Predicting the Stability of Base‐mediated C─H Carboxylation Adducts Using Data Science Tools
Maike Eckhoff, Shubham Deolka, Aleria Garcia‐Roca, Lilly Meynberg, Liudmila Seidel, Matthew S. Sigman, Jonny Proppe

TL;DR
This paper introduces a computational method combining quantum chemistry and machine learning to predict the stability of CO2 adducts in C–H carboxylation reactions.
Contribution
A novel predictive workflow integrating quantum chemistry and statistical modeling for CO2 adduct stability.
Findings
The workflow was applied to 60 nucleophiles, identifying reactions that yield stable carboxylation adducts.
Experimental validation confirmed predictions for five carbanions, including three stable and two unstable adducts in DMSO.
The method was extended to assess structurally distinct carbanions for broader applicability.
Abstract
Base‐mediated C–H carboxylation is a versatile pathway for utilizing carbon dioxide (CO2) as a C1 building block in organic synthesis. However, CO2 constitutes a notorious thermodynamic sink, which restricts this approach to activated or intrinsically reactive nucleophiles. To qualitatively assess the stability of CO2 adducts, we present a computational approach that integrates quantum chemistry with statistical modeling to build a predictive workflow. The target property is the CO2 affinity, specifically the negative Gibbs free reaction energy. This predictive workflow has been applied to 60 novel carbon‐centered nucleophiles, suggesting reactions that yield stable carboxylation adducts. The results have been validated through experimental methods for five carbanions, which include three stable and two unstable adducts in DMSO according to our predictions. In addition, we examined two…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1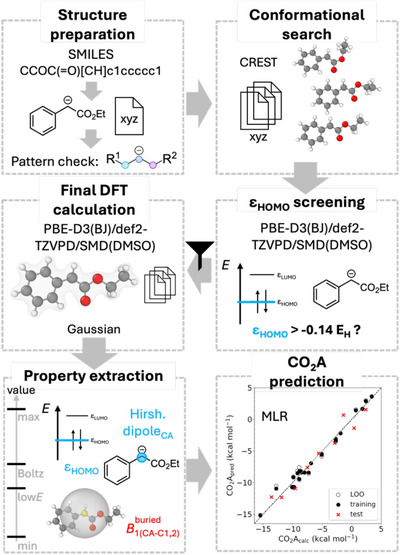 Figure 2
Figure 2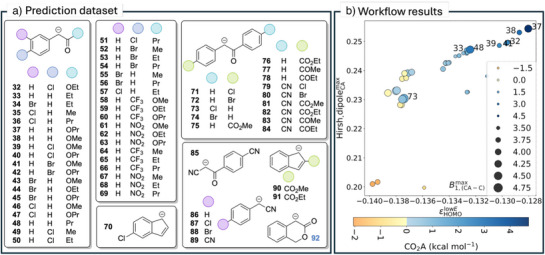 Figure 3
Figure 3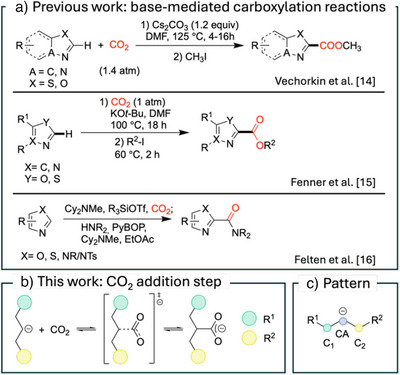 Figure 4
Figure 4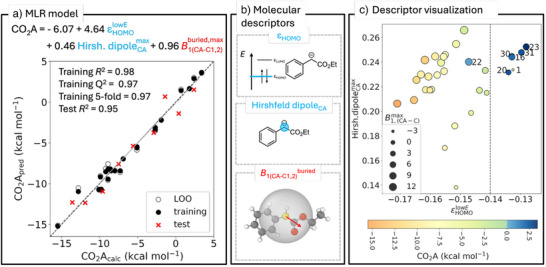 Figure 5
Figure 5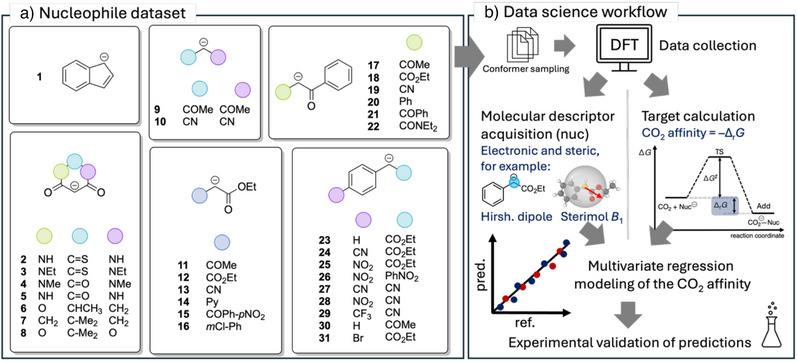 Figure 6
Figure 6- —Deutscher Akademischer Austauschdienst10.13039/501100001655
- —National Science Foundation10.13039/100000001
- —Federal Ministry of Research, Technology and Space (BMFTR), Germany
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCarbon dioxide utilization in catalysis · CO2 Reduction Techniques and Catalysts · Catalytic C–H Functionalization Methods
Carbon dioxide (CO_2_) serves as a valuable C1 building block in organic synthesis.^[^ 1, 2, 3, 4, 5 ^]^ Carboxylation products find numerous applications, including CO_2_‐binding strategies^[^ 6, 7, 8, 9 ^]^ and serving as key components in pharmaceuticals, particularly in prodrugs, as esters and amides.^[^ 10 ^]^ Of the many synthetic tactics for incorporating CO_2_ into molecules, C─H carboxylation is particularly relevant due to its high atom and step economy.^[^ 11, 12 ^]^ When carrying out base‐mediated C─H carboxylation under mild and transition‐metal‐free conditions, the resulting processes are potentially both more environmentally friendly and cost‐efficient.^[^ 13 ^]^ Previous studies (Scheme 1a) have highlighted base‐mediated carboxylation reactions promoted by Cs_2_CO_3_ for electron‐deficient aromatic heterocycles, as reported by Vechorkin et al.,^[^ 14 ^]^ Fenner and Ackermann demonstrated that these reactions are achievable using KOtBu as the stoichiometric base.^[^ 15 ^]^ The resulting highly nucleophilic carbanion facilitates the subsequent CO_2_ capture step at low to moderate temperatures and atmospheric CO_2_ pressure. In the work of Felten et al., base‐mediated carboxylation reactions of azoles were activated and stabilized by silyl triflate reagents.^[^ 16 ^]^
