The Function of RcAG2 and RcFUL in the Flower Shape Change of Rosa chinensis ‘Viridiflora’
Jinfeng Zhang, Hui Liao, Yipeng Yang, Xixi Zhang, Caijie Yi, Lina Song, Zijing Li, Hua Zhang, Peng Ji

TL;DR
This study identifies two genes, RcAG2 and RcFUL, that influence flower shape changes in Rosa chinensis 'Viridiflora', offering insights into floral development.
Contribution
The study reveals the functional roles of RcAG2 and RcFUL in floral organ identity and development in Rosa chinensis.
Findings
RcAG2 and RcFUL are involved in floral organ development and exhibit distinct phenotypic effects when overexpressed in Arabidopsis.
The genes lack signal peptides and transmembrane domains but contain MADS-box domains and phosphorylation sites.
Overexpression of RcFUL resulted in leaf-like petals and increased sepal number, while RcAG2 caused petal reduction and stamen exposure.
Abstract
The floral morphology of Rosa chinensis significantly influences its ornamental value. However, the molecular mechanisms underlying specific floral types remain poorly understood. Viridiflora, a stable genetic variant of R. chinensis, exhibits homeotic transformation of floral organs into sepal-like structures, providing a valuable model for studying floral organ identity and development. In this study, Viridiflora was compared with Old Blush to elucidate floral development through morphological observation, transcriptomic