The Diverse Neuromuscular Spectrum of VPS13A Disease
Anne Buchberger, Evamaria Riedel, Marie Hackenberg, Alexander Mensch, Stefanie Beck‐Woedl, Joohyun Park, Tobias B. Haack, Bernhard Haslinger, Jan Kirschke, Holger Prokisch, Andreas Hermann, Christian Mawrin, Adrian Danek, Benedikt Schoser, Kevin Peikert, Marcus Deschauer

TL;DR
This study explores the neuromuscular symptoms of VPS13A disease, revealing a wider range of severity than previously recognized.
Contribution
The study highlights the underrecognized neuromuscular spectrum in VPS13A disease and identifies a novel copy-neutral inversion variant.
Findings
Neuromuscular signs in VPS13A disease range from hyporeflexia to severe muscle wasting.
Electromyography showed chronic neurogenic changes in all patients.
Muscle MRI revealed fatty atrophy, particularly in the calves.
Abstract
VPS13A disease (chorea‐acanthocytosis) is a rare neurodegenerative disorder caused by biallelic variants in VPS13A, typically presenting with hyperkinetic movement disorders, while neuromuscular signs are often mild. The aim of the project was to investigate the frequency and severity of neuromuscular impairment in VPS13A disease. We systematically assessed the neuromuscular involvement in six patients with VPS13A disease. Our evaluation included genetic and clinical data, blood tests, electrophysiological studies, muscle MRI, and tissue samples from muscle and nerve. Age at clinical onset was 14 to 38 years (median: 37.5). Age at onset of paresis was 27 to 29 years (median: 29). Initial symptoms included seizures (5/6), hyperkinesia (2/6), and muscle weakness (1/6). Neuromuscular signs ranged from hyporeflexia (5/6) to progressive muscle wasting (3/6). Nine VPS13A variants were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
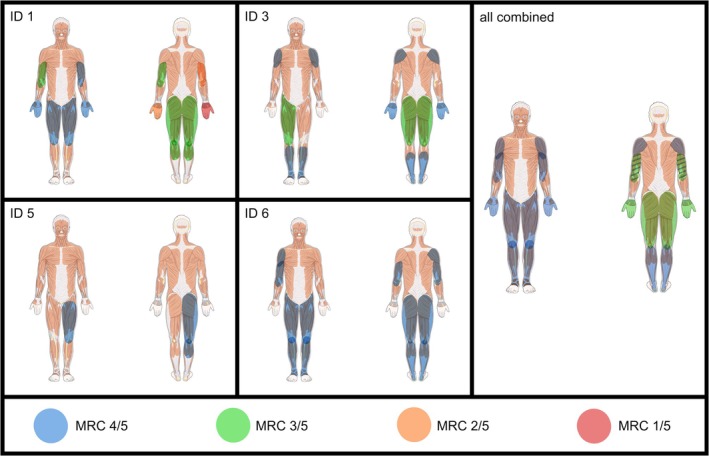 FIGURE 1
FIGURE 1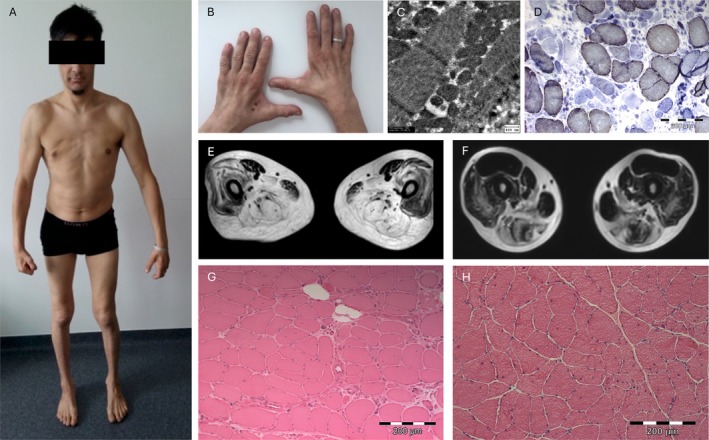 FIGURE 2
FIGURE 2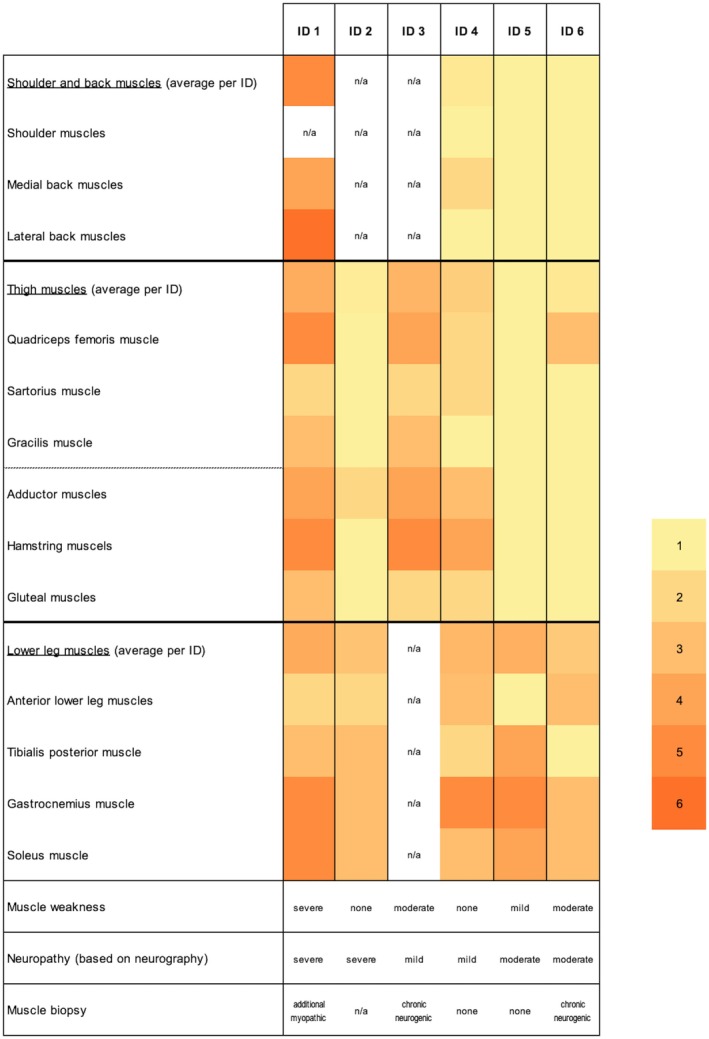 FIGURE 3
FIGURE 3| ID | Age at onset (years) | Age at onset of muscle weakness (years) | Age at last examination (years) | Symptoms of onset | Psychiatric findings | Muscle weakness (years since onset of muscle weakness) | Ambulatory (non‐ambulatory onset age [years]); devices | Muscle atrophy | Signs of peripheral neuropathy | Neuromuscular syndrome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 29 | 29 | 45 | Seizures, progressive paresis | Impulse control disorder |
Severe: distal UL 1/5, proximal UL and LL 3/5 (16) |
No (32); wheelchair |
Severe: proximal and distal UL, proximal LL | Areflexia | ALS mimic |
| 2 | 38 | n/a | 42 | Seizures, orofacial hyperkinesia | Impulse control disorder | None | Yes | None | Areflexia, reduced vibration sense (4–5/8) | Subclinical polyneuropathic syndrome |
| 3 | 23 | 27 | 37 | Seizures, low physical endurance |
Moderate: proximal UL 4+/5 proximal LL 4−/5 distal LL 4+/5 (10) |
Yes; foot orthotics wheelchair (part‐time) |
Severe: proximal LL Mild: distal LL | Areflexia, reduced vibration sense (5/8) | Limb‐girdle syndrome | |
| 4 | 14 | n/a | 32 | Seizures | Reduced attention span, disinhibition, psychosis | None | Yes | None | Reduced vibration sense (4–5/8) | Subclinical polyneuropathic syndrome |
| 5 | 18 | 29 | 31 | Seizures | Memory impairment, disinhibition |
Mild: proximal LL 4+/5 distal LL 4+/5 (2) | Yes | Mild: proximal UL | Areflexia | Motor polyneuropathic syndrome |
| 6 | 25 | 29 | 38 | Motor restlessness | Reduced attention span, disinhibition |
Moderate: proximal UL 4/5 proximal LL 3/5 distal LL 4/5 (9) |
No (34); wheelchair | None | Areflexia | Limb‐girdle syndrome |
