Presentation, Diagnosis, and Acute Treatment of Secondary Hemophagocytic Lymphohistiocytosis: A Case Report
Alexandra Bartholomew, Michael Connick, Catherine Loehr, Shane Sanne

TL;DR
This case report describes the presentation, diagnosis, and treatment of a rare immune disorder called secondary hemophagocytic lymphohistiocytosis in a 53-year-old woman.
Contribution
The paper provides a detailed clinical case to improve understanding and management of secondary HLH.
Findings
The patient exhibited symptoms like fever and fatigue, leading to a diagnosis of secondary HLH.
Clinical and laboratory criteria were used to confirm the diagnosis.
The case highlights the importance of recognizing HLH for timely treatment.
Abstract
Secondary hemophagocytic lymphohistiocytosis (HLH) is a rare, immunologically driven disorder with a high mortality rate. It is typically diagnosed on the basis of clinical and laboratory criteria. We present the case of a 53-year-old woman with multiple previous hospitalizations for fever of unknown origin and fatigue. Her workup ultimately led to a diagnosis of HLH. We discuss the presentation, diagnostic criteria, and clinical treatment of secondary HLH to guide workup and management for future patients affected by this rare disease.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
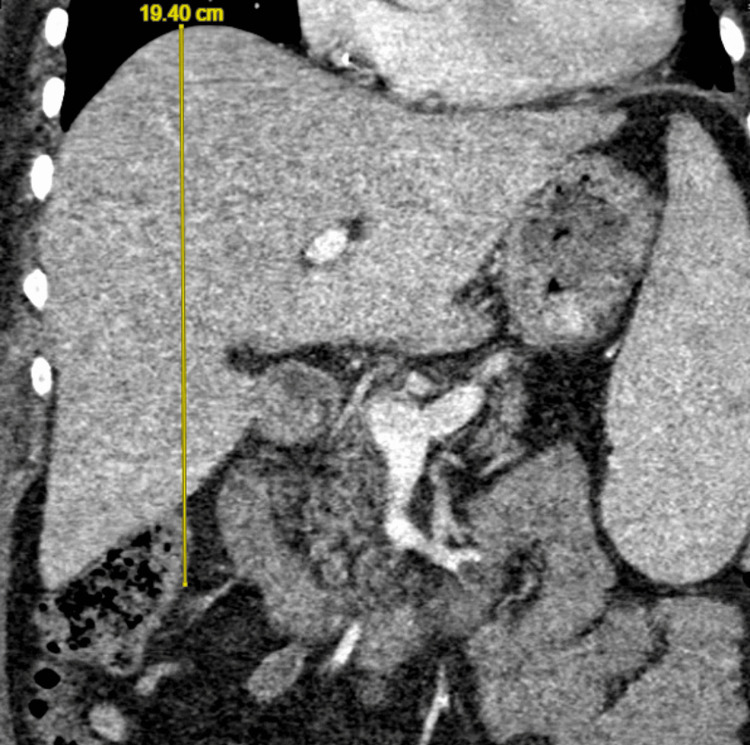 Figure 1
Figure 1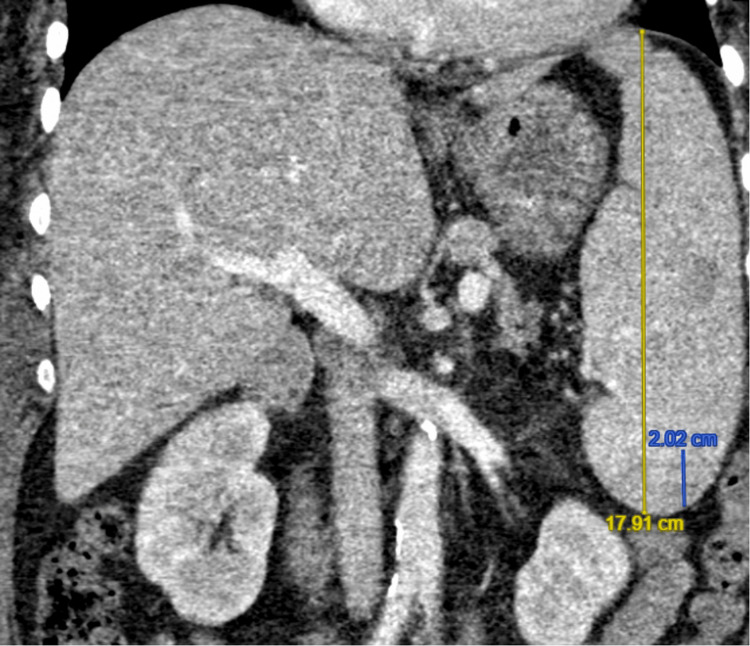 Figure 2
Figure 2| Parameters | Patient Values | Reference Range |
| Temperature (°C) | 38.9 | 36.5-37.3 |
| Heart rate (beats per minute) | 132 | 60-100 |
| Respiratory rate (breaths per minute) | 20 | 12-18 |
| Blood pressure (mmHg) | 76/47 | 90-120/60-80 |
| MAP (mmHg) | 57 | 70-100 |
| Oxygen saturation (%) | 100 (on room air) | 95-100 |
| Lactate (mmol/L) | 3.6 | 0.3-2.0 |
| Parameters | Patient Values | Reference Range | HLH-2024 Criteria (≥5 of 7 for diagnosis) |
| 1. Temperature (°C) | 38.9 | 36.5-37.3 | ≥38.5 |
| 2. Extension of spleen below the costal margin (cm) | ≥2 | <2 | ≥2 |
| 3. Hemophagocytosis in spleen/bone marrow/lymph node | absent | absent | present |
| 4a. Hemoglobin (g/L) | 89 (8.9 g/dL) | 120-160 (12.0-16.0 g/dL) | <90 (<9.0 g/dL) |
| 4b. Platelets (x 109 /L) | 27 | 130-400 | <100 |
| 4c. Neutrophils (x 109 /L) | 3.1 | 1.8-8.0 | < 1.0 |
| 5a. Fasting triglycerides (mmol/L) | 6.1 | <1.7 | ≥3 |
| 5b. Fibrinogen (g/L) | 6.7 | 2-6 | ≤1.5 |
| 6. Ferritin (μg/L) | 4,331 | 20-210 | ≥500 |
| 7. Soluble interleukin-2 receptor (U/mL) | ≥4000 | 175-850 | ≥2400 |
| Parameters | Patient Value | HScore |
| Known underlying immunosuppression | No (0 points) | No (0 points); Yes (18 points) |
| Temperature, °F (°C) | 102.0 (38.9) (33 points) | <101.1 (<38.4) (0 points); 101.1–102.9 (38.4-39.4) (33 points); >102.9 (>39.4) (49 points) |