profiling, and functional genetics. Four distinct developmental stages were defined, encompassing the formation of sepal, petal, stamen, and pistil primordia. Transcriptome analysis identified candidate genes associated with the Viridiflora phenotype, among which RcAGAMOUS2 (RcAG2) and RcFRUITFULL (RcFUL) were selected for in-depth functional characterization. The…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1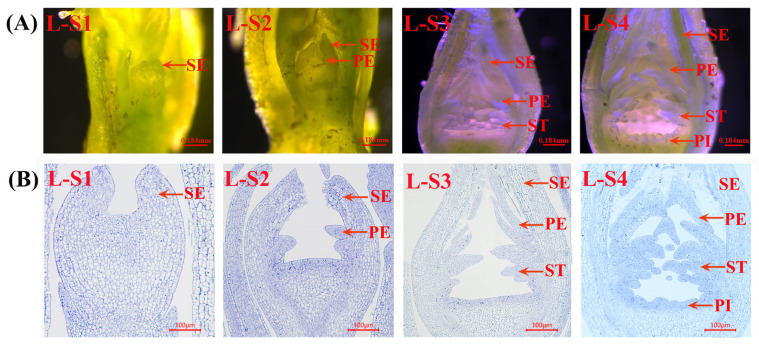 Figure 2
Figure 2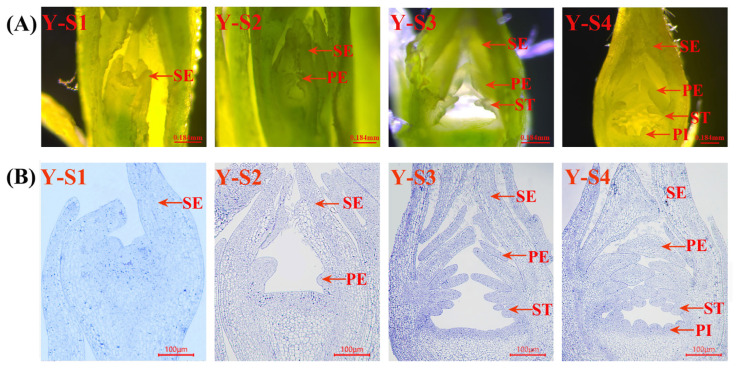 Figure 3
Figure 3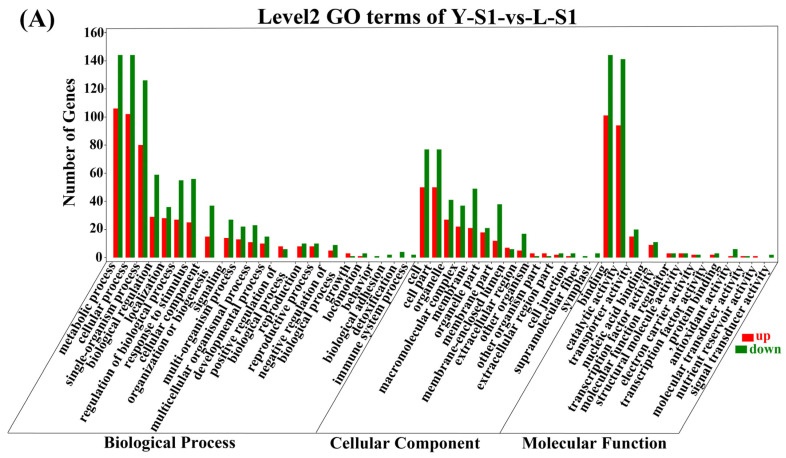 Figure 4
Figure 4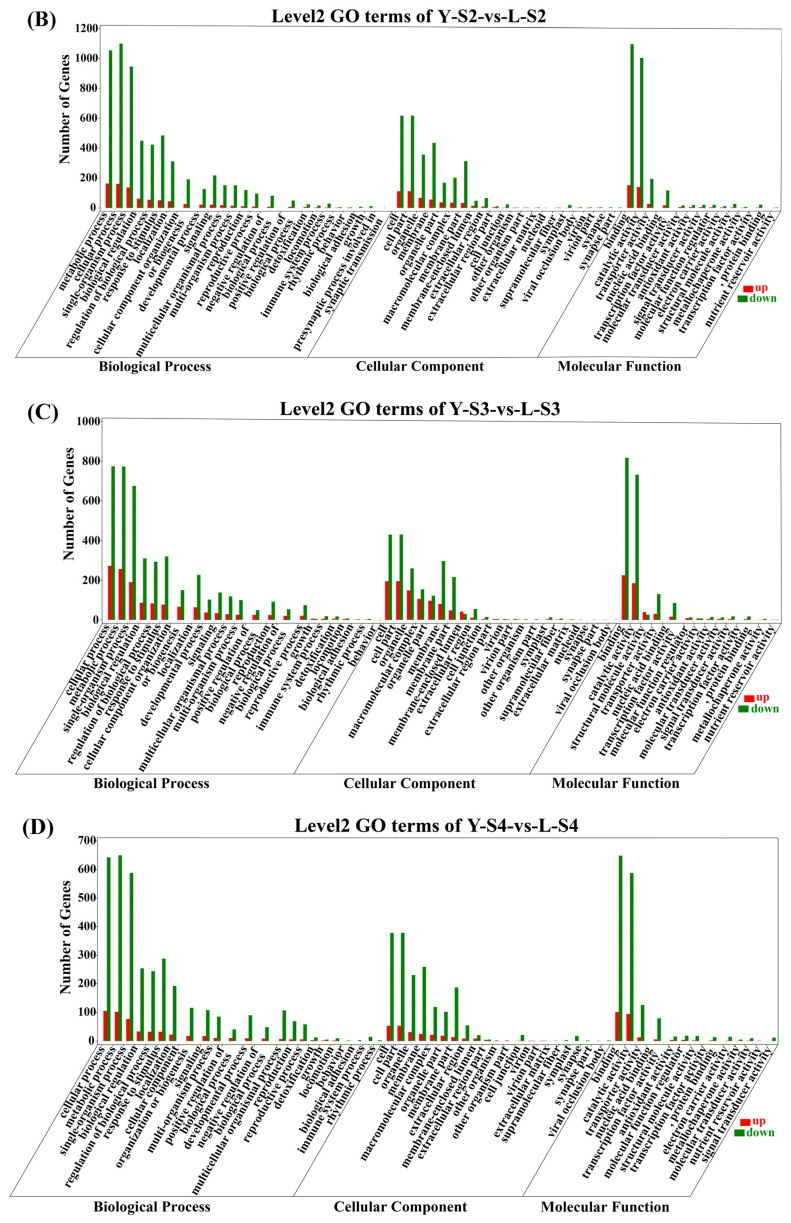 Figure 5
Figure 5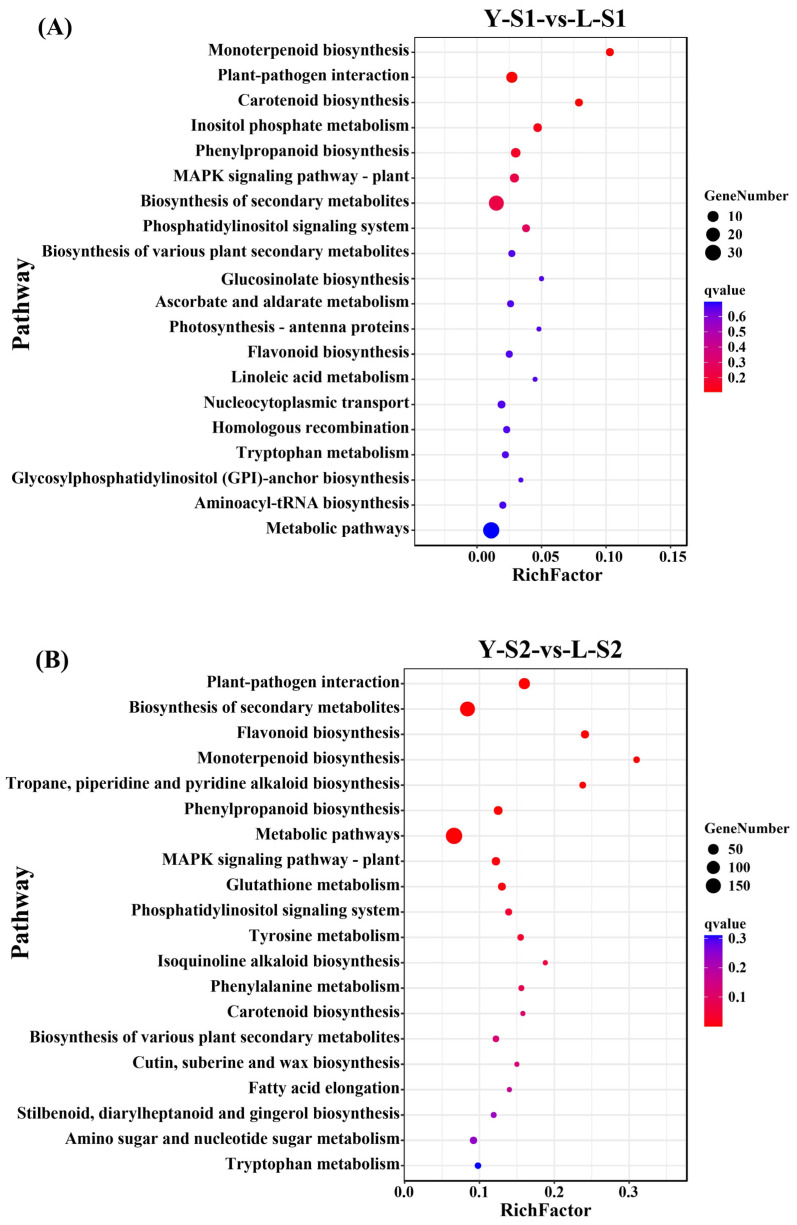 Figure 6
Figure 6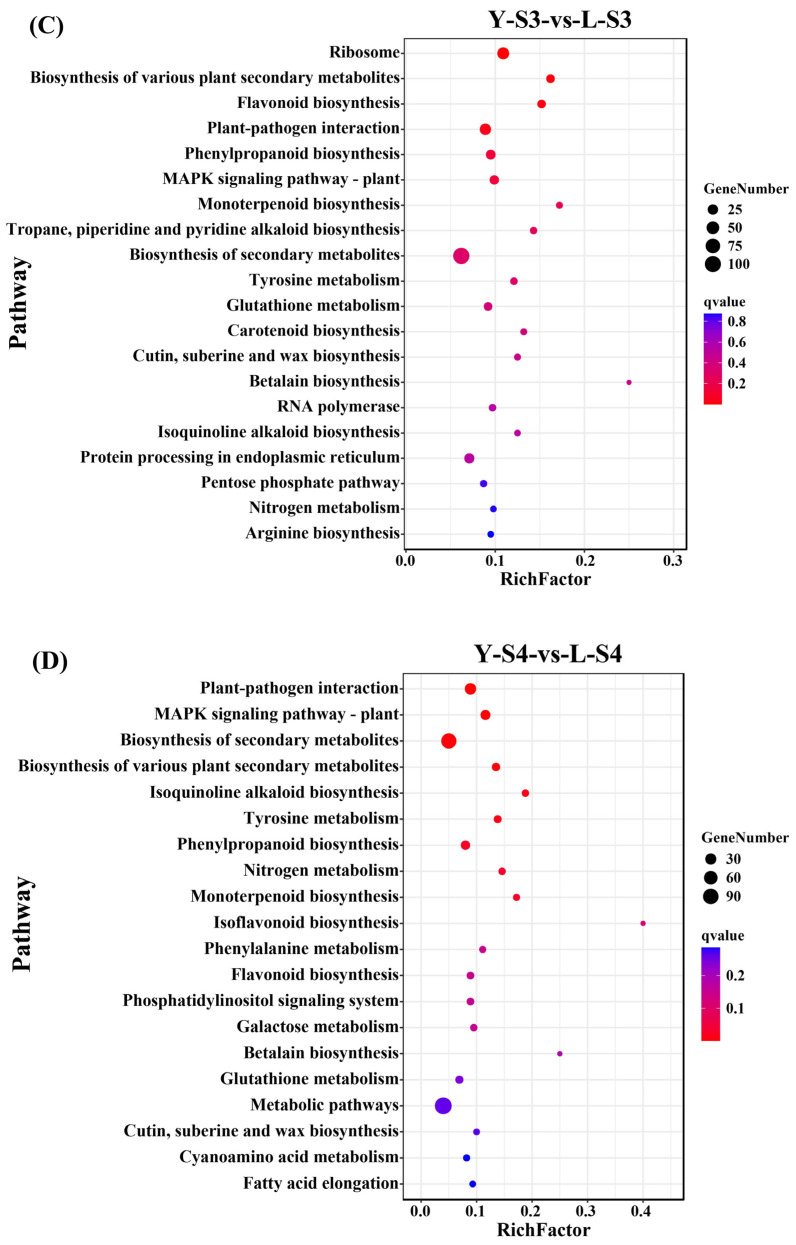 Figure 7
Figure 7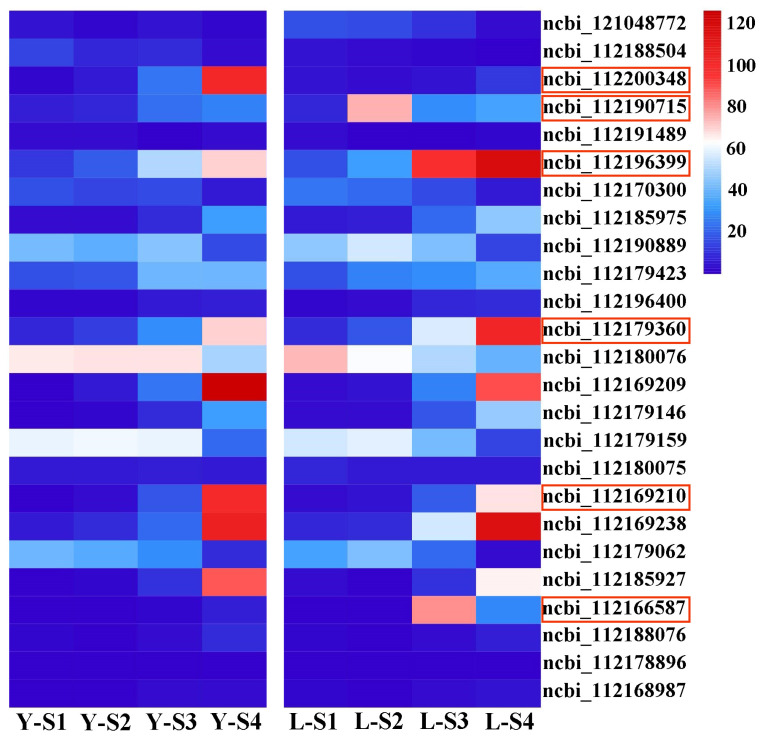 Figure 8
Figure 8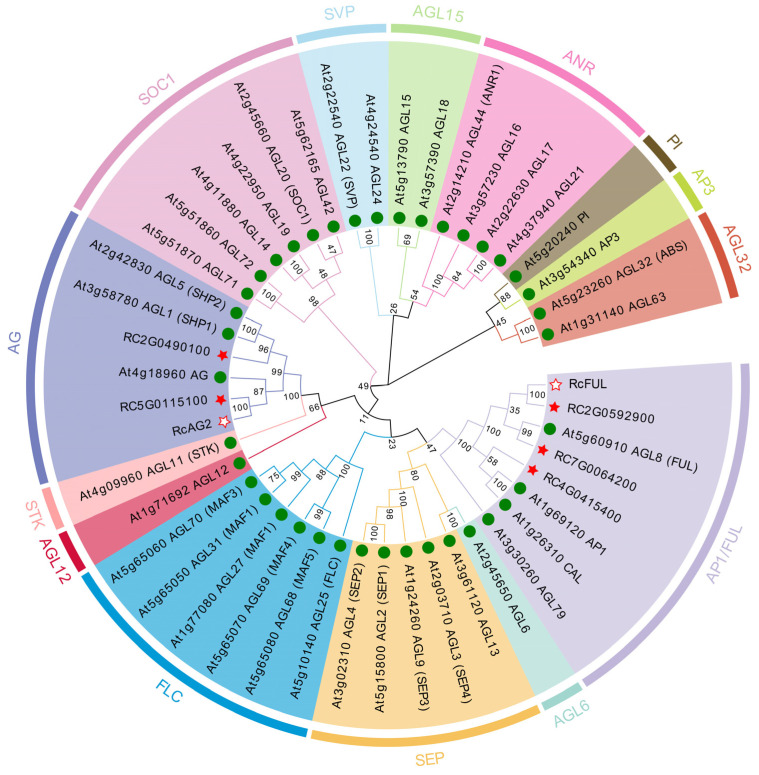 Figure 9
Figure 9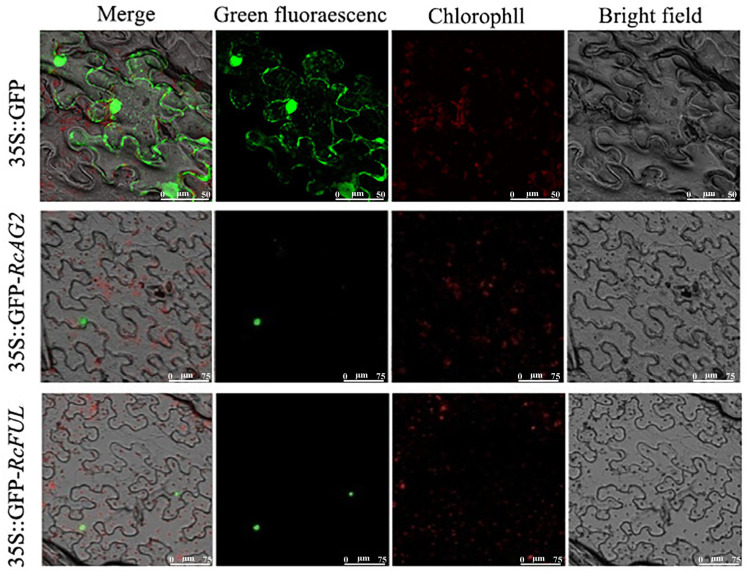 Figure 10
Figure 10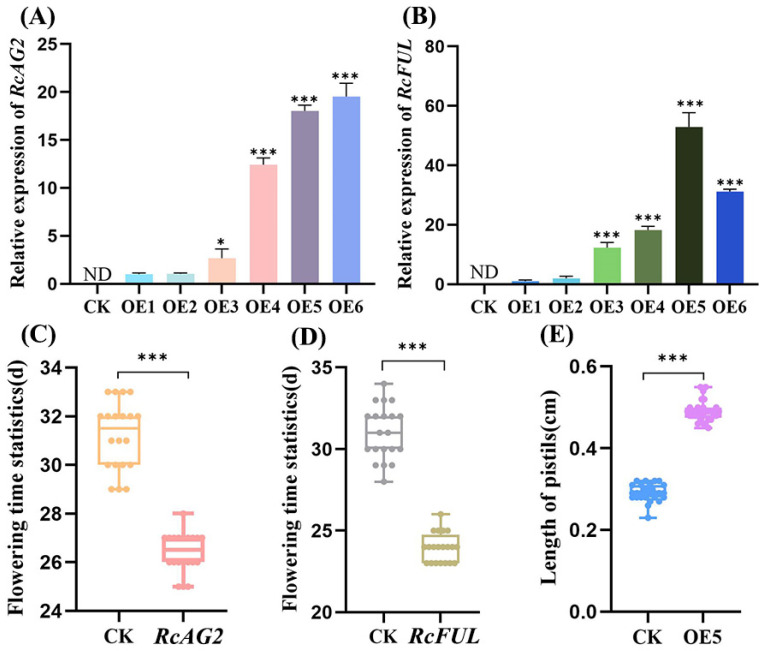 Figure 11
Figure 11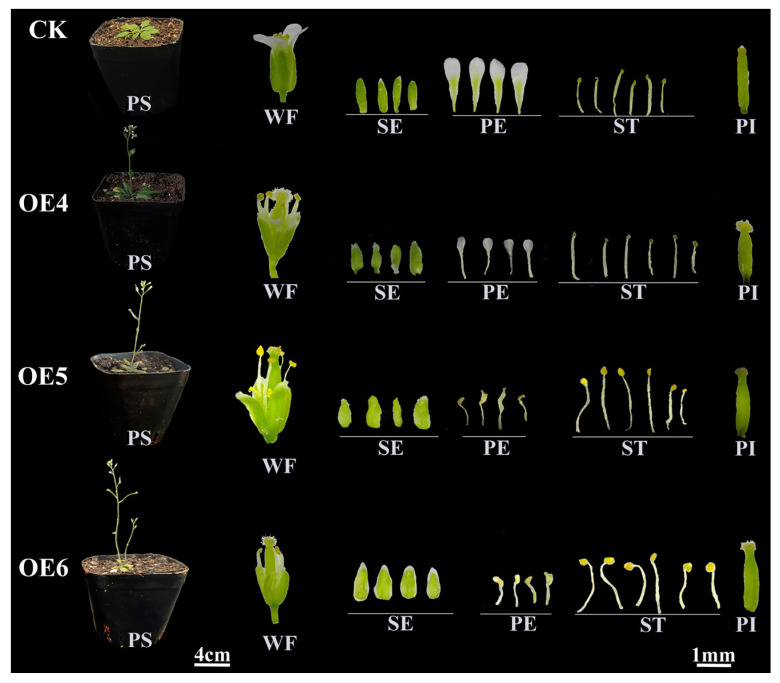 Figure 12
Figure 12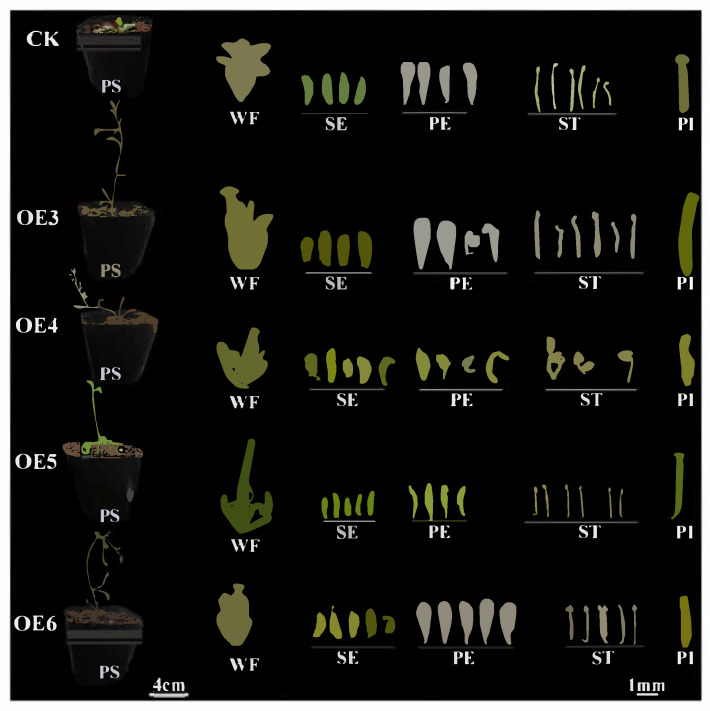 Figure 13
Figure 13- —Natural Science Foundation of Heilongjiang Province
- —open progect of Beijing Key Laboratory of Greening Plants Breeding
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Molecular Biology Research · Plant biochemistry and biosynthesis · Plant Gene Expression Analysis