| ID | Genotype (variant first allele/variant second allele) | Chorein (RBC) | ACA (%) | CK range (U/L) | NfL (pg/mL) | NCS | EMG | Affected muscles in MRI | Muscle biopsy (age at biopsy) | ECG | Echo | Bicaudate index in MRI | EEG |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.8190_8191dup | Absent | 41 | 900–5083 | n/a | Axonal sensorimotor |
Neurogenic Myopathic | Shoulder, back, thighs, posterior lower legs |
Neurogenic Myopathic 40% COX‐deficiency (35) | Left axis deviation | LVEF mildly reduced | 1.57 | Generalized slowing |
| 2 | c.8743G>T/c.5171T>G | Reduced | 14 | 587–1643 | n/a | Axonal sensorimotor | Neurogenic | Medial thighs, lower legs | n/a | Normal | n/a | 1.52 | Normal |
| 3 | c.6059del/chr9:g.79925985_79932966inv | Absent | 20–40 | 2490‐12,420 | 29 | Axonal sensory (ulnar and median nerve only) | Neurogenic | Gluteus, thighs |
Neurogenic 2%–3% COX‐deficiency (25) | Sinus arrhythmia | Normal | 1.64 | Signs of drowsiness |
| 4 | c.6059del/chr9:g.79925985_79932966inv | Absent | 20–40 | 2538‐2817 | 16.5 | Axonal motor (peroneal nerve only) | Neurogenic | Back, legs, posterior>anterior |
Structurally normal Minimal mitochondrial accumulation (15) | Pathological waves in II, aVL | Normal | 1.61 | Generalized seizure, focal onset |
| 5 | c.283+1G>A | Absent | 18 | 608–1061 | n/a | Axonal sensorimotor | Neurogenic | Anterior thigh, posterior lower legs |
Normal (26) | Sinus brady‐cardia, left axis deviation | Normal | 1.61 | Mild signs of drowsiness |
| 6 | c.556‐290_697‐483del/c.990‐272_1161+1379del | Reduced | + | 498–1442 | 33 | Axonal sensorimotor | Neurogenic | Lower legs |
Neurogenic COX‐deficiency (35) | Normal | Normal | 1.57 | Normal |
| ID | NCS motor nerves (mV) | NCS sensory nerves (μV) | EMG | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median left/right | Ulnar left/right | Radial left/right | Tibial left/right | Peroneal left/right | Median left/right | Ulnar left/right | Radial left/right | Sural left/right | Neurogenic | Myopathic | PSA (intensity) | |
| 1 | 7.6 | 0.5/No signal | 5.3 | 7.1 | Normal | 8.9/7.0 | 6.5/7.3 | 6.4 | Normal |
Proximal, distal UL Proximal, distal LL | Proximal, distal UL | Distal UL (+) |
| 2 | 7.7 | Normal | Normal | 5.4/3.5 | 3.8/5.1 | 6.3 | 4.1 | n/a | Normal |
Proximal, distal UL Proximal, distal LL | (Sporadic) |
Proximal (+), distal UL (+) Proximal (+), distal LL (++) |
| 3 | Normal | Normal | n/a | Normal | Normal | Normal | 9.7/11.2 | n/a | Normal |
Proximal UL Proximal, distal LL | None | None |
| 4 | Normal | Normal | n/a | Normal | 4.9 | Normal | Normal | n/a | Normal |
Proximal, distal UL Proximal, distal LL | None |
Distal UL (+) Distal LL (++) |
| 5 | 6.8 | 7.3 | n/a | 8.8 | 2.3 | 7.1 | 9.1 | n/a | 5 |
Proximal, distal UL Proximal, distal LL | None | None |
| 6 | Normal | Normal | n/a | 8.4 | 5.7/3.8 | 9.1 | 4.2 | n/a | Normal | Proximal, distal LL | None | None |
| ID | Muscle | Neurogenic | Myopathic | Lipid staining (Oil Red, Sudan Orange) | COX deficiency (%) | Respiratory chain activities | Electron microscopy | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Angulated atrophic fibers | Atrophic fiber grouping | Fiber type grouping | Fiber type predominance | Muscle fiber necrosis | Regenerating fibers | Internalized nuclei (%) | Fiber size variation | Endo−/peri‐mysial fibrosis | ||||||
| 1 | Vastus lateralis | + | + | − | + (type I) | + (5%) | + | + (2%) | + | + | Multiple fibers with lipid storage | 40 | Mildly reduced I and IV activities | Structurally normal mitochondria, autophagic vacuoles, lipid deposits |
| 3 | Vastus medialis | + | + | + | + (type II) |
+ (6%) (myo‐phago‐cytes) | + | − | + | − | Normal | 2–3 | n/a | n/a |
