (PCT) in the α-thalassemia groups were significantly higher than those in the normal group, particularly in the Hb H disease subgroup. However, all these differences remain within the normal clinical reference range and lack significant clinical relevance. This observation may be attributed to the correlation between disease severity and age. Symptoms during childhood are generally mild, and no significant clinical abnormalities have been observed thus far, suggesting a state of clinical sub-health.
Despite the valuable insights provided by this study regarding the multisystemic effects of different α-thalassemia genotypes in children and adolescents, several limitations warrant acknowledgment. Our cross-sectional study, employing an age- and sex-stratified approach, has established a robust framework for evaluating the multifaceted impact of α-thalassemia across pediatric populations. We have elucidated genotype-specific differences, particularly the pronounced severity of non-deletional Hb H disease. However, certain limitations must be recognized. Potential gender imbalances within subgroups may have influenced sex-specific comparisons, and the absence of longitudinal data restricts our ability to fully characterize long-term disease trajectories. To address these gaps, we propose that future research prioritize larger-scale, longitudinal studies that incorporate comprehensive clinical and biochemical markers while controlling for treatment-related variables. Such studies would more effectively delineate the progression of growth impairments and iron overload from childhood into adulthood.
Conclusion
Our analysis highlights the significant clinical and biochemical impacts of different types of α-thalassemia, particularly Hb H disease. We observed critical differences in hematological indices, iron metabolism, and biochemical indicators among patients, which varied according to the severity of their condition. These findings underscore the necessity for regular monitoring and tailored management strategies to mitigate complications and improve the quality of life for these patients. Therefore, it is essential to pay attention to the anemia, growth and development, and ferritin levels of affected children and to intervene promptly to promote their normal growth and development, especially in patients with Hb H disease. By enhancing our understanding of the clinical heterogeneity of α-thalassemia and its management, we can better address the specific needs of this patient population.
Supplemental Information
10.7717/peerj.20586/supp-1Supplemental Information 1Raw dataAll the information of the collected data and contains four tables, each of which is for different groups of information. The baseline information represented by numbers is described below, and the rest are the numerical values of clinical testing items, whose units have been shown in detail in the table 1–7. Note: Nation, Han = 1,Hui = 2, Li = 3, Miao = 4 Gender: boy = 1, girl = 2
10.7717/peerj.20586/supp-2Supplemental Information 2Hematological and biochemical characteristics of children aged 1–5 years, boys and girls, with α-thalassemia
10.7717/peerj.20586/supp-3Supplemental Information 3Supplementary table 2. Hematological and biochemical characteristics of children aged 6–11 years (boys and girls) with α-thalassemia
10.7717/peerj.20586/supp-4Supplemental Information 4Hematological and biochemical characteristics of children aged 12–18 years (boys and girls) with α-thalassemia
10.7717/peerj.20586/supp-5Supplemental Information 5Hb H disease blood characteristics of children
10.7717/peerj.20586/supp-6Supplemental Information 6Biochemical and ferritin characteristics of children with deletional vs. non-deletional Hb H disease
10.7717/peerj.20586/supp-7Supplemental Information 7Coagulation function characteristics of children with deletional vs. non-deletional Hb H disease
10.7717/peerj.20586/supp-8Supplemental Information 8Hematologic and biochemical characteristics among deletional and non-deletional Hb H Disease difference genotypes
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Delvecchio M Cavallo L 2010 Growth and endocrine function in thalassemia major in childhood and adolescence Journal of Endocrinological Investigation 33616810.1007/bf 0334655120203539 · doi ↗ · pubmed ↗
- 2De Onis M Onyango AW Borghi E Siyam A Nishida C Siekmann J 2007 Development of a WHO growth reference for school-aged children and adolescents Bulletin of the World Health Organization 8566066710.2471/blt.07.04349718026621 PMC 2636412 · doi ↗ · pubmed ↗
- 3Fucharoen S Viprakasit V 2009 Hb H disease: clinical course and disease modifiers Hematology American Society of Hematology. Education Program 2009263410.1182/asheducation-2009.1.2620008179 · doi ↗ · pubmed ↗
- 4Hamid M Keikhaei B Galehdari H Saberi A Sedaghat A Shariati G Mohammadi-Anaei M 2022 Genotype-phenotype correlation in patients with deletional and nondeletional mutations of Hb H disease in Southwest of Iran Scientific Reports 12485610.1038/s 41598-022-08986-435319015 PMC 8941133 · doi ↗ · pubmed ↗
- 5Harteveld CL Achour A Arkesteijn SJG Ter Huurne J Verschuren M Bhagwandien-Bisoen S Schaap R Vijfhuizen L El Idrissi H Koopmann TT 2022 The hemoglobinopathies, molecular disease mechanisms and diagnostics International Journal of Laboratory Hematology 44Suppl 1283610.1111/ijlh.1388536074711 PMC 9542123 · doi ↗ · pubmed ↗
- 6Higgs DR Engel JD Stamatoyannopoulos G 2012 Thalassaemia Lancet 37937338310.1016/s 0140-6736(11)60283-321908035 · doi ↗ · pubmed ↗
- 7Horvei P Mac Kenzie T Kharbanda S 2021 Advances in the management of α-thalassemia major: reasons to be optimistic Hematology. American Society of Hematology. Education Program 202159259910.1182/hematology.202100029534889445 PMC 8791144 · doi ↗ · pubmed ↗
- 8Kattamis A Kwiatkowski JL Aydinok Y 2022 Thalassaemia Lancet 3992310232410.1016/s 0140-6736(22)00536-035691301 · doi ↗ · pubmed ↗