1. Introduction
Rosa chinensis, a semi-evergreen shrub or climbing plant of the genus Rosa (Rosaceae), is revered as the “queen of flowers” and ranks among China’s top ten famous flowers. It is also one of the world’s most widely cultivated ornamental plants, prized for its diverse flower forms, fragrances, and colors [1]. While current research on R. chinensis flower morphology has largely focused on petal number, less attention has been given to the floral-organ variant cultivar Rosa chinensis ‘Viridiflora’ (hereafter Viridiflora). In Viridiflora, petals, stamens, and pistils are transformed into sepal-like structures, resulting in entirely green flowers. This unique morphology and coloration make it highly valued in ornamental horticulture and appreciated in international markets. Rosa chinensis ‘Old Blush’ (hereafter Old Blush), a recurrent-flowering ancient Chinese rose with high adaptability, is a key genetic resource in breeding and a major ancestor of modern roses. Viridiflora, a natural mutant of ancient Chinese roses, is closely related to Old Blush [1,2]. Due to its genetic stability and representative traits, Old Blush is frequently used as a control in rose developmental studies and has been proposed as a model cultivar [3,4].
Flower structure consists of a peduncle—an extension of the branch—topped by a receptacle from which sepals, petals, stamens, and carpels arise as modified leaves, illustrating the developmental plasticity of floral organs [5]. In some species, sepals are often mistaken for petals due to their size and color, as seen in Hydrangea macrophylla [6], Clematis florida [7], and Aquilegia viridiflora [8]. In other plants, such as R. chinensis [9], Paeonia lactiflora [10], Camellia japonica [11], stamen or pistil conversion into petals underlies the formation of double flowers. Unusual petal morphologies—such as slender or curled forms—also contribute to floral diversity in species like Chrysanthemum morifolium [12] and Phalaenopsis aphrodite [13]. The ABC model of floral organ identity, proposed by Coen and Meyerowitz in 1991 based on studies in Arabidopsis thaliana and Antirrhinum majus describes how combinatorial gene functions specify sepal (A-class), petal (A + B), stamen (B + C), and carpel (C-class) development [1,14]. However, ectopic expression of ABC genes outside floral whorls typically fails to convert leaves into floral organs [15,16]. This model was later expanded to the “ABCDE model”, incorporating D-class genes essential for ovule development and E-class genes (SEPALLATA) required for the identity of all floral organs [17,18,19]. Mutations or misregulation of these genes often lead to homeotic transformations. Notably, co-expression of the A, B, C, and E-class genes can convert leaves into floral organs in Arabidopsis [18,20].