| 4 | Thigh | − | − | − | − | − | − | − | − | − | n/a | − | Normal | n/a |
| 5 | Gastro‐cnemius | − | − | − | − | − | − | − | − | − | n/a | n/a | n/a | n/a |
| 6 | Gluteus medius | + | − | + | − | − | − | + | + | − | Normal | < 10 | Normal |
Structurally normal mitochondria, few small autophagic vacuoles |
- —Advocacy for Neuroacanthocytosis Patients10.13039/100012178
- —EMINA network
- —Else Kröner‐Fresenius‐Stiftung10.13039/501100003042
- —German Research Foundation (Deutsche Forschungsgemeinschaft) (DFG)10.13039/501100001659
- —European Research Council (ERC)
- —Federal Ministry of Education and Research (BMBF)
- —Hermann and Lilly Schilling‐Foundation (“Hermann und Lilly Schilling‐Stiftung für medizinische Forschung”)
- —Federal Ministry of Education and Research (BMBF)/VDI
- —Hertie Network of Excellence in Clinical Neuroscience
- —TUM clinician scientist program KKF
- —Advocacy for Neuroacanthocytosis Patients10.13039/100012178
- —EMINA network
- —Else Kröner‐Fresenius‐Stiftung10.13039/501100003042
- —German Research Foundation (Deutsche Forschungsgemeinschaft) (DFG)10.13039/501100001659
- —European Research Council (ERC)
- —Federal Ministry of Education and Research (BMBF)
- —Hermann and Lilly Schilling‐Foundation (“Hermann und Lilly Schilling‐Stiftung für medizinische Forschung”)
- —Federal Ministry of Education and Research (BMBF)/VDI
- —Hertie Network of Excellence in Clinical Neuroscience
- —TUM clinician scientist program KKF
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeurogenetic and Muscular Disorders Research · Genetic Neurodegenerative Diseases · Blood disorders and treatments
Introduction
1
VPS13A disease, formerly referred to as chorea‐acanthocytosis, is an autosomal recessive disorder due to biallelic variants in VPS13A, which encodes for VPS13A (chorein) [1, 2]. The disease mechanism is loss of function [3]. While the detailed pathomechanism is under investigation, it is known that VPS13 proteins are involved in bulk lipid transfer at membrane contact sites [4], with VPS13A binding the endoplasmic reticulum to both mitochondria and lipid droplets as well as to the plasma membrane [4, 5, 6, 7, 8]. Besides possible impairment of lipid transport and mitochondrial function, there is evidence for regulatory functions of VPS13A in the cytoskeleton and for cell survival via two pathways, the Lyn kinase and the PI3K pathway, that affect membrane protein phosphorylation and cortical actin expression [9]. Affected individuals characteristically, but not necessarily, show red blood cell acanthocytosis in association with neurological dysfunction [3, 10]. Although clinical findings are typically related to degeneration of the basal ganglia and involve both hyper‐ and hypokinetic movement disorders, including generalized chorea and orofaciolingual dystonia [3, 10], phenotypes can vary drastically. Clinical manifestation may also range from behavioral and cognitive changes to epilepsy [3, 9]. Mild neuromuscular involvement, characterized by elevated phosphocreatine kinase (CK) levels and areflexia, is common [3]. In addition, muscle weakness, fasciculations, and atrophy may occur, with lower limb‐girdle muscular weakness observed in seven of nine cases of a recent study [11]. While one earlier case was described as mimicking motor neuron disease [12], there was evidence of myopathy in others [11, 13]. Individuals suffering from the closely related X‐linked McLeod syndrome (XK disease) [10, 11, 14, 15] appear to show myopathy more often [16], although prominent neurogenic changes occur in patients with XK disease as well [15, 17] and may be considered similarly pronounced as in VPS13A disease [17].
