Site-Specific Recruitment, Localization of Ionized Monomer to Macromolecular Crowded Droplet Compartments Can Lead to Catalytic Coacervates for Photo-RAFT in Dilution
Wenjing Niu, Xiyu Wang, Ran Zhang, Yuanli Cai

TL;DR
Researchers developed catalytic droplets that mimic biological systems by using macromolecular crowding to enable efficient reactions in dilute conditions.
Contribution
A new method for site-specific monomer recruitment in catalytic coacervates using macromolecular crowding and confinement.
Findings
Macromolecular crowding enables 97% monomer conversion in dilute conditions within 12 minutes.
Salt-bridging molecular recognition localizes monomers to dense-phase compartments.
Hierarchical complexation leads to compartmentalized droplets with distinct phases.
Abstract
Catalytic coacervates, or droplet reactors, represent a forefront research area in chemistry and materials science. Despite advancements in this field, challenges persist in achieving liquid–liquid phase separation (LLPS) droplet compartmentalization and site-specific reactant recruitment/localization for reaction catalysis, similar to those within biological systems. Herein, we describe the catalytic coacervates for aqueous photo-RAFT in dilution, focusing on the site-specific recruitment/localization of ionized monomer with the aid of macromolecular crowding and confinement. Cooperative hydrogen-bonded interpolymer complexation (IPC) of imidazolium-copolymers initiates the ion-cluster formation. Further hierarchical inter-cluster complexation (ICC) leads to the LLPS droplet compartmentalization into charged dense-phase and neutral dilute-phase compartments. Site-specific recruitment…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
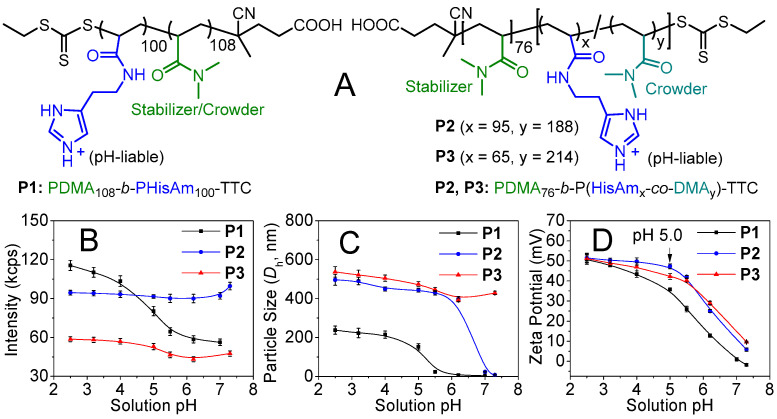 Figure 1
Figure 1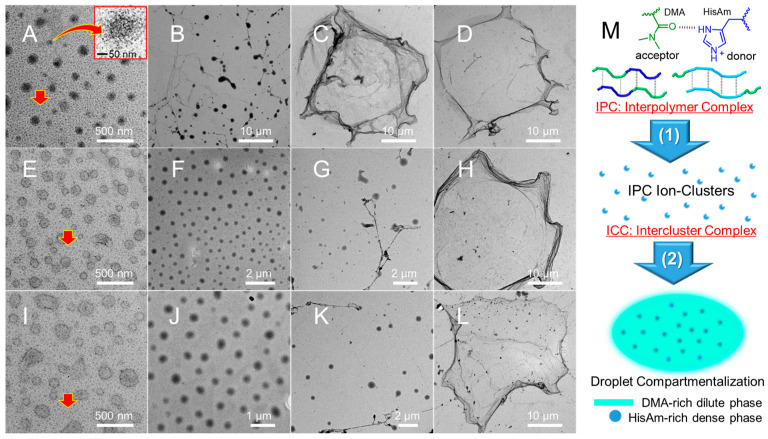 Figure 2
Figure 2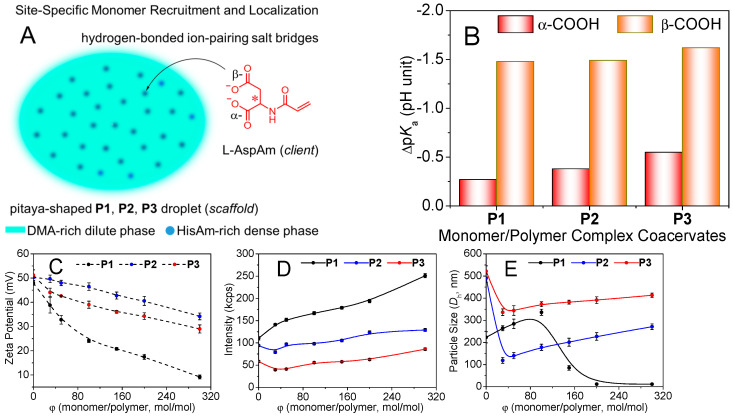 Figure 3
Figure 3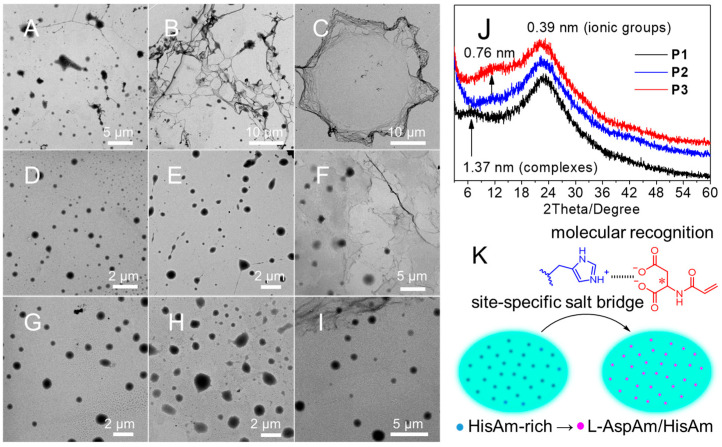 Figure 4
Figure 4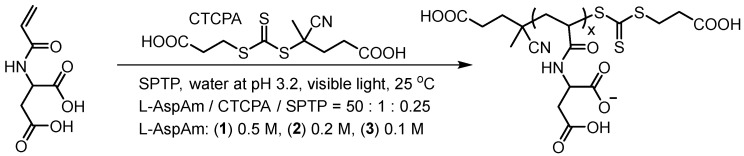 Figure 5
Figure 5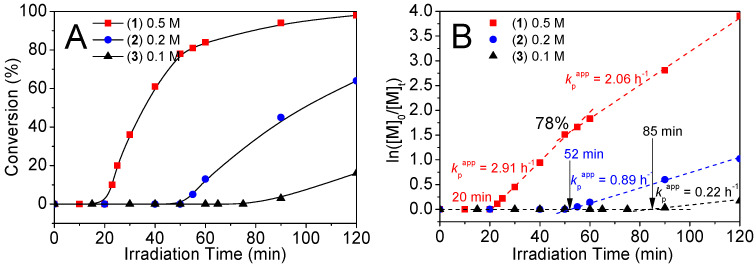 Figure 6
Figure 6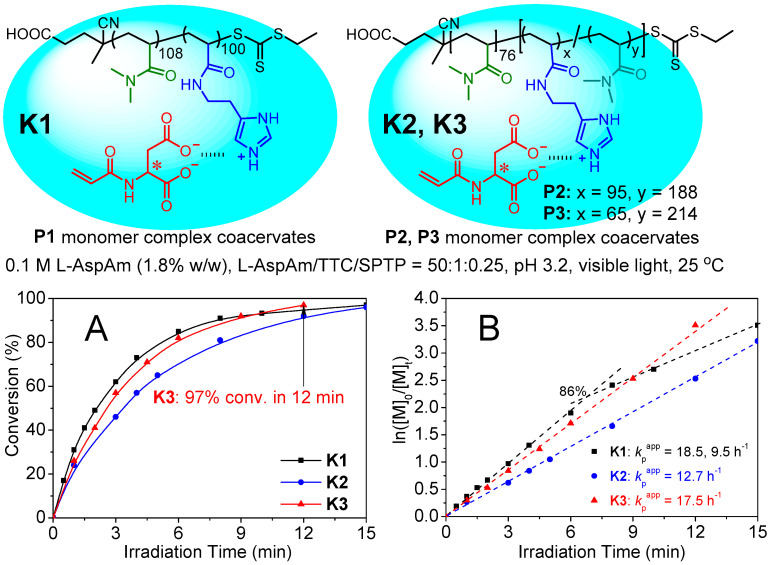 Figure 7
Figure 7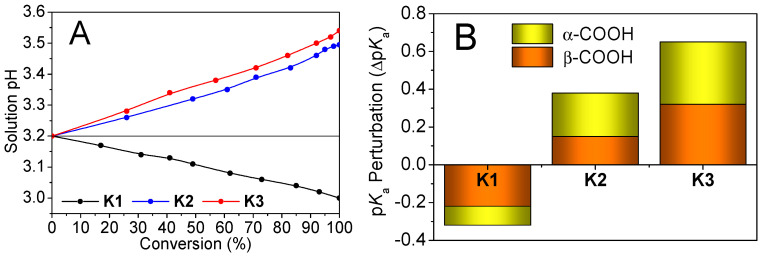 Figure 8
Figure 8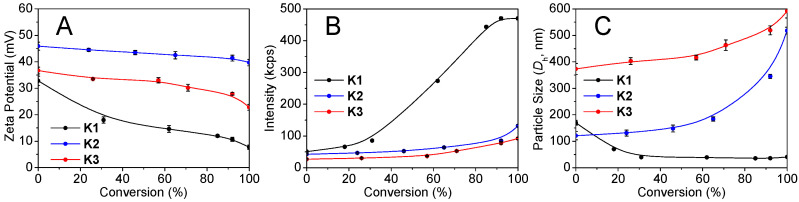 Figure 9
Figure 9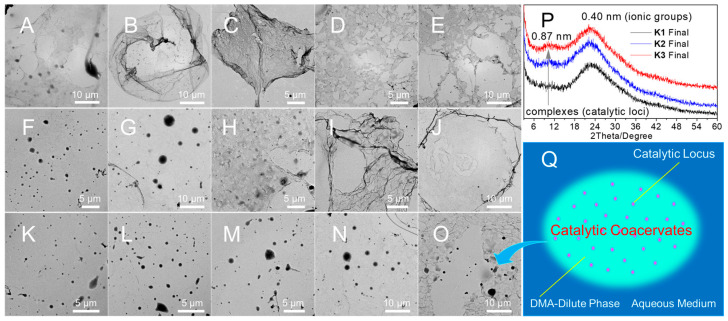 Figure 10