Abnormal floral phenotypes in R. chinensis are generally attributed to mutations or altered expression of floral organ identity genes, often accompanied by changes in phytohormone signaling. Northern blot analyses revealed that Viridiflora and Rosa hybrid ‘Motrea’ (hereafter Motrea) exhibit suppressed expression of AGAMOUS (AG), a C-class gene essential for stamen and carpel formation, which also influences meristem activity and gibberellin biosynthesis [21]. RcAG is expressed throughout bud development in normal roses but is silent in early bud stages in both Viridiflora and Motrea [22]. Furthermore, expression levels of RcAG are significantly lower in Viridiflora compared to Old Blush, likely due to methylation at four CpG sites within its promoter region [23]. Similar phenotypes occur in Thalictrum thalictroides, where ThtAG1 suppression leads to stamen and pistil conversion into sepals [24]. In addition, RcAG is mainly expressed in the third and fourth whorls of rose flower and has the function of terminating the development process. Suppression of RcAG expression could lead to increase the number of rose petals [25]. The MADS-box transcription factor FRUITFULL (FUL) belongs to the APETALA1/FRUITFULL (AP1/FUL) subfamily, which includes three evolutionary lineages: euAP1, euFUL, and Ful-like. Among them, euFUL and FUL-like proteins are functionally redundant and have conserved C-terminal motifs (L/MPPWML) [26,27]. In A. thaliana, AtFUL plays a role in regulating leaf, flower, and fruit development. Transient silencing RcFUL in the rose could lead to a delay in flowering time, indicating that this gene was a positive regulatory factor for flowering [28].
RcAG and RcFUL play significant roles in the organ development of flowers, but there are differences in their expression and function. At present, RcAG is mostly documented in studies investigating double-flowered roses, affecting the termination of the second whorl of flower organs. RcFUL is mostly related to the flowering time of roses and the development of carpels. Comparing MIKC^C^-type MADS-box gene expression between Viridiflora and Old Blush, revealing up-regulation of RcFUL2, RcFUL3, and RcFUL5 during carpel differentiation in Viridiflora, absence of RcAP3.3 expression in early developmental stages, and organ-specific expression of RcAP1.2, RcAG2, and RcSEP4.3 exclusively in Viridiflora leaves [1]. Studies have shown that AtFUL can have a direct or indirect impact on miRNA172 [29,30]. Small RNA sequencing identified significant down-regulation of miR172 in Viridiflora petals, stamens, and pistils, coupled with up-regulation of its target RcAP2, suggesting miR172 may negatively regulate RcAP2 to promote phyllody [31,32]. In addition, it was found that AtFUL can directly bind to the promoter of AP2 expressed in inflorescence meristems and pistil tissues [33].
To further investigate the molecular mechanisms underlying the unique floral morphology of Viridiflora, this study combined microscopic observation of floral bud development with transcriptome analysis to identify candidate genes. Functional characterization via subcellular localization and heterologous overexpression in A. thaliana was performed to elucidate their roles in floral organ development. These findings provide new insights into the genetic regulation of ornamental traits in R. chinensis and establish a theoretical foundation for targeted breeding strategies.
2. Results
2.1. Morphological Dynamics of Floral Bud Differentiation in Viridiflora and Old Blush Roses
All The experimental materials consisted of flower buds from Rosa chinensis ‘Old Blush’ and ‘Viridiflora’ (Figure 1), with Old Blush serving as the control. Floral bud differentiation in Viridiflora and Old Blush was systematically characterized through four distinct developmental stages, designated as L-S1–L-S4 for Viridiflora (Figure 2), and Y-S1–Y-S4 for Old Blush (Figure 3). Comparative analysis revealed a highly consistent organogenesis pattern between the two cultivars, with floral organs initiating in successive whorls from the periphery toward the center of the floral meristem.
Y-S1/L-S1: Sepal Primordium Formation. The earliest morphological change involved the emergence of five triangular protrusions along the flanks of the floral apex, indicating sepal primordia initiation. These structures exhibited convex morphology with centripetal cellular expansion and typical meristematic organization.
Y-S2/L-S2: Petal Primordium Initiation. Following sepal differentiation, minute protuberances became visible adaxial to the sepal whorls, marking the onset of petal formation. Concurrently, the receptacle region expanded markedly, exhibiting upward growth and a concave basal morphology. Repeated cycles of primordia generation resulted in multilayered petal development.
Y-S3/L-S3: Stamen Primordium Differentiation. Multiple protrusions emerged proximal to the petal-receptacle junction, indicating stamen initiation. These primordia elongated progressively, eventually forming distinct staminal structures. A notable morphological divergence was observed between cultivars: Old Blush developed elliptical stamens, whereas Viridiflora exhibited elongated, taper-shaped structures.
Y-S4/L-S4: Pistil Primordium Development. The differentiation process concluded with the formation of central protrusions within the innermost floral zone, which subsequently underwent radial elongation and lateral expansion to form the pistil group. By the termination of this phase, all floral organ categories had been established, indicating complete bud differentiation.
2.2. Transcriptome Profiling Reveals Differential Gene Expression Pattern and Regulatory Networks During Floral Development
The quality and reliability of the transcriptomic data were assessed using total RNA from all 24 samples, which exhibited high integrity as confirmed by 1% agarose gel electrophoresis (Figure S1). CleanReads ratios ranged from 99.75% to 99.83% for Viridiflora and 99.75% to 99.86% for Old Blush, indicating high sequencing quality. Correlation analysis among three biological replicates showed intra-group coefficients of ≥0.87, demonstrating strong reproducibility and confirming the suitability of the data for further analysis. Differentially expressed genes (DEGs) between Old Blush and Viridiflora were identified using thresholds of |log_2_(FC)| > 1 and FDR < 0.05 to uncover genes potentially associated with divergent floral organ development. A total of 6535 DEGs were detected across four developmental stages, comprising 1523 up-regulated and 5012 down-regulated genes. Early stages showed limited transcriptomic divergence, with similar numbers of up- and down-regulated genes. In contrast, later stages exhibited a substantial increase in DEGs, particularly in down-regulated genes, suggesting enhanced negative regulation that may contribute to abnormal development in Viridiflora (Figure S2).