Given the sparse and heterogeneous data available so far, there is a critical need for a systematic exploration of the nature and severity of clinical, paraclinical, imaging, and myopathological neuromuscular involvement in VPS13A disease. Here, we report six cases of VPS13A disease presenting a wide range of neuromuscular involvement, including three patients with moderate to severe paresis. Our study highlights the importance of thorough and repeated phenotyping to ensure early diagnosis, optimal disease management, and to enhance understanding of the underlying pathophysiological mechanisms of VPS13A disease.
Patients and Methods
2
Six biological males (five German, one Turkish) with genetically confirmed VPS13A disease were recruited from the University Hospitals of Munich (TUM and LMU) and Rostock and systematically assessed for neuromuscular involvement, regardless of clinically manifest neuromuscular symptoms. Two patients were siblings (IDs 3, 4). Genetic and clinical details of one individual (ID 6) and the cardiac manifestation of three patients (IDs 1, 3, 4) are already on record [18, 19]. All patients gave informed written consent to the scientific use of their clinical and paraclinical data (ethics vote: Biobank Munich 9/15 s; Rostock A 2022‐0058). Clinical, paraclinical, and genetic data were collected during the diagnostic process. Chorein (VPS13A) Western blot in red blood cell (RBC) membranes and acanthocyte counts (wet blood smear) were performed as previously described [20, 21]. Muscle magnetic resonance imaging (MRI) scans were performed on all patients using 3T Philips Ingenia MRI scanners. T2 Dixon sequences [22] were generated, producing in‐phase, out‐of‐phase, fat‐only, and water‐only images, along with quantitative images for assessing muscle infiltration in the thighs. The Morrow scale [23] was employed for evaluating edema, while the Mercuri scale [24] was used for grading fatty infiltration. To derive a disease‐characteristic muscle pattern with respect to our patients' progression stages, we added the two scales to create a combined severity score (1: no involvement; 6: severe involvement). Muscle biopsies (IDs 1, 3, 4, 5) and one sural nerve biopsy (ID 1) were taken during the initial work‐up for CK elevation or muscle weakness, and in one case (ID 6) during surgery for hip arthrosis in the context of known VPS13A disease. Available slides had been stained for hematoxylin and eosin (H&E), Trichrome‐Gomori (IDs 1, 3, 4, 6), and lipid staining (Oil Red or Sudan Orange; IDs 1, 3, 6). In one case (ID 5), new sections from frozen material were prepared and stained accordingly. Immunohistochemistry, including adenosine triphosphatase (ATPase) and cytochrome c oxidase/succinate dehydrogenase (COX/SDH) (IDs 1, 3, 4, 6), respiratory chain analysis (IDs 1, 4, 6), and electron microscopy (IDs 1, 6) were examined where available. Cryopreserved muscle samples were used to assess the respiratory chain activities spectrophotometrically as previously published for IDs 1, 4 [25] and 6 [26]. Statistical data analysis and visualization were performed using RStudio version 1.0.153.
Results
3
Clinical, Paraclinical, and Genetic Assessment
3.1
Clinical, paraclinical, and genetic findings are summarized in Tables 1 and 2. All individuals were biologically male, all but one (ID 2, Turkish) of German heritage. Age at onset ranged from 14 to 38 (median = 23; SD = 8) years. Age at the last examination ranged from 31 to 45 (median = 38; SD = 5) years. First presenting signs were seizures (5/6; IDs 1–5), hyperkinesia or motor restlessness (2/6; IDs 2, 6), and muscle weakness (1/6; ID 1). Age at onset of neuromuscular signs was 27 to 29 (median = 29; SD = 1) years.
Regarding neuromuscular symptoms, 4/6 reported muscle weakness, while none had sensory complaints. Concerning neuromuscular signs, 4/6 (IDs 1, 3, 5, 6) had mild (1/4; ID 5), moderate (2/4; IDs 3, 6), or severe (1/4; ID 1) muscle weakness, leading to gait impairment in 3/4 (IDs 1, 3, 6; for visualization of pattern and severity of paresis, see Figure 1). Trendelenburg's sign was positive in 2/6 (IDs 3, 6). Gower's sign was observed in half of the cohort (3/6; IDs 1, 3, 6). Split hand sign was present in 1/6 (ID 1) (Figure 2). Hints of the “rubber person gait” were visible in 1/6 (ID 3), but could not be easily differentiated from the paresis. DTR were absent or reduced in 4/6 (IDs 1, 2, 3, 5, 6). Clinical assessment of sensory functions included light touch, vibration, pain, temperature, and proprioception, whereby reduced sense of vibration in 4/6 (IDs 1–4) was the only sign of sensory neuropathy.