Figure 10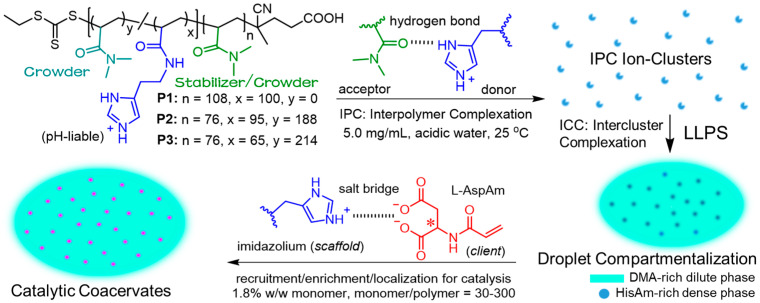 Figure 11
Figure 11- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced Polymer Synthesis and Characterization · Innovative Microfluidic and Catalytic Techniques Innovation · Pickering emulsions and particle stabilization
1. Introduction
Complex coacervation [1,2,3,4] of charged proteins and nucleic acids via liquid–liquid phase separation (LLPS) [5] is a key mechanism underlying intracellular organization and compartmentalization [6,7]. The transiently ordered systems exhibit extremely dynamic liquid-like properties [4,8]. As artificial mimics, complex coacervates [9] are a novel class of liquid materials showing low interfacial tension against water [10], semipermeability [11], and living properties [12,13]. Catalytic coacervates [14,15,16], in particular, have been broadly explored for their biosensor [17], nanoreactor [18], and protocell [19] applications. Despite significant advancements [14], achieving lifelike LLPS droplet compartmentalization and site-specific reactant recruitment/localization into the droplet compartments for reaction catalysis under dilute conditions remains a challenge [3,8,14,20].