Functional characterization of the DEGs was carried out through GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analyses. GO analysis revealed consistent functional profiles across stages (Figure 4), with dominant molecular functions including binding, catalytic activity, and transporter activity. Major cellular components consisted of cell, cell part, organelle, and membrane, while key biological processes involved metabolic, cellular, and single-organism processes. KEGG analysis indicated significant enrichment in secondary metabolite biosynthesis and general metabolic pathways across all stages (Figure 5), underscoring central metabolic mechanisms underlying the developmental differences.
To pinpoint key regulatory genes, MADS-box family genes with low expression (FPKM (Fragments Per Kilobase of exon model per Million mapped fragments) < 0.5) were filtered out, and the remaining genes were analyzed via heatmap (Figure 6). Notable up-regulation in Viridiflora was observed for NCBI Gene IDs 112166587 (RcAG2), 112196399 (RcAP1), 112190715 (RcFUL), and 112179360 (RcMADS2), whereas NCBI Gene IDs 112200348 (RcMADS4) and 112169210 (RcMADS9) were significantly down-regulated. Notably, during petal primordium development, RcFUL expression is significantly upregulated, and this gene may influence the development of the second whorl of floral organs; during stamen primordium development, the expression level of RcAG2 is significantly upregulated, and this gene may play a role in the abnormal development of the third whorl of floral organs. Based on functional annotation and previous studies, to explore the causes of abnormal development of petals and stamens, RcAG2 and RcFUL were selected as candidate genes due to their high expression in Viridiflora. Validation using qRT-PCR on six DEGs confirmed expression trends consistent with the transcriptome data (Figure S3).
2.3. Phylogenetic and Protein Structural Analysis of RcAG2 and RcFUL
To elucidate the evolutionary relationships of RcAG2 and RcFUL, their protein sequences were subjected to analysis against the MADS-box gene family of A. thaliana. The results shown that the clustering branches of RcAG2 and RcFULL differ. RcAG2 clustered with the AG gene of A. thaliana in a single branch, categorizing it within the AG subfamily as a C-class gene essential for flower development. RcFUL and the FUL gene of A. thaliana were grouped within the same family, belonging to the AP1/FUL subfamily, and were classified as Class A genes involved in flower development (Figure 7).
To characterize the biochemical attributes of RcAG2 and RcFUL, their physicochemical properties were computationally predicted (Table S1). The molecular formulas were determined as C_1241_H_2006_N_380_O_388_S_8_ and C_1264_H_2065_N_375_O_395_S_11_, respectively. Both proteins exhibited theoretical isoelectric points (pI) > 7, classifying them as alkaline proteins. With instability indices exceeding 40, both were predicted to be unstable, though RcAG2 demonstrated greater stability than RcFUL. As show in Figures S4 and S5, negative grand average of hydropathicity (GRAVY) scores indicated hydrophilic characteristics for both proteins. Neither protein contained transmembrane domains or signal peptides, suggesting extracellular localization without secretory capacity. Secondary structure composition was predicted to further understand protein folding. RcAG2 consisted of 56.05% α-helices, 3.23% β-turns, 8.87% extended strands, and 31.85% random coils. RcFUL contained 51.36% α-helices, 2.33% β-turns, 8.17% extended strands, and 38.13% random coils. Homology modeling via SWISS-MODEL provided three-dimensional structural validation, while conserved domain analysis confirmed that both proteins contain K-box and MADS-MEF2-like domains, with variations only in domain arrangement.
2.4. Functional Characterization of RcAG2 and RcFUL
To determine the subcellular localization of RcAG2 and RcFUL proteins, their CDS sequences were cloned from Viridiflora flower bud cDNA. Sequencing confirmed CDS lengths of 756 bp for RcAG2 and 774 bp for RcFUL (Figure S6). Recombinant plasmids were constructed and verified via colony PCR and restriction digestion, then transformed into Agrobacterium for transient expression in Nicotiana benthamiana leaves. Subcellular localization confirmed the nuclear localization of both RcAG2 and RcFUL, supporting their predicted roles as transcription factors (Figure 8).
To investigate the functional effects of RcAG2 and RcFUL, heterologous overexpression was performed in A. thaliana. Positive transgenic plants were selected using antibiotic resistance and confirmed by detecting target gene expression (Figure S7). A total of 12 positive lines were successfully generated. The expression levels of RcAG2 and RcFUL in transgenic lines were quantified by qRT-PCR using empty vector lines as controls. RcAG2 expression was undetectable in controls but significantly elevated in transgenic plants, with OE4, OE5, and OE6 showing approximately 13-, 18-, and 20-fold higher expression than OE1, respectively (Figure 9A). Similarly, RcFUL expression was absent in controls but strongly induced in transgenics, with OE5 and OE6 exhibiting about 50- and 30-fold increases compared to OE1 (Figure 9B).
Both RcAG2 and RcFUL transgenic plants displayed early flowering phenotypes. While empty vector plants flowered around 30 days after sowing, RcAG2 and RcFUL transgenics flowered approximately 4 days and 7 days earlier, respectively (Figure 9C,D). Notable alterations in floral organ morphology were observed. In RcAG2-overexpressing lines (OE4–OE6), sepals failed to fully enclose reproductive structures at early developmental stages, and petals exhibited abnormal morphologies such as reduced size, fan-shaped or stamen-like appearance, and light-yellow pigmentation (Figure 10 and Table S2). The inner whorl organs remained largely unaffected. RcFUL transgenics (OE3–OE6) also exhibited substantial floral abnormalities, including increased sepal and petal counts (up to five), greenish or leaf-like petals, reduced stamen number, and elongated pistils (~1.7-fold longer in OE5; Figure 9E and Figure 11 and Table S3). Severe phenotypic disruptions in some lines led to loss of self-pollination capacity and failure to produce seeds. Collectively, these findings indicate that heterologous expression of RcAG2 and RcFUL not only promotes early flowering but also markedly disrupts floral organ development in A. thaliana, supporting their functional involvement in floral morphology and development.
3. Discussion
Floral development in roses involves a precisely timed sequence of morphological changes. The developmental process of rose flower buds can be broadly categorized into six stages: pre-differentiation, sepal primordium formation, petal primordium formation, stamen primordium formation, pistil primordium formation, and completion of floral bud differentiation [34]. Among these, the periods during which sepal, petal, stamen, and pistil primordia develop are of particular interest, as they represent critical phases for the formation of key floral organs. In alignment with these findings, the present study focused specifically on these four stages to elucidate key mechanisms underlying floral organ formation. In this study, the RNA-seq was utilized to analyze the DEGs of Old Blush and Viridiflora during the flower bud differentiation stages. The number of down-regulated genes is more than that of up-regulated genes, indicating that most genes are involved in the abnormal development of Viridiflora flowers in the form of negative regulation.