However, an underlying aim in this area is to determine when carbanions will form stable CO_2_ adducts as they are generally unstable due to the high thermodynamic stability of CO_2_.^[^ 17 ^]^ While a reactive nucleophile can facilitate adduct formation, stabilizing this adduct poses a different challenge, although kinetic barriers may correlate with reaction energies under certain conditions.^[^ 18 ^]^ Recent studies have explored various approaches to address these thermodynamic challenges. For instance, Li et al. successfully synthesized a carboxylation product using indene through an acidic workup.^[^ 19 ^]^ Additionally, employing reagents that further react with the carboxylation adducts has proven effective to circumvent decomposition.^[^ 14, 15, 16 ^]^ To achieve a comprehensive understanding of the carboxylation process, it is also essential to consider the kinetics of carboxylating carbanions as well. Recent studies^[^ 19, 20 ^]^ have indicated that CO_2_ is suitable to react with a diverse set of nucleophiles in DMSO, including succinimide^[^ 21 ^]^ or piperidine.^[^ 22 ^]^ An overarching goal would be to establish structural guidelines through the development of a predictive model for when stable CO_2_ adducts will form. Such models could guide synthetic chemists in CO_2_ capture and valorization while also providing fundamental knowledge. Toward this goal, we report herein a predictive model of the formation of stable CO_2_ adducts arising through base‐mediated C─H carboxylation focused on the specific reaction step depicted in Scheme 1b. The resultant model was used to both understand the structural features required to form stable adducts and was applied prospectively to identify new structures that were validated experimentally.
As the first step, we generated a dataset based on 31 structurally diverse nucleophiles (Scheme 2a). These nucleophiles were selected for their kinetic suitability to react with CO_2_, as indicated by Mayr and Patz's reactivity scale.^[^ 19, 20 ^]^ To produce physical organic molecular descriptors, conformer ensembles were obtained using the GFN2‐xTB^[^ 23 ^]^ method implemented in CREST,^[^ 24, 25 ^]^ followed by density functional theory (DFT) calculations with Gaussian.^[^ 26 ^]^ Structure optimizations were performed with PBE‐D3(BJ)/def2‐TZVPD^[^ 27, 28, 29, 30, 31 ^]^ including implicit solvation using the SMD model^[^ 32 ^]^ for DMSO. From the resultant structures, a range of steric and electronic descriptors were extracted.^[^ 33, 34, 35 ^]^ For each descriptor, ensemble‐based values were collected, including the minimum (min), maximum (max), and the lowest‐energy conformer (lowE) value, as well as the Boltzmann‐weighted average (Boltz) values of the ensemble. To train a model, the affinity of CO_2_ for binding to the carbon‐centered nucleophile (see structure pattern in Scheme 1c) was assessed by computing the negative Gibbs free reaction energy as the target property (CO_2_A_calc_) at a higher computational level using B3LYP^[^ 36, 37 ^]^ as exchange–correlation functional instead of PBE (see computational details and benchmark as well as modeling details in Sections S1–S5).