Polymerization-induced self-assembly (PISA) [21,22,23] including photo-PISA [24,25,26] is robust for scalable synthesis of polymer nanoparticles at high solids contents up to 50% w/w [27,28]. Charged nanoparticles can be produced via aqueous dispersion RAFT polymerization of ionic liquid monomers [29], and via polymerization-induced electrostatic self-assembly (PIESA) [30,31,32,33] through the polyion complexation of charged growing chains with oppositely charged polyelectrolytes. LLPS-PIESA [34] was exploited for the compartmentalization of dynamic coacervate droplets. However, these coacervates showed a solidification trend, rendering them detrimental for the purposes of catalysis due to the loss of dynamic properties, enabling kinetically-frozen reaction inactivation inside dodecyl-modified polyelectrolyte droplet compartments [35]. In other words, the liquid-like dynamic properties should be maintained during the compartmentalization and reactant recruitment and localization into droplet compartments for sustainable catalytic coacervates [6,14,36].
Biological evidence indicates that macromolecular crowding and confinement [37,38,39] are fundamental mechanisms that are closely tied to chemical organization in biological systems [6]. In this work, we aim to explore catalytic coacervates for photo-RAFT in dilution, focusing on site-specific monomer recruitment/localization with the aid of macromolecular crowding and confinement. As shown in Scheme 1, we synthesized the ionized histamine acrylamide (HisAm) and nonionic N,N-dimethylacrylamide (DMA) copolymers. In spite of the water-miscible homopolymers, the block and statistical block copolymers underwent hydrogen-bonded interpolymer complexation (IPC) [40] resulting in complex ion-cluster formation. Hierarchical intercluster complexation (ICC) resulted in pitaya-shaped droplets with cationic HisAm-rich dense-phase and neutral DMA-rich dilute-phase compartments. These multiphase droplets acted as the scaffolds for site-specific recruitment/localization of anionic L–aspartate acrylamide (L–AspAm) monomer into droplet dense-phase compartments via salt-bridging molecular recognition [41,42,43,44]. Furthermore, the “substantial DMA-dilution” (that is, macromolecular crowding [37,38,39]) leads to the sustainable dense-phase catalytic sites confined within liquid apolar dilute-phase crowding surroundings. Distinct from solvent-miscible macromolecular chain transfer agent (macro-CTA) used for classical PISA formulations, these droplets functioned as the scaffolds and catalytic sites for the reaction catalysis in <2% w/w monomer dilution, leading to 97% conversion in 12 min. These active coacervates highlighted a high catalytic efficiency toward lifelike reaction functionalities [45].
2. Results and Discussion
2.1. Spontaneous LLPS Droplet Compartmentalization of Imidazolium-Copolymer in Dilution
First, we synthesized the nonionic poly(N,N-dimethylacrylamide) PDMA_108_-TTC (^1^H NMR: DP = 108; SEC: Mn = 12.1 kDa, Đ = 1.12; TTC: the trithiocarbonate chain-end) and PDMA_76_-TTC (^1^H NMR: DP = 76; SEC: Mn = 9.2 kDa, Đ = 1.17) macro-CTA through visible light initiated RAFT (photo-RAFT) polymerization [46]. Subsequently, the photo-RAFT of HisAm using PDMA_108_-TTC macro-CTA yielded poly(N,N-dimethylacrylamide)-b-poly(histamine acrylamide) (P1, PDMA_108_-b-PHisAm_100_-TTC; SEC: Mn = 34.7 kDa, Đ = 1.18), and the statistical copolymerization of HisAm and DMA using PDMA_76_-TTC macro-CTA afforded the statistical block copolymers P2 (PDMA_76_-b-P(HisAm_95_-co-DMA_188_)-TTC; SEC: Mn = 45.3 kDa, Đ = 1.25) and P3 (PDMA_76_-b-P(HisAm_65_-co-DMA_214_)-TTC; SEC: Mn = 43.6 kDa, Đ = 1.28), with the molecular structures shown in Figure 1A. The chain-extension experiments confirmed the high degree of the RAFT chain-end fidelity (Figures S1–S5). Unlike the P1 PHisAm homoblock, substantial DMA units were inserted in the P2 and P3 PHisAm segments. This composition character was termed as “substantial DMA-dilution” for the macromolecular crowding and confinement discussed below.