| Organomegaly | Hepatomegaly and splenomegaly (38 points) | No (0 points); Hepatomegaly or splenomegaly (23 points); Hepatomegaly and splenomegaly (38 points) |
| Number of cytopenias; defined as hemoglobin ≤9.2 g/dL (≤5.71 mmol/L) and/or WBC ≤5,000/mm³ and/or platelets ≤110,000/mm³ | Hemoglobin 8.9 g/dL; WBC 3,100/mm³; platelets 27,000/mm³ (34 points) | 1 lineage (0 points); 2 lineages (24 points); 3 lineages (34 points) |
| Ferritin, ng/mL (or μg/L) | 4,331 (35 points) | < 2,000 (0 points); 2,000-6,000 (35 points); >6,000 (50 points) |
| Triglycerides, mg/dL (mmol/L) | 6.1 mmol/L | <132.7 (<1.5) (0 points); 132.7-354 (1.5–4) (44 points); >354 (>4) (64 points) |
| Fibrinogen, mg/dL (g/L) | 6.7 g/L | >250 (>2.5) (0 points); ≤250 (≤2.5) (30 points) |
| AST, U/L | 19 U/L | <30 (0 points); ≥30 (19 points) |
| Hemophagocytosis features on bone marrow aspirate | No (0 points) | No (0 points); Yes (35 points) |
| 204 points = 88-93% probability of HLH | Optimal cutoff is 169 points |
| Parameters | Admission | Discharge |
| Fever curve (°C) | 38.9 | 36.6 |
| Hemoglobin (g/L) | 89 (8.9 g/dL) | 99 (9.9 g/dL) |
| Platelets (x 109/L) | 27 | 72 |
| WBC (x 109/L) | 3.1 | 3.8 |
| Timeline | Events |
| Prior admission (Day 0) | Patient admitted for generalized weakness, abdominal pain, fever, and pancytopenia requiring multiple transfusions. Bone marrow biopsy unremarkable. Started on IV dexamethasone and transitioned to oral. |
| Day 19 | Patient discharged with dexamethasone taper (starting at 10mg daily) and hematology/oncology follow-up due to concern for underlying hematologic or oncologic process. |
| First (current) admission (Day 46) | Patient admitted for pancytopenia and shock with abdominal pain. Negative infectious and malignant workup. Comprehensive HLH workup resulted in HLH diagnosis. Patient started high-dose steroid therapy and etoposide infusions with subsequent clinical and laboratory improvement. |
| Day 60 | Patient discharged with dexamethasone taper (starting at 21mg daily) and outpatient etoposide infusions. |
| Second admission (Day 67) | Patient admitted for fever, hypotension, and sepsis. Workup revealed ESBL bacteremia, cellulitis, and invasive aspergillosis treated with antibiotics (vancomycin and meropenem) and anti-fungals (voriconazole). Also found to have AKI likely due to hypoperfusion injury. Etoposide treatments stopped. Continued on steroids. |
| Day 86 | Patient discharged to long-term acute care facility (LTAC) in stable condition with continued dexamethasone taper (starting at 5mg daily) and outpatient voriconazole treatment. |