Pattern and severity of paresis in the individual patients (IDs 1, 3, 5, 6; unaffected not shown), and schematic representation of the overall paresis distribution in this VPS13A disease cohort (all combined). Paresis is indicated by color overlay. For the combined pattern, the median MRC score was calculated across all affected muscle groups. MRC = Medical Research Council grading of muscle strength. The artwork used in this figure was adapted from Servier Medical Art (http://https://smart.servier.com/). Servier Medical Art by Servier is licensed under Creative Commons Attribution 4.0. Alterations were color overlay and resizing.
Neuromuscular manifestations of VPS13A disease. Generalized muscle atrophy with severe atrophy of the small hand muscles with split hand sign in one individual (ID 1) (A, B). Muscle MRI of the same patient showed severe fatty degeneration of the posterior thigh muscles (E), similarly seen in another representative patient (ID 3) (F). The muscle biopsy of one patient (ID 1) revealed neurogenic and vacuolar myopathic changes with moderate hypertrophic fibers and mild internalized nuclei in H&E staining (G), multiple COX‐negative fibers in COX/SDH staining (predominantly affecting atrophic fibers, not distributed in a mosaic pattern) (D), and subsarcolemmal accumulated mitochondria of variable sizes, but normal ultrastructure in electron microscopy (C). A muscle biopsy of patient ID 6 showed neurogenic changes with grouping of atrophic and scattered angulated atrophic fibers in H&E staining. Additionally, internalized nuclei were mildly increased (H).
The most pronounced sensory involvement was present in cases with mild muscle weakness (IDs 2, 4). In contrast, the patient with the most severe paresis (ID 1) exhibited only a slightly reduced sense of vibration.
Biallelic pathogenic and/or likely pathogenic VPS13A variants were detected in 6/6 (Table 2). Six of the nine detected variants were novel. Four cases were sporadic, and two were brothers (IDs 3, 4). Most patients (5/6) carried biallelic null variants (IDs 1, 3, 4, 5, 6). One individual (ID 1) carried a novel duplication in a homozygous state. The two brothers (IDs 3, 4) were compound heterozygotes for a previously described deletion [12, 27, 28], and a novel copy‐neutral inversion spanning exons 36 to 40 that disrupts the open reading frame. The intronic inversion breakpoints were missed by exome sequencing and detected only by genome analysis. In 1/6 (ID 5), two novel splice‐site variants were identified. One individual (ID 6) carried a previously reported deletion of exons 8 and 9 [27], together with a compound heterozygous novel deletion affecting exon 13, which was published upon discovery [18]. In only 1/6 (ID 2), biallelic missense variants were identified. Both variants were novel and classified as likely pathogenic based on multiple lines of evidence, including functional studies showing markedly reduced chorein levels on Western blot, extremely low frequency in control population databases, and computational predictions supporting a deleterious effect (loss of a splice site for variant 1).
Western blot confirmed the diagnosis on the protein level in all patients (6/6) and acanthocytes were significantly increased (6/6; Table 2). CK levels were elevated in 6/6 (498–12,420 U/L; median of highest values: 2230 U/L). Neurofilament light chain (NfL) levels were elevated in 3/3 (16.5–33 pg/mL; median: 29 pg/mL; normal range: < 12 pg/mL; IDs 3, 4, 6).
Electrophysiology
3.2
NCS and EMG studies were performed in all patients (6/6, Table 3). Sensory neurography was compatible with neuronopathy or axonal neuropathy in 4/6 (IDs 1, 2, 3, 5, 6), most pronounced in the ulnar nerve. Similarly, motor neurography was compatible with neuronopathy or axonal neuropathy in 4/6 (IDs 1, 2, 4, 5, 6), most frequently in the tibial and peroneal nerves.
In EMG, all patients (6/6) showed chronic neurogenic changes with high amplitudes, polyphasic potentials, and reduced interference patterns. In 5/6 (IDs 1–4, 6), this equally affected proximal and distal muscles. Additional myopathic changes with short and small potentials and early recruitment were detected in 1/6 (ID 1). Pathological spontaneous activity (positive sharp waves and/or fibrillations) was present in 3/6 (IDs 1, 2, 4) with equal severity in proximal and distal muscles.
Comparing NCS findings with clinical signs, in cases with moderate to severe sensorimotor neuropathy (IDs 1, 2, 5, 6), 2/4 (IDs 1, 2) presented with reduced vibration sense as the only sensory impairment. Additionally, 2/4 showed no (ID 2) or very mild (ID 5) muscular weakness (MRC 4+/5).
MRI
3.3
MRI scans of the shoulder and/or back muscles were available for 4/6 (IDs 1, 4, 5, 6), thigh muscles for 6/6, and lower leg muscles for 5/6 (IDs 1, 2, 4, 5, 6). To assess muscle involvement patterns, edema (Morrow score 0–2) and fatty degeneration (Mercuri score 1–4) were evaluated and combined into a combined severity score (1–6; Figure 3). The calf muscles were most frequently impaired (5/5; IDs 1, 2, 4, 5, 6) and showed the highest combined severity score (gastrocnemius muscle: 3–5, median 5; soleus muscle: 3–5, median 3; n = 5). They also represented the predominant imaging changes in 2/2 patients with mild MRI findings (IDs 2, 6). The gracilis muscle appeared relatively spared (2/6; IDs 1, 3).