Potentiometric titration results (Figure S6) indicate a decrease in the acid dissociation constant (pKa) of the imidazole group from 7.25 (monomer) to 6.26 (P1, ΔpKa = −1.0 pH unit), 6.50 (P2, −0.75 unit), and 6.42 (P3, −0.83 unit). Upon solution neutralization, the P1 dispersion turns bluish (Figure S7) due to phase separation. ^1^H NMR analysis confirmed the (de)ionization and hydration of the phase-separated P1 imidazole/imidazolium motifs (Figure S8).
Figure 1B exhibits that the light scattering intensity of the 5 mg/mL dispersion at pH 2.5–7.3 (43–116 kcps) is notably higher than that of pure water (0.66 kcps), as determined by dynamic light scattering (DLS), indicating the phase separation in charged imidazolium form. Distinct from the anti-sigmoidal intensity plot of P1 dispersion, the intensities of the P2, P3 dispersions are relatively constant due to the “substantial DMA-dilution” -induced swelling. Their number- and intensity-average hydrodynamic diameters (Dh) are different (Figure S9) due to the hierarchical nanostructures discussed below. Solution neutralization leads to the decreased P1 and P2 particle sizes (Figure 1C). However, the Dh of P3 particles decreases slightly due to the swelling that was caused by the “substantial DMA-dilution”. Zeta potential (ζ) analysis (Figure 1D) illustrates the charged particles with ζ > 35 mV in water at pH 2.5–5.0, above which the absolute values of ζ-potential decrease due to imidazolium deionization. The particles show excellent colloidal stability due to the steric stabilization of the hydrophilic PDMA block.
The nanostructures were examined using transmission electron microscopy (TEM), using a cryofixation technique that primarily involved immersing the sample in liquid nitrogen and freeze-drying under high vacuum. Figure 2 displays P1 droplets/dots A (pH 2.5, inset: dots in droplet), droplets/ribbons B (pH 5.0), and lamellae C,D (pH 5.5, pH 7.0). Additionally, Figure S10 exhibits numerous dots A. TEM statistical analysis (Figure S11) gave a number-average particle diameter (Dn) of 116 ± 43 nm for droplets and 10 ± 5 nm for dots. These dots were identified as chain-collapsed ion-clusters. Given that both nonionic DMA and ionized HisAm [47] homopolymers are water-soluble, the phase separation of their copolymers can thus be ascribed to (1) interpolymer complexation (IPC) [40] driven by cooperative hydrogen bonding between the DMA (acceptor) and HisAm (donor) segments, resulting in complex ion-clusters; (2) intercluster complexation (ICC) by a delicate balance of the charge repulsion and multivalent hydrogen bonds, leading to the LLPS droplet compartmentalization into pitaya-shaped droplets (Figure 2M). Therefore, the imidazolium-block copolymer underwent phase separation via a unique hierarchical IPC-to-ICC LLPS droplet compartmentalization mechanism.
The dynamic properties of P2 and P3 droplets are enhanced by the “substantial DMA dilution”. Even given the droplet Ostwald ripening and transition to lamellae (Figure 2E–L), the coexisting light gray-colored dots and bubble-like droplets E, I (pH 2.5) indicate the notable swelling. The droplet fluidity is indicated by the bubble images reminiscent of the droplet fusion/dripping/surface wetting [48] and new formations (Figure S12). Specifically, at pH 2.5, P2 bubbles (Dn = 95 ± 26 nm)/clusters E (Dn = 11 ± 6 nm, Figure S13), along with swelling P3 bubbles (Dn = 100 ± 34 nm)/clusters (16 ± 6 nm, Figure S15) I, demonstrate the IPC-to-ICC LLPS droplet compartmentalization. At pH 5.0, uniform droplets F (Dn = 277 ± 77 nm, Figure S14) and J (Dn = 320 ± 73 nm, Figure S16) indicate the ICC-driven LLPS and droplet Ostwald ripening. At pH 7.0, lamellae H, L signify droplet aging caused by imidazole deionization. The aged compartments are unsuitable for the catalytic purpose due to the loss of dynamic properties. Both the “substantial DMA-dilution” and the “imidazoliumionization” facilitate the hierarchical IPC-to-ICC LLPS droplet compartmentalization, leading to the catalytic coacervates for photo-RAFT in dilution discussed below.