| Third admission (Day 203) | Patient admitted for abdominal pain, pancytopenia, shock, and sepsis requiring pressor support and ICU stay. Presentation consistent with HLH relapse. Patient started on anakinra and dexamethasone. Anakinra was stopped due to little response. Patient’s clinical condition continued to worsen. Goals of care discussion with patient and family. Decision made to withdraw life-prolonging treatment and to focus on comfort care. |
| Day 238 | Patient discharged home with family on hospice. |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAutoimmune and Inflammatory Disorders Research · Immunodeficiency and Autoimmune Disorders · Otitis Media and Relapsing Polychondritis
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a potentially fatal condition linked to an excessively active immune system. The pathophysiology of both the primary (familial) and secondary (acquired) forms of HLH is commonly attributed to immunologic dysregulation, which can result in overactivity of the cell-mediated immune system [1]. The resulting hyperinflammatory state can lead to cytopenias and ultimately to severe infection, sepsis, end-organ damage, and not infrequently death.
The primary form of HLH largely affects children and is associated with several genetic mutations [2]. The secondary form of HLH has been associated with infection, autoimmunity, immune suppression, organ transplant, and malignancy; while the disease process of this potentially fatal illness is not completely understood, its pathophysiology is thought to be multifactorial [1-3]. Moreover, HLH in adults previously has been estimated to account for less than 1% of admissions to tertiary care centers, though the scarcity of data makes incidence difficult to assess accurately [3]. Diagnostic challenges of HLH derive largely from its widely variable and non-specific presentation. Delays in diagnosis and treatment may further exacerbate the effects of HLH, resulting in deterioration of clinical status. This medical instability may make certain treatment options, such as chemotherapy and stem cell transplant, more difficult to pursue.
The most widely recognized set of diagnostic criteria for both primary and secondary HLH is based upon the HLH-2004 clinical trial [4]. These criteria were recently updated in 2024. Based on the 2024 criteria, a diagnosis of HLH is made through a molecular diagnosis consistent with HLH or by meeting five out of seven established criteria [5]. The HLH-2024 criteria remain the preferred diagnostic guidelines today. Additionally, an adult individual's risk of having HLH can be estimated using the HScore, which consists of a set of weighted clinical and laboratory criteria such as temperature, number of cytopenias, and evidence of hemophagocytosis features on bone marrow aspirate. This tool is recognized to be highly sensitive but less specific.