Muscular involvement in MRI according to a combined severity score based on the sum of the Morrow scale [23] for edema and the Mercuri scale [24] for fatty infiltration (1: no involvement; 6: severe involvement). Muscle involvement in MRI is visualized in a heatmap and in relation to other clinical and paraclinical findings. Colors represent the extent of involvement of each muscle by patient. The average extent of muscle involvement by muscle group is shown as a mean colored value. Clinical muscle weakness, neuropathy in NCS, and muscle biopsy results are quantified from normal to severe by patient. MRI = magnetic resonance imaging; n/a = not available; NCS = nerve conduction studies.
Comparing MRI findings with clinical assessments, the prominent involvement of the anterior (4/6; IDs 1, 3, 4, 6) and posterior (3/6; IDs 1, 3, 4) thigh muscles on MRI corresponded with the clinical presentation. In the lower legs, while imaging revealed frequent and severe involvement (5/5), muscular weakness was only mild. In contrast, 6/6 displayed changes in the EMG and NCS of the lower legs, matching the imaging results.
Muscle and Nerve Biopsy
3.4
Myopathological analysis was available for 5/6 (IDs 1, 3–6; Table 4; Figure 2). In 3/5 (IDs 1, 3, 6), neurogenic changes were detected, including angulated atrophic fibers (3/3; IDs 1, 3, 6), atrophic fiber grouping (2/3; IDs 1, 3), fiber type grouping (2/3; IDs 3, 6), and fiber type predominance (2/3; IDs 1, 3), of both type 1 (ID 1) and type 2 (ID 3) fibers. Occasionally, muscle fibers in various stages of necrosis (5%–6%) and regenerating fibers were seen (2/5; IDs 1, 3). Additional myopathic features such as internalized nuclei and fiber size variation were present in 2/5 (IDs 1, 6) and 3/5 (IDs 1, 3, 6), respectively. In 2/3 (IDs 3, 6), those findings were accompanying marked chronic neurogenic changes and were therefore most likely secondary to denervation. In 1/6 (ID 1), myopathic changes including fiber size variation (9–125 μm diameter; normal: 40–80 μm) with hypertrophic fibers, internalized nuclei (2%), and endo‐ and perimysial fibrosis with fatty replacement were more prominent. Lipid staining showed multiple fibers with lipid storage in 1/3 (ID 1). COX deficiency was present in 3/5 (IDs 1, 3, 6). Respiratory chain activity was normal in 2/3 (IDs 4, 6) and demonstrated nonspecific reduction of complex I and IV activities in 1/3 (ID 1). Electron microscopy revealed structurally normal mitochondria in 2/2 (IDs 1, 6; Figure 2), some autophagic vacuoles and lipid deposits (1/2; ID 1).
All individuals with myopathological changes showed clinical muscle weakness, with the most prominent biopsy abnormalities found in the clinically most severely affected case (ID 1). In the two cases with no or mild paresis (IDs 4, 5), no significant myopathological changes were observed.
Histopathological analysis of the sural nerve was available for one individual (ID 1) and showed no significant changes.
Discussion
4
Our findings further contribute to understanding the neuromuscular phenotype of VPS13A disease. The spectrum of neuromuscular involvement in our cohort ranged from subclinical changes, which we defined as abnormalities in (para‐)clinical assessments without perceived neuromuscular symptoms, to severe atrophic paresis. Four of six patients showed at least mild paresis, slightly emphasized on the dorsal lower limb girdle, with age at onset of muscle weakness between 27 and 29 years, while two remained only subclinically affected. Muscular weakness in VPS13A disease has been previously reported [3, 11, 12], typically as lower limb‐girdle muscular weakness with onset age ranging from 29 to 53 years [11], with sporadic involvement of distal lower limbs [11, 12]. Isolated reports of severe muscle weakness included one mimicking motor neuron disease [12] and one with initial misdiagnosis of inflammatory myopathy [29], although diagnosis in these cases was not confirmed by Western blot. In our cohort, muscle weakness was one of the most debilitating symptoms in half of the patients. Upper limbs were affected up to split hand sign, which is typically associated with amyotrophic lateral sclerosis [30], proving involvement far beyond the lower limb girdle. Amplitude reduction in sensorimotor ENG was common, reinforcing the previously documented prevalence in literature [3, 11, 31, 32], and highlighting the presence of subclinical neuromuscular changes in patients without neuromuscular symptoms. We found no evidence for reduced nerve conduction velocity, as reported in a small cohort of three [33]. While in earlier studies, EMG changes were solely neurogenic [31], we observed additional myopathic patterns, which have been less recognized [11, 33]. The range of hyperCKemia from ~500 U/L to ~12,400 U/L notably exceeded earlier reports of CK elevations between 500 U/L and 5000 U/L [11], suggesting a broader spectrum of CK changes in VPS13A disease. Muscle MRI consistently demonstrated fatty degeneration or edema, particularly in the calf muscles. While systematic imaging data of VPS13A disease patients is lacking, some heterogeneous involvement of lower leg muscles has been described, with sparing of the soleus muscle reported in one case [34]. As sparing of the soleus muscle was not confirmed in our larger cohort and the anterior and posterior thigh muscles showed regular changes, we were able to revise the pattern.