In summary, we achieved IPC-to-ICC LLPS droplet compartmentalization of block and statistical block imidazolium-copolymers. It is well-known that cooperative hydrogen bonding between complementary polymers results in stimulus-responsive interpolymer complexes (IPCs) [40,49,50,51]. However, to the best of our knowledge, this is the first report on hydrogen-bonded hierarchical LLPS droplet compartmentalization. The charged dense-phase and neutral dilute-phase compartments endow droplets with a high efficiency in the site-specific recruitment of oppositely charged L–AspAm monomers discussed below.
2.2. Site-Specific Monomer Recruitment and Localization into Droplet Compartments
It is well-documented that the liquid-like coacervate droplets can create an intriguing microenvironment for reactant recruitment/enrichment/localization, and destabilizing the reactants and lowering transition states to enhance reaction catalysis [14,36]. Motivated by these compelling findings, we sought to explore the reaction catalysis through the site-specific recruitment/localization of monomers into droplet dense-phase compartments under dilute conditions (Figure 3A). The process was driven by the COO^−^/imidazolium^+^ ion-pairing [43] biologically known as salt bridges [41,44].
We first examined the pKa shift of ionizable groups when mixing L−AspAm monomer with the copolymer dispersions at a monomer/polymer molar ratio (φ) of 50:1 at 30 mM (L−AspAm + HisAm units). This combination caused modest decreases in the pKa of α-COOH (L−AspAm) from 2.25 to 1.98, 1.87, and 1.70, but significant decreases in the pKa of β-COOH from 4.13 to 2.65, 2.64, and 2.51 (Figure S17). Figure 3B illustrates the pKa shifts indicating a notable increase in acidity. Additionally, imidazole buffer zooms (see those of precursor droplets in Figure S6) vanished due to an elevation in imidazole basicity. The concurrent enhanced COOH acidity and imidazole basicity enable dominant COO^−^•••imidazolium^+^ salt bridges for site-specific monomer recruitment and localization to oppositely charged droplet dense-phase compartments.
This observation well aligns with the notion that apolar environments (that is, DMA-rich dilute phase) strengthen charge–charge interactions and destabilize transition states involving charge separation. This mechanism is commonly seen in enzyme active sites [52]. Generally, protein folding induces the pKa perturbations of ionizable motifs [53,54,55]. For example, a salt bridge forms between Asp-70 and His-31 during T4 lysozyme folding [53], leading to notable pKa shifts of COOH from 3.5–4.0 to 0.5 and of imidazole from 6.8 to 9.1. Biologically, pKa perturbations play key roles in enzyme solubility and stability and functions such as kinetic properties [54]. The following study therefore focused on salt-bridged monomer recruitment at pH 3.2. Theoretically, the monomer/polymer complex coacervates contain 100% imidazolium and >90% COO^−^ motifs (>98% α-, ~80% β-COOH ionized, Figure S18), facilitating the salt-bridging molecular recognition.
To elucidate the composition (φ) impacts on the complex coacervation, we prepared a range of the mixtures at 5.0 mg/mL polymer at pH 3.2, with φ ranging from 30 to 300. The dispersions are transparent with perfect long-term storage stability. Figure 3C shows a ζ-shift of P1 parent droplets from ζ = 48 mV close to isoelectric point (9.2 mV) at φ = 300, but P2 and P3 droplets show a slight ζ-shift due to the “substantial DMA-dilution”. The P1 dispersion exhibits a dramatic increase in light scattering intensity, but P2, P3 dispersions show a mild increase in intensity (Figure 3D). Figure 3E exhibits an anti-sigmoidal variation of P1 particle size, which is associated with the charge-balanced morphological transition to the solid-like lamellae, as discussed below. Nevertheless, the sizes of the “substantial DMA-dilution” -featured P2 and P3 particles decrease sharply at φ = 0–30 but increase mildly at 30–300 due to the droplet shrinkage and Ostwald ripening.