Treatment for secondary HLH can include chemotherapy (such as dexamethasone and etoposide) as well as immune therapy or even hematopoietic stem cell transplant [2]. While treatment regimens and survival rate have improved, the prognosis for secondary HLH remains poor, with the mortality rate reaching up to 80% [6].
This report describes the case of a 53-year-old woman with multiple previous hospitalizations for fever of unknown origin and fatigue. She was eventually diagnosed with secondary HLH and treated for the condition. However, she later re-presented with recurrent HLH, and her health rapidly declined thereafter. This case demonstrates the importance of prompt workup and treatment in patients suspected of suffering from HLH.
This case report was previously presented as a poster at Medicine Research Day in New Orleans in April 2025 and at Greater New Orleans American Physician Science Association Day in New Orleans in April 2025.
Case presentation
A 53-year-old woman with a past medical history of hypertension and heart failure with recovered ejection fraction presented to the emergency department with weakness, fatigue, fever, chills, nausea, vomiting, and poor appetite for two months. She had been admitted to other hospitals in the months prior for similar presentations. Previous workups before this presentation had revealed severe anemia with a hemoglobin of 5.9 g/dL (59 g/L). Due to her lack of diagnosis and concern for an immunological cause of her illness, she had already completed a high-dose dexamethasone taper. Upon presentation, her vital signs were notable for fever, tachycardia, and hypotension, and she had an abnormally high lactate (Table 1).
On physical exam, the patient was ill-appearing with purpura and pitting edema of her bilateral lower extremities up to the knees, as well as scattered ecchymoses on all extremities. Due to concern for pancytopenia, sepsis, and heart failure, she was admitted to the medical intensive care unit; she was also started on broad-spectrum antibiotics and pressors. Infectious diseases, gastroenterology, rheumatology, endocrinology, and hematology/oncology services were consulted.
Extensive infectious workup (including but not limited to chest X-ray, transthoracic echocardiogram, blood cultures, urine cultures, respiratory viral panel, and cytomegalovirus and human immunodeficiency virus testing) revealed no source of infection, and antibiotics were subsequently discontinued. Over the following days, her condition worsened. The patient underwent a comprehensive workup to evaluate for HLH. Computed tomography (CT) scan of the abdomen revealed hepatosplenomegaly; her liver measured 19.4 cm (Figure 1), and her spleen measured 17.9 cm (Figure 2). A right iliac crest bone marrow biopsy was without definitive evidence of hemophagocytosis. Flow cytometry was performed without evidence of a B- or T-cell lymphoproliferative disorder or acute leukemia.
CT scan of the abdomen demonstrating hepatomegaly.
CT scan of the abdomen demonstrating splenomegaly, meeting one of the seven HLH-2024 criteria.HLH: hemophagocytic lymphohistiocytosis
Several other tests were ordered as part of the patient's medical assessment. The patient met HLH-2024 diagnostic criteria (Table 2) and also had an 88-93% probability of having HLH according to the HScore (Table 3). Given that her overall clinical and laboratory workup was suspicious for HLH, her hematology/oncology team made the decision to start etoposide and dexamethasone on day six of admission. Her regimen was based on the HLH-94 protocol [4]. She received IV hydrocortisone (50 mg every six hours) and was transitioned to oral prednisone (60 mg daily). She subsequently was switched to IV dexamethasone (20 mg daily) and then to oral dexamethasone (20 mg daily). She also received three IV etoposide treatments (150 mg/m^2^) in accordance with the recommendation of etoposide treatments twice weekly for two weeks and then weekly.
The patient clinically improved (Table 4), and she was discharged on day 14 with an oral steroid taper (oral dexamethasone 21 mg for another six days, followed by 9 mg for 14 days and then 6 mg for 14 days) and outpatient etoposide infusions (150 mg/m^2^ once weekly for another six weeks) in accordance with HLH-directed therapy. Despite an extensive diagnostic workup, no underlying etiology, including malignancy or viral infection, could be determined.