The MADS-box family is a class of important transcriptional regulatory factors related to the regulation of flower development. This study mainly screened out the DEGs of MADS-box family and obtained six genes involved in the development of floral organs: RcAG2, RcAP1, RcFUL, RcMADS4, RcMADS9, and RcMADS2. The number of DEGs is the largest during the primordial development stage of petals and stamens. The DEGs is discovered mostly during the primordial development stages of petals and stamens, and RcAG2 and RcFUL were significantly expressed during these two periods were selected for research. Phylogenetic tree analysis indicates that RcAG2 may play the role of a Class C gene, and RcFUL may play the role of a class A gene.
RcAG2 gene identified as a C-class MADS-box transcription factor, plays a conserved role in floral organ development. Previous studies have shown that AG homologs not only regulate the differentiation and development of stamens and carpels but also regulate the termination of the floral meristem [35,36,37]. In this study, heterologous overexpression of RcAG2 in A. thaliana significantly altered petal morphology, leading to reduced petal size and eventual degradation, resulting in premature exposure of stamens. Similar phenotypic changes were observed when the AG gene from Prunus mume was overexpressed in A. thaliana [38]. Additionally, loss of AG function has been reported to cause stamens to transform into petal-like structures, resulting in double flowers [39]. Although AG overexpression generally induces stamen petaloidy and reduces stamen number, such phenotypes were not observed in the RcAG2-transgenic A. thaliana lines in this experiment.
RcFUL, also part of the MADS-box family, is primarily involved in petal and sepal development. Heterologous overexpression of RcFUL in A. thaliana resulted in leaf-like petals, increased sepal number, reduced petal size, elongated stigmas, and defects in pistil and stamen development. One of the transgenic lines with low transgene expression exhibited partially normal flowers, suggesting that the constitutive 35S promoter may not fully restrict gene expression to floral organs. The use of a native promoter in future studies may help achieve more specific spatial expression. These findings are consistent with earlier reports indicating that the FRUITFULL (FUL) gene promotes inflorescence meristem formation in early development and is later expressed in carpels and siliques, also influencing leaf development [40]. Furthermore, expression of RhFUL in tobaccos led to smaller leaves, greenish flowers, and increased sepal number, resembling the phenotypes observed in RcFUL-transgenic A. thaliana in this study [41].
The distinct functions of RcAG2 and RcFUL illustrate how divergent expression patterns and functional properties of transcription factors contribute to the complexity of floral morphology. In summary, the above results reinforce the central roles of MADS-box genes in floral development and highlight the value of rose mutants such as Viridiflora for probing the genetic regulation of floral organ identity. The functions of RcAG2 and RcFUL still require further research. In the future, gene silencing technology can be utilized to directly verify their roles in roses. Searching for the upstream and downstream genes of these two genes to construct a regulatory network will enable a more in-depth analysis of the specific regulatory pathways. Meanwhile, the other genes involved in the abnormal development of Viridiflora flower organs will be further explored and identified, and the functional verification and analysis should be conducted to reveal the mechanism of rose flower organ variations.
4. Materials and Methods
4.1. Plant Materials
The all plants were cultivated in the greenhouse of the Beijing Academy of Forestry and Landscape Architecture. Flower buds at specific developmental stages, determined via microscopic observation, were collected and immediately stored at −80 °C for subsequent use. Arabidopsis thaliana and Nicotiana benthamiana, employed in later experimental steps, were maintained under controlled laboratory conditions. Healthy, disease-free Viridiflora and Old Blush plants were selected for cutting propagation. Vigorously growing branches were excised, with the basal end cut obliquely and the apical end trimmed flat, retaining 1–3 buds. Cuttings were immersed in rooting solution for approximately 3 h before being transferred into a 1:1 (v/v) mixture of vermiculite and perlite. After 60 days of rooting, the plantlets were potted, and newly developed flower buds from the following year were used for experimental analyses.
4.2. Morphological Analysis of Floral Bud Differentiation
Stereoscopic microscope observation: The buds of Old Blush and Viridiflora were cut in half. One-half was observed under a stereomicroscope and photographed at different stages of flower bud differentiation, while the other half was quickly stored in liquid nitrogen.
Semi-thin section method observation: Flower buds were cut from the branches of Old Blush and Viridiflora plants, then quickly put into FAA fixative and placed in a vacuum pump. The pumping process should be monitored continuously until no more bubbles appear and all the flower buds sink to the bottom of the tube; then, the vacuum was slowly released. This step was repeated 3~4 times. After the final cycle, the fixative was replaced, and samples were kept at 4 °C overnight. The fixed flower bud materials were sent to the Institute of Botany, Chinese Academy of Sciences, for subsequent operation.
4.3. Transcriptomic Analysis
The floral buds of Old Blush and Viridiflora at different developmental stages were collected as experimental materials, with three biological replicates per stage. Total RNA was extracted from all samples using RNAprep Pure Plant Kit (DP441) (Tiangen, Beijing, China), and transcriptomic libraries were constructed. Sequencing was performed by Beijing Qiyuan Biotechnology Co., Ltd. (Beijing, China). After quality assessment, raw reads were processed to obtain clean reads for subsequent analysis. Gene expression levels were quantified using HISAT2 [42] for alignment to the reference genome, and FPKM values were calculated. Differential gene expression analysis was conducted with DESeq2, applying thresholds of |log_2_(FC)| > 1 and FDR < 0.05 to identify significantly differentially expressed genes (DEGs). Functional enrichment analyses, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses, were performed using the Bioconductor package 3.22 “clusterProfiler 4.0” [43] to interpret the biological roles and pathways associated with the DEGs.
4.4. Quantitative Real-Time PCR Analysis
Gene-specific primers were designed using multiPrime [44], with RcTCTP [45,46] of R. chinensis serving as the internal reference gene (Table S4). cDNA synthesized from reverse-transcribed total RNA was used as the template. qPCR was performed using the ChamQ SYBR qPCR Master Mix kit with the following reaction system: 0.4 μL each of forward and reverse primers, 1 μL cDNA, 10 μL of 2 × ChamQ SYBR qPCR Master Mix (Sigma Aldrich, St. Louis, MO, USA), and 8.2 μL ddH_2_O, making a total reaction volume of 20 μL. The thermal cycling conditions were as follows: initial denaturation at 94 °C for 2 min; 50 cycles of denaturation at 94 °C for 5 s, annealing at 60 °C for 30 s; and a final extension at 72 °C for 10 min. Each sample was run in three technical replicates and three biological replicates. Gene expression levels were analyzed using the 2^−∆∆Ct^ method to determine relative expression changes of target genes across different developmental stages.