Initially, a multivariate linear regression (MLR) model was developed by correlating the CO_2_A_calc_ to the molecular features of the nucleophiles.^[^ 34, 38 ^]^ Model performance was measured by statistical metrics, including the coefficient of determination (R ^2^) and the mean absolute error (MAE, in kcal mol^−1^) for both training and test sets. A leave‐one‐out (LOO) analysis and k‐fold cross‐validation (k = 5) were performed to evaluate the overall robustness of the model (see SI for details).
The best three‐parameter MLR model (Scheme 3a) performs well on the test set (R ^2^ = 0.95) and in five‐fold cross‐validation (R ^2^ = 0.97). This model is based on two electronic descriptors and one steric descriptor: the energy of the highest occupied molecular orbital (HOMO), ε_HOMO_, the Hirshfeld dipole moment at the carbanionic site (CA, see Scheme 1b), and the buried Sterimol^[^ 40 ^]^ B 1 value defined at CA and C_1_/C_2_ (see Scheme 3b). In Scheme 3c, the electronic properties are plotted against each other and the magnitude of the steric parameter is reflected by the size of datapoints. The CO_2_ affinity is visualized as a heat map. The plot reveals a clear trend: the lower ε_HOMO_, the higher the CO_2_ affinity. A threshold of ε HOMO = –0.140 E_h_ can be identified for a CO_2_ affinity of approximately zero. The two additional parameters improve the model, as ε HOMO alone does not fully capture the observed relationship. Two nucleophiles appear to be outliers: 22 (classified as false negative) and 1 (classified as false positive). This highlights a limitation of the model: both nucleophiles feature uncommon structural motifs that are not well captured by linear regression.
The model parameters provide valuable insight into the CO_2_ addition step. A higher ε HOMO indicates a more favorable electron transfer to CO_2_, facilitating bond formation in the adduct. Additionally, a larger Hirshfeld dipole moment at the carbanionic site suggests a stronger electrostatic attraction with CO_2_. The buried Sterimol B 1 value represents the nucleophile's bulkiness: as it increases, steric shielding enhances adduct stabilization.
In the next step, we constructed an automated and user‐friendly workflow that integrates both the classification and MLR models to screen nucleophiles and focus the search for potentially stable CO_2_ adducts (see Scheme 4 and https://git.rz.tu‐bs.de/proppe‐group/co2_affinity_prediction). To initiate the workflow, 60 potential nucleophiles were combinatorially designed based on core structural features found in nucleophiles forming the most stable adducts of the initial dataset (see Scheme 5a). The only required input is a set of SMILES strings. To quickly assess the structures after an initial DFT calculation but before full computational analysis, they were screened based on the ε HOMO threshold. Of those evaluated, 39 nucleophiles met the criterion, and their CO_2_ affinities were predicted using the MLR model (see Scheme 5b). The predicted CO_2_ affinities of the top candidates identified for stable product formation are summarized in Table 1. Of these, five nucleophiles—11, 12, 38, 39, 41—were subjected to experimental validation. To balance the validation, three nucleophiles (38, 39, and 41) were selected that have high predicted CO_2_ affinities, while two (11 and 12) were calculated to have negative CO_2_ affinities in DMSO at 20 °C. Consistent with the predictions, nucleophiles 38, 39, and 41 reacted with CO_2_ to form the desired adducts (RCOO^−^) as detected by direct injection HRMS analysis (see the ESI). Nucleophiles 11 and 12 did not form adducts in agreement with our model. However, NMR identification of the detected adducts in DMSO was not possible, presumably due to poor stability in the experimental process, consistent with results from the Mayr/Ofial group (see the ESI for details).^[^ 19 ^]^ Therefore, we evaluated different conditions, wherein we found that when toluene was used as solvent at 80 °C that all three of the positively predicted nucleophiles yielded the desired carboxylated products (38% for 38, 35% for 39, and 33% for 41, confirmed by ^1^H NMR spectroscopy and HRMS (Table 1)). Quantum‐chemical calculations under the experimental conditions (toluene, 80 °C) likewise reproduced the observed outcome, predicting three stable and two unstable adducts. The identification of products in toluene could be attributed to the better stability of in‐situ generated carbanions.