Unfortunately, the patient was readmitted multiple times in the subsequent months due to neutropenic fever, shock, and sepsis (Table 5). A week after discharge from her current admission, she presented to the hospital in shock. Her workup revealed extended-spectrum beta-lactamase (ESBL) bacteremia, an acute kidney injury (AKI), and invasive aspergillosis. Her presentation was highly suspicious for recurrent HLH. Her etoposide treatments were thus discontinued early due to these life-threatening side effects. She was treated with antibiotics, including vancomycin and meropenem, as well as voriconazole. She was discharged on voriconazole for 11 weeks and an oral dexamethasone taper (5 mg daily for 10 days followed by 2.5 mg daily for seven days).
The patient’s next admission occurred nearly four months later. She presented with abdominal pain, pancytopenia, and shock similar to her previous HLH flare. She temporarily required pressors and an ICU stay but was eventually stabilized.
Given that she had previously developed significant and life-threatening toxicities leading to early termination of her etoposide medication regimen, pursuing etoposide was not a viable option. Thus, an IL-1 receptor antagonist, anakinra (10 mg/kg per day), was started as a temporizing agent. Unfortunately, she showed little response, as she continued to have fevers, fluctuating hypotension, pancytopenia, and rising ferritin levels. Her anakinra treatments were stopped after less than a week. She was also started on hydrocortisone (50 mg every six hours) and then transitioned to dexamethasone (10 mg every 12 hours).
Due to her worsening clinical condition, the patient wished to discontinue further therapy in favor of concentrating on comfort care. With the help of her palliative care team, she was discharged with plans for home hospice less than eight months after her initial diagnosis.
Discussion
The exact etiology of secondary HLH is still not fully understood. Indeed, its clinical presentation can be non-specific and perplexing, leading to possible delays in diagnosis and treatment. As previously noted, early diagnosis can influence patient outcome [7], thus making reliable diagnostic criteria even more important.
While the HLH-2004 criteria have historically been the most widely accepted diagnostic tool for HLH, their use in diagnosing secondary HLH in particular has become somewhat controversial. This controversy stems from their origins as the inclusion criteria for a pediatric clinical trial that mostly examined patients afflicted by primary HLH [8] rather than secondary HLH, which is the more common form in adults.
As noted previously, the HLH-2004 criteria were recently updated to the HLH-2024 criteria, which now exclude natural killer (NK)-cell activity from the recommended laboratory measurements. Given the differences in pathogenesis between primary and secondary HLH [3], it would be beneficial to investigate the development of a revised set of guidelines and validated diagnostic criteria specific to secondary HLH. Some work has already been done on this front, including the development of a diagnostic score known as the HScore [9,10]. Currently, the HLH-2024 criteria are still generally preferred.
This patient met six out of seven HLH-2024 criteria, including those for fever, splenomegaly, cytopenias of two cell lines, hypertriglyceridemia, elevated ferritin, and elevated interleukin-2 receptor. The only HLH-2024 criterion that this patient did not fulfill was evidence of hemophagocytosis. However, despite the fact that she met diagnostic criteria, her case was a perplexing one, likely owing in part to the rarity of this illness as well as its non-specific presentation. Greater awareness of secondary HLH and its presentation may be beneficial in mitigating delayed recognition of the disease.
Furthermore, this patient’s recurrence complicated an already challenging case. She was no longer a candidate for etoposide, one of the mainstays of HLH treatment, and showed a minimal response to other attempted therapies. A firmer understanding of the underlying etiology in this case could have led to opportunities for more targeted therapy. Further work needs to be done to understand the varied etiology underlying secondary HLH.
Finally, modified treatment algorithms specific to secondary HLH in adults would be worth investigating. Current treatment approaches are largely adapted from pediatric guidelines; proposed adjustments for treatment of HLH in adults include dose reductions and individualized tailoring [11]. Treatment modifications should also be considered in patients for whom typical treatments are incompatible or those who show signs of relapsed disease.
Few case reports on recurrent adult HLH exist. A report by Machaczka et al. details the successful treatment of recurrent malignancy-associated HLH in a young woman, which included a stem cell transplant and donor lymphocyte infusion [12]. However, in this case, the patient’s HLH was known to be associated with malignancy, whereas for our patient, the etiology was unknown.