Molecular genetic testing revealed six novel VPS13A variants. These included one frameshift variant, two missense variants, two splice site variants, and a copy‐neutral inversion. Exome sequencing often fails to detect structural variants with intronic breakpoints, as demonstrated in our case. In contrast, genome sequencing offers a more comprehensive view, enabling the detection of complex structural variants. By identifying the first reported VPS13A inversion, our findings underscore the superior sensitivity of genome sequencing in detecting disease‐causing structural variants and expand the known mutational spectrum in VPS13A disease. The inversion resulted in a lack of chorein in Western blot analysis in both brothers. However, significant muscle weakness was present in only one of them, indicating no genotype–phenotype correlation in this family. Matching observations were made in siblings with homozygous splice site variants, where only one sibling showed marked muscle weakness [11]. Similar to the homozygous frameshift variant detected in our most severely affected patient, most VPS13A disease‐associated variants are predicted to be loss‐of‐function variants [3] regardless of neuromuscular findings, while missense variants are rare [35]. Thus, no conclusions about a “muscle genotype” can be drawn. In line with this, no clear genotype–phenotype correlation has yet been identified in VPS13A disease [3].
Our cohort of six patients of this rare disease was exclusively male. Given the autosomal recessive mode of inheritance, a more balanced sex distribution would be expected, as observed in other studies.
Regarding the pathomechanism of neurogenic changes, our electrophysiological findings suggest either a neuronopathy or proximally emphasized axonopathy independent of nerve length. Motor nerves were considerably more affected in the lower extremities, while sensory damage was more common in the upper extremities. The sural nerve was notably spared, which has not been reported in VPS13A disease so far. Sural sparing is traditionally associated with immune‐mediated neuropathy [36, 37], with single reports in cases of hereditary neuropathy [38, 39]. One explanation for sural sparing neuropathies is the more proximal measurement site of the sural nerve, reducing sensitivity to distal pathologies [40]. Alternatively, its anatomical protection may limit pressure‐induced damage to the blood‐nerve barrier and subsequent immune‐mediated impairment [41]. Since we observed length‐independent neuropathy without conduction blocks in our cohort, we cannot confirm either theory. In line with our data, sural nerve biopsies showed no significant pathology [28], while in phenotypically [14] and likely pathologically [42] related XK disease, nerve pathology supported axonal sensorimotor neuropathy [17]. The pathophysiological pathway of VPS13A disease likely involves bulk lipid transfer [10, 43]: Since VPS13A has been shown to bind at membrane contact sites between the endoplasmic reticulum and both mitochondria and lipid droplets as well as the plasma membrane (where it forms a complex with XK [7, 8, 44]), biallelic variants in VPS13A may lead to impairment of lipid transfer and possibly mitochondrial dysfunction [5, 6, 45, 46]. Pluripotent stem cell‐derived midbrain/hindbrain neurons from VPS13A disease patients showed a distinct mitochondrial trafficking phenotype in proximal axons, suggesting a proximal axonopathy mechanism [47]. As length‐independent axonal impairment may also occur in primary neuronopathy with secondary axonal damage, neuropathological data of spinal neurons are currently lacking to determine the primary site of damage and definitively distinguish between neuronopathy and proximal axonopathy. We confirmed that elevated NfL levels are consistently observed in VPS13A disease, aligning with previous reports of elevated NfL in VPS13A and XK disease [48], therefore supporting the presence of neuro‐axonal damage. Although not specific, this may aid in the diagnostic process, particularly in cases without overt neuromuscular symptoms. However, further studies are needed to explore the potential of NfL as a biomarker for prognosis, correlation with disease course, and phenotypic stratification.
In addition to the evident neurogenic changes, myopathic signs were also present. In half of our cohort, CK levels were highly elevated (above 10‐fold), which is unusual for a neurogenic process. Proximally emphasized muscle weakness with Gower's sign and myopathic EMG changes were indicative of myopathy. One patient's muscle biopsy revealed marked myopathic changes with internalized nuclei and fiber size variation, as well as multiple fibers with lipid storage in Oil Red staining, which has not yet been reported in VPS13A disease but is typical for lipid storage myopathies [49]. In the second most affected individual, subtle myopathic changes accompanied a chronic neurogenic pattern, consistent with secondary myopathic changes after denervation. Lipid staining was normal in this case. Clinical and histopathological changes in VPS13A disease have been previously described as predominantly neurogenic [12, 31, 50]. Nonetheless, some overlapping neuropathic and myopathic muscle biopsy changes have been observed [11, 28, 50, 51, 52, 53, 54]. Myopathic features, such as internalized nucleation and fiber splitting, have been postulated to be secondary to chronic denervation [28, 50, 52, 53]. Other studies interpreted myopathic biopsy patterns as primary [11, 13, 51, 54], for example, due to the detection of nemaline rods in myofibers or the observation of whorled and bizarrely shaped fibers [11, 13, 51]. In our cohort, neurogenic changes were more frequent than myopathic findings, which is compatible with secondary changes in the context of a neurogenic pathology. However, a single patient showed changes that could be indicative of a primary myopathy. The increased lipid storage and marked increase of COX‐deficient fibers in the muscle of this patient with severe atrophic paresis is striking. While lipid accumulation and increase of COX‐negative fibers may suggest bulk lipid transfer and/or mitochondrial dysfunction, our electron microscopy and respiratory chain activities did not provide clear evidence. COX‐deficiency was mainly seen in atrophic fibers without the typical mosaic pattern of mitochondrial myopathy. Therefore, we could not histopathologically confirm a causative mitochondrial or bulk lipid transport disorder in VPS13A disease in muscle. However, we cannot fully exclude the coexistence of a second competing pathology such as lipid storage myopathy or mitochondrial myopathy in this patient, although exome sequencing revealed no indicative variants. In XK disease, similar observations were made: A series of 10 patients found neurogenic changes in all cases, but less prominent myopathic changes in only four cases, which might therefore be classified as secondary to the neurogenic process; however, highly elevated CK levels in some patients and rhabdomyolysis in one patient pointed towards an additional myopathy [17].