4.5. Gene Cloning and Vector Construction
Total RNA was extracted from flower buds of Viridiflora and Old Blush at various developmental stages and reverse-transcribed into cDNA. Gene-specific primers were designed using Primer 6.0 (Table S5). The target genes were amplified using Viridiflora cDNA as the template under the following PCR conditions: initial denaturation at 95 °C for 3 min; 30 cycles of denaturation at 95 °C for 15 s, annealing at 58 °C for 20 s, and extension at 72 °C for 1 min; followed by a final extension at 72 °C for 5 min. The 50 μL reaction system consisted of: ddH_2_O, 2 μL each of forward and reverse primers, 10 pg–30 ng cDNA, and 25 μL of 2× Phanta Max Master Mix (Dye Plus) (Vazyme, Nanjing, China). After confirming the correctness of the target gene sequences by sequencing, primers containing appropriate restriction sites were designed. The amplified fragments were ligated into the overexpression vector (pCAMBIA2300-35S) and the subcellular localization vector (pRTL2-GFP). The recombinant plasmids were then transformed into Agrobacterium tumefaciens GV3101 competent cells. Positive colonies were selected via PCR verification, and validated strains were cultured and stored at −80 °C for further use.
4.6. Bioinformatics Analysis
The homologous sequences of RcAG2 and RcFUL genes in A. thaliana were identified by using the TAIR online website (https://www.arabidopsis.org/, accessed on 6 April 2025) [47], and the phylogenetic tree was constructed by using MEGA11.0 software. The physicochemical properties of RcAG2 and RcFUL proteins were analyzed by Expasy-ProtParam (https://web.expasy.org/protparam/, accessed on 6 April 2025). The secondary and tertiary structures of proteins were predicted by SOPMA and SWISS-MODEL (https://npsa.lyon.inserm.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html, https://swissmodel.expasy.org/interactive, accessed on 6 April 2025). The signal peptide sites of protein sequences were predicted by SignalP 4.1 (https://services.healthtech.dtu.dk/services/SignalP-4.1/, accessed on 6 April 2025). The transmembrane regions of proteins were predicted by TMHMM 2.0 (https://services.healthtech.dtu.dk/services/TMHMM-2.0/, accessed on 6 April 2025). The protein domains of RcAG2 and RcFUL were predicted by NCBI CD-Search (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 6 April 2025); and the subcellular localization of proteins was predicted by WoLF-PSORT (https://wolfpsort.hgc.jp/, accessed on 6 April 2025).
4.7. Subcellular Localization
The Agrobacterium strains of the empty vector (pRTL2-GFP) and the re-engineered vector (pRTL2-RcAG2-GFP and pRTL2-RcFUL-GFP) were shaken, collected, and resuspended. The OD600 of the bacterial solution was adjusted to 0.6–1.0, and incubated in the dark at 28 °C for 3 h. The leaves of N. benthamiana growing for 4 weeks were selected, injected, and immersed until the whole leaves were moist, then dark cultured for 48~72 h. The lower epidermis of the leaves was torn off and made into temporary pieces. The slides were placed on a laser confocal microscope to observe GFP fluorescent.
4.8. Heterologous Overexpression
The wild-type Arabidopsis seeds were disinfected and seeded on 1/2 MS medium. Following vernalization, they were cultured under light at 25 °C. After two true leaves were grown, they were transplanted into the substrate and cultured at room temperature. The Agrobacterium liquid of the empty vector (pCAMBIA2300-35S) and the re-engineered vector (pCAMBIA2300-35S-RcAG2 and pCAMBIA2300-35S-RcFUL) was propagated, respectively. After resuspension, the OD600 was adjusted to 0.6–0.8, and Silwet L-77 (Beijing, China) with 0.075% concentration was added for activation. A. thaliana was infected by inflorescence infection method (absorbed the infection solution drops onto the unflowering inflorescences and cultivated them for 1 d in the dark, then repeated the infection once after 7 d), and conventional light culture was carried out after 24 h of dark treatment. After obtaining the T1 transgenic Arabidopsis plants, the positive plants were screened, the RNA was extracted from the inflorescence, and the cDNA was obtained by reverse transcription. The specific primers with restriction sites were used for PCR identification, and the expression of the target gene in the positive Arabidopsis plants was detected by real-time fluorescence quantitative detection. AtACT7 (F: GTATGCTCTTCCTCATGCTATCCTT; R: TTCCCGTTCTGCGGTAGTG) was used as the internal reference gene.
5. Conclusions
In conclusion, RcAG2 and RcFUL were screened and cloned in this study. Both genes have typical MADS-box family domains. Subcellular localization showed that all of them function in the nucleus. Overexpression of RcAG2 showed petal shrinkage like stamen phenomenon; overexpression of RcFUL showed sepal number increase and could not bear fruit normally. These results suggest that both genes can affect the normal development of flower organs, but the regulation of flower organs in R. chinensis is unknown. The results of this study laid a foundation for exploring the special flower type of green calyx and provided a certain theoretical basis for rose flower type breeding.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Liu J. Fu X. Dong Y. Lu J. Ren M. Zhou N. Wang C. Mikcc-type mads-box genes in Rosa chinensis: The remarkable expansion of abcde model genes and their roles in floral organogenesis Hortic. Res.201852510.1038/s 41438-018-0031-429736250 PMC 5928068 · doi ↗ · pubmed ↗
- 2Chmelnitsky I. Azizbekova N. Khayat E. Zieslin N. Morphological development of normal and phyllody expressing Rosa hybrida cv. Motrea flowers Plant Growth Regul.20023721522110.1023/A:1020819123385 · doi ↗
- 3Bendahmane M. Dubois A. Raymond O. Bris M.L. Genetics and genomics of flower initiation and development in roses J. Exp. Bot.20136484785710.1093/jxb/ers 38723364936 PMC 3594942 · doi ↗ · pubmed ↗
- 4Dubois A. Carrere S. Raymond O. Pouvreau B. Cottret L. Roccia A. Onesto J. Sakr S. Atanassova R. Baudino S. Transcriptome database resource and gene expression atlas for the rose BMC Genom.20121363810.1186/1471-2164-13-638PMC 351822723164410 · doi ↗ · pubmed ↗
- 5Wang H. Zhang R.C.J. Duan X. Zhao H. Shan H. Kong H. Diversity of flowers in basic structure and its underlying molecular mechanisms Sci. Sin. (Vitae)20194929230010.1360/N 052018-00219 · doi ↗
- 6Yoshida K. Oyama K. Kondo T. Insight into chemical mechanisms of sepal color development and variation in hydrangea Proc. Jpn. Acad. Ser. B 202197516810.2183/pjab.97.00333563878 PMC 7897900 · doi ↗ · pubmed ↗
- 7Liu S. Song W. Pan W. Wu G. Transcriptome sequencing and SSR prediction of clematis calyx based on SMRT sequencing platform Sci. Rep.2024142894910.1038/s 41598-024-80504-039578612 PMC 11584640 · doi ↗ · pubmed ↗
- 8Sharma B. Kramer E.M. Aquilegia b gene homologs promote petaloidy of the sepals and maintenance of the c domain boundary Evodevo 201782210.1186/s 13227-017-0085-729209492 PMC 5704387 · doi ↗ · pubmed ↗