Overall, relapsed and refractory HLH has poor outcomes in the adult population. Hematopoietic stem cell transplant (HSCT) is a mainstay of treatment in these cases, regardless of the underlying cause of HLH, and may result in response rates up to nearly 50% [13]. However, stabilizing and medically optimizing patients prior to HSCT remains challenging. New drugs such as emapalumab and ruxolitinib are emerging as potential bridges to HSCT, with partial responses ranging from 14.2% to 100% [11,13,14]. Both of these medications were considered for our patient prior to her decision to pursue comfort care.
Other emerging therapies include alemtuzumab, tocilizumab, and anakinra, which have shown promising results [11,13,14]. One study showed that anakinra, a medication that is generally tolerated well, was effective in 90.5% of patients, with patients showing fever resolution within a median of one day as well as decreased ferritin and C-reactive protein levels [15]. Anakinra was trialed in our patient during her last admission, but unfortunately, she showed little response to the drug, resulting in discontinuation.
Overall, HLH continues to present a significant challenge for patients and providers from both a diagnostic and treatment perspective. Despite its often non-specific presentation, providers should maintain a high index of suspicion for HLH in patients with recurrent fevers of unknown origin accompanied by cytopenias. Thereafter, providers should quickly begin an HLH workup as well as look into potential causes and individualized treatments.
Conclusions
HLH is a complex and potentially deadly proinflammatory disorder. Our patient fulfilled six of the seven HLH-2024 diagnostic criteria. The exact cause of HLH in this patient remains elusive and likely never will be fully elucidated. Although a diagnosis was reached and a management plan initiated, her condition continued to lead to extended hospital admissions and an unfavorable outcome. Due to the life-threatening nature of this disease, timely diagnosis is imperative to improving patient outcomes. Clinicians should consider HLH as a potential diagnosis in patients with recurrent fevers and cytopenias, prompting hematological workup. Additionally, clinicians who suspect HLH should investigate possible underlying etiologies, such as malignancy or infection.
This patient suffered multiple adverse effects not only from her illness but also from her treatment, and eventually she chose to pursue comfort care rather than further therapy. Attaining overall more favorable prognoses will require continued research into this disease, including the development of both validated diagnostic criteria for secondary HLH as well as improved guidelines for therapy regimens.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Clinical features and diagnosis of hemophagocytic lymphohistiocytosis Up To Date Mc Clain K Eckstein O Rosée P Wellesley, MA Wolters Kluwer 2025 https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-hemophagocytic-lymphohistiocytosis
- 2Merck Manual: Hemophagocytic lymphohistiocytosis (HLH) 4 2025 2024 https://www.merckmanuals.com/professional/hematology-and-oncology/histiocytic-syndromes/hemophagocytic-lymphohistiocytosis-hlh
- 3Hemophagocytic lymphohistiocytosis Annu Rev Pathol Al-Samkari H Berliner N 27491320182893456310.1146/annurev-pathol-020117-043625 · doi ↗ · pubmed ↗
- 4HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis Pediatr Blood Cancer Henter JI Horne A AricóM 1241314820071693736010.1002/pbc.21039 · doi ↗ · pubmed ↗
- 5Diagnostic guidelines for familial hemophagocytic lymphohistiocytosis revisited Blood Henter JI Sieni E Eriksson J 2308231814420243904677910.1182/blood.2024025077 PMC 11619794 · doi ↗ · pubmed ↗
- 6HLH: diagnostics revisited and improved Blood La Rosée P La Rosée F 2274227514420243960771410.1182/blood.2024026243 · doi ↗ · pubmed ↗
- 7Hemophagocytic lymphohistiocytosis Arch Pathol Lab Med Ponnatt TS Lilley CM Mirza KM 50751914620223434785610.5858/arpa.2020-0802-RA · doi ↗ · pubmed ↗
- 8Can we truly diagnose adult secondary hemophagocytic lymphohistiocytosis (HLH)? A critical review of current paradigms Pathol Res Pract Naymagon L 15332121820213341834610.1016/j.prp.2020.153321 · doi ↗ · pubmed ↗