Next, to further test the transferability of the predictions and to assess the robustness of the experimental protocol (DMSO, 20 °C), we examined two additional secondary carbanions, 1 (indene) and 92 (isochromanone). Compound 1 was a particularly interesting case, as quantum‐chemical calculations predicted it to be unstable although the HOMO energy is indicative of a stable structure (Table 1), whereas the ML workflow suggested stability. Prior results from the Mayr/Ofial group likewise indicated that 1 forms a stable adduct, which we were able to confirm experimentally. Compound 92, which differs structurally from the other nucleophiles and was suggested by the Ofial group in personal communication, was also tested. Here as well, a stable adduct was observed. Upon deprotonation with KO^t^Bu and subsequent reaction with CO_2_ in DMSO, the desired protonated adducts were successfully identified (see Table 1 and the Supporting Information).
To further contextualize these results, we established a correlation between kinetic barriers and reaction energies using Mayr's nucleophilicity scale to estimate the CO_2_ affinity of carbanions and potentially other nucleophiles.^[^ 18 ^]^ Indeed, we observed a modest correlation of the reactivity, expressed as nucleophilicity N, with the CO_2_ affinity (here, R ^2^ = 0.74). In short, higher nucleophilicity corresponds to lower kinetic barriers and more stabilized products, leading to increased CO_2_ affinity (see details in Section S8). Because the N parameter is available only for nucleophiles that have been kinetically characterized and are listed in Mayr's Reactivity Database, it cannot be incorporated into the model for design purposes. However, existing N values serve well as benchmarks.
In conclusion, we have developed a workflow for assessing the stability of CO_2_ adducts formed with carbon‐centered nucleophiles. Our approach employs a three‐parameter multivariate linear regression model that integrates both electronic and steric factors to accurately estimate the CO_2_ affinity. While the current study focused on a set of well‐characterized nucleophiles to establish a robust and reproducible modeling framework, the workflow is readily transferable to larger and more diverse libraries, including literature‐derived nucleophile candidates, as data availability and consistency improve. Future extensions of this framework are aimed at uncovering design‐relevant structural motifs that can guide the selection and modification of nucleophiles across a broader chemical space. Experimental validation of stable and unstable adduct formation reinforces the reliability of our model. The prediction workflow is user‐friendly for users with some experience in running Python‐based workflows that interface with quantum chemistry software, and requires only SMILES strings as input, making it a potentially useful tool for those planning CO_2_ addition experiments. By integrating the thermodynamic stability findings from this study with kinetic data from previous research, we can determine nucleophiles that are likely to successfully undergo carboxylation reactions from both kinetic and thermodynamic perspectives. This insight facilitates designing novel prodrugs and carbamates, thereby advancing pharmaceutical development and improving strategies for CO_2_ binding.
Supporting Information
The authors have cited additional references within the Supporting Information.^[^ 41, 42, 43, 44, 45, 46, 47 ^]^ Computational and experimental details are provided. The computationally optimized structures, and statistical modeling scripts of this communication are available at https://git.rz.tu‐bs.de/proppe‐group/co2_affinity_prediction.
Conflict of Interests
The authors declare no conflict of interest.
Supporting information
Supporting Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1G. Fiorani , W. Guo , A. W. Kleij , Green Chem. 2015, 17, 1375–1389, 10.1039/C 4GC 01959 H. · doi ↗
- 2Q. Liu , L. Wu , R. Jackstell , M. Beller , Nat. Commun. 2015, 6, 5933, 10.1038/ncomms 6933.25600683 · doi ↗ · pubmed ↗
- 3J. Artz , T. E. Müller , K. Thenert , J. Kleinekorte , R. Meys , A. Sternberg , A. Bardow , W. Leitner , Chem. Rev. 2018, 118, 434–504, 10.1021/acs.chemrev.7b 00435.29220170 · doi ↗ · pubmed ↗
- 4S. Dabral , T. Schaub , Adv. Synth. Catal. 2019, 361, 223–246, 10.1002/adsc.201801215. · doi ↗
- 5X.‐F. Liu , K. Zhang , L. Tao , X.‐B. Lu , W.‐Z. Zhang , Green Chem. Eng. 2022, 3, 125–137.
- 6A. Demessence , D. M. D'Alessandro , M. L. Foo , J. R. Long , J. Am. Chem. Soc. 2009, 131, 8784–8786, 10.1021/ja 903411 w.19505094 · doi ↗ · pubmed ↗
- 7D.‐H. Nam , O. Shekhah , G. Lee , A. Mallick , H. Jiang , F. Li , B. Chen , J. Wicks , M. Eddaoudi , E. H. Sargent , J. Am. Chem. Soc. 2020, 142, 21513–21521, 10.1021/jacs.0c 10774.33319985 · doi ↗ · pubmed ↗
- 8A. C. Forse , P. J. Milner , Chem. Sci. 2021, 12, 508–516, 10.1039/D 0SC 06059 C.PMC 817897534163780 · doi ↗ · pubmed ↗