Conclusion
5
In summary, neuromuscular involvement in VPS13A disease ranges from subclinical changes in NCS, EMG, and MRI to severe atrophic paresis. It is characterized by length‐independent predominant motor neuropathy or neuronopathy, and in severe cases, additional myopathic features may also be observed. Similar to XK disease [17], VPS13A disease frequently leads to muscle weakness. It should therefore not be considered solely a primary movement disorder but a multidimensional condition encompassing a spectrum of neurological signs with varying manifestations, even with muscle weakness as the first and most prominent clinical sign. Thorough phenotyping regarding neuromuscular signs is crucial to support early diagnosis and optimal clinical management of VPS13A disease. Furthermore, understanding the natural history of neuromuscular involvement, which seems to be slowly progressing with frequent clinical manifestation in the late second life decade, is essential for identifying meaningful subgroups and potential biomarkers, ultimately facilitating a comprehensive understanding of the condition and supporting trial readiness.
Author Contributions
A.B. contributed to the conception of the project, acquisition and interpretation of data, analysis of data, and the drafting, editing, and revision of the manuscript. E.R. contributed to the acquisition of data, analysis and interpretation of data, and the drafting and revision of the manuscript. M.H. contributed to the acquisition of data, analysis and interpretation of data, and the drafting and revision of the manuscript. A.M., S.B.‐W., J.P., T.B.H., B.H., J.K., H.P., A.H., and C.M. contributed to the acquisition of data, and the editing and revision of the manuscript. A.D. contributed to the acquisition of data, interpretation of data, and the editing and revision of the manuscript. B.S. contributed to the acquisition of data, analysis and interpretation of data, and the editing and revision of the manuscript. K.P. contributed to the conception of the project, acquisition of data, interpretation of data, and the editing and revision of the manuscript. M.D. contributed to the conception and supervision of the project, acquisition of data, analysis and interpretation of data, and the editing and revision of the manuscript. I.C. contributed to the conception and supervision of the project, acquisition of data, analysis and interpretation of data, and the drafting, editing, and revision of the manuscript. All authors discussed the results and approved the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1L. Rampoldi , C. Dobson‐Stone , J. P. Rubio , et al., “A Conserved Sorting‐Associated Protein Is Mutant in Chorea‐Acanthocytosis,” Nature Genetics 28, no. 2 (2001): 119–120.11381253 10.1038/88821 · doi ↗ · pubmed ↗
- 2S. Ueno , Y. Maruki , M. Nakamura , et al., “The Gene Encoding a Newly Discovered Protein, Chorein, Is Mutated in Chorea‐Acanthocytosis,” Nature Genetics 28, no. 2 (2001): 121–122.11381254 10.1038/88825 · doi ↗ · pubmed ↗
- 3K. Peikert , C. Dobson‐Stone , L. Rampoldi , et al., “VPS 13A Disease,” in Gene Reviews(®), ed. M. P. Adam , J. Feldman , G. M. Mirzaa , et al. (University of Washington, Seattle, 2002).
- 4M. Hanna , A. Guillén‐Samander , and P. De Camilli , “RBG Motif Bridge‐Like Lipid Transport Proteins: Structure, Functions, and Open Questions,” Annual Review of Cell and Developmental Biology 16, no. 39 (2023): 409–434.10.1146/annurev-cellbio-120420-01463437406299 · doi ↗ · pubmed ↗
- 5N. Kumar , M. Leonzino , W. Hancock‐Cerutti , et al., “VPS 13A and VPS 13C Are Lipid Transport Proteins Differentially Localized at ER Contact Sites,” Journal of Cell Biology 217, no. 10 (2018): 3625–3639.30093493 10.1083/jcb.201807019 PMC 6168267 · doi ↗ · pubmed ↗
- 6M. Leonzino , K. M. Reinisch , and P. De Camilli , “Insights Into VPS 13 Properties and Function Reveal a New Mechanism of Eukaryotic Lipid Transport,” Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids 1866, no. 10 (2021): 159003.34216812 10.1016/j.bbalip.2021.159003 PMC 8325632 · doi ↗ · pubmed ↗
- 7A. Guillén‐Samander , Y. Wu , S. S. Pineda , et al., “A Partnership Between the Lipid Scramblase XK and the Lipid Transfer Protein VPS 13A at the Plasma Membrane,” Proceedings of the National Academy of Sciences of the United States of America 119, no. 35 (2022): e 2205425119.35994651 10.1073/pnas.2205425119 PMC 9436381 · doi ↗ · pubmed ↗
- 8J. S. Park , Y. Hu , N. M. Hollingsworth , G. Miltenberger‐Miltenyi , and A. M. Neiman , “Interaction Between VPS 13A and the XK Scramblase Is Important for VPS 13A Function in Humans,” Journal of Cell Science 135, no. 17 (2022): jcs 260227.35950506 10.1242/jcs.260227 PMC 9482346 · doi ↗ · pubmed ↗