The droplets were freeze-dried and examined using TEM and X-ray diffraction (XRD) analyses. Figure 4 displays Ostwald ripened P1 droplets A (φ = 30), and lamellae B (50)/C (100), suggesting a strong solidification trend, which halt the diffusion-limited reaction due to reduced fluidity. However, the “substantial DMA-dilution” leads to P2 droplets D, E (30–50), droplets/lamellae F (100), and P3 droplets G, H, I (30–100). Figure 4J reveals the amorphous structures with features at d = 0.39 nm corresponding to COO^−^/Na^+^ and imidazolium^+^/Cl^−^ salts [56,57]. Moreover, the d-spacing of ion-pairing complexes decreases from 1.37 (P1) to 0.76 nm (P2, P3), suggesting the “substantial DMA-dilution” -induced size reduction. All of these results demonstrate that the salt-bridging molecular recognition facilitates site-specific monomer recruitment/localization to the oppositely charged dense-phase compartments maintaining apolar DMA-rich dilute-phase crowding environments, as depicted in Figure 4K.
2.3. Kinetic Properties of Catalytic Coacervates for Photo-RAFT in Dilution
As mentioned above, we achieved the site-specific monomer recruitment/localization into the scaffold droplet dense-phase compartments, resulting in droplets at low φ in water at pH 3.2. These intriguing results inspired us to explore coacervate reaction catalysis [14]. Accordingly, we studied the kinetic properties of these active coacervates, under aqueous photo-RAFT conditions, in <2% w/w monomer dilution at 25 °C.
As a control experiment (Figure 5, top scheme), we studied the dilution effects on the photo-RAFT polymerization varying (1) 0.5, (2) 0.2, and (3) 0.1 M L–AspAm concentration. Conversion was determined using ^1^H NMR (Figure S19), and the growing polymers were studied by aqueous size exclusion chromatography (SEC, Figure S20). Figure 5A shows remarkable concentration impacts. Conversion decreased from 98% (0.5 M L–AspAm) to 64% (0.2 M), 16% (0.1 M) after the 2-h light irradiation. Figure 5B shows the distinctive dilution-prolonged induction time. Linear semi-logarithmic plots suggesting the constant concentration of growing-chain radicals. The clear shift of SEC trace and low dispersity (Đ ≤ 1.17) of the growing polymers (Figure S20) suggest the well-controlled photo-RAFT polymerization. The results demonstrate that the dilution not only increase the induction time but also decrease the chain propagation rate of this well-controlled polymerization, which is unfavorable for the dilution-compulsory practical applications.
Thereafter, we sought to explore the catalytic coacervates to speed up aqueous photo-RAFT in dilution. As shown in top scheme of Figure 6, the kinetic properties were studied through the following steps: (1) Monomer Recruitment involved mixing L−AspAm monomer with copolymer dispersions at φ = 50 (target DP) at 0.10 M monomer (1.8% w/w) at pH 3.2, and stirring overnight. (2) Heterogeneous Polymerization involved transferring the stock solution (1.0 mL) to a reaction flask, adding the SPTP photoinitiator (TTC/SPTP = 1:0.25), deoxygenating with argon gas in darkness, and finally exposing to visible light at 25 °C. Conversion was determined using ^1^H NMR (Figure S21). Reaction kinetics of the P1, P2, and P3 monomer complex coacervates are denoted as K1, K2, and K3 respectively.
Figure 6A shows immediate polymerization without the induction period observed in the discussed control experiment. Moreover, a rapid chain propagation is evidenced by a 97% conv. in 12 min. Figure 6B shows a rapid–slow K1 kinetic plot with apparent chain propagation rate constant (kp^app^) of 18.6, 9.5 h^−1^, dramatically higher than 0.22 h^−1^ of the 0.1 M counterpart (Figure 5B). The onset of the reaction deceleration occurred at 86% conv., exceeding 78% conversion in the 0.5 M counterpart (Figure 5B). Generally, in traditional RAFT polymerization, dilution slows down reaction, and low target DPs lead to notable reaction retardation by the intermediate reversible addition-fragmentation reactions [58]. Nevertheless, in this reaction in 1.8% w/w monomer dilution at low target DP of 50, the monomer was consumed promptly and swiftly. This reaction catalysis can thus be ascribed to site-specific monomer recruitment/localization to the droplet dense-phase compartments.
Figure 6B shows K2, K3 linear semi-logarithmic kinetic plots, where K3 reached 97% conversion in 12 min (see Figure 6A). The K3 (kp^app^ = 17.5 h^−1^), faster than the K2 (12.7 h^−1^), highlights the critical role of the liquid-like dynamic properties for the reaction catalysis. All of these results demonstrate that the reaction catalysis is driven by site-specific monomer recruitment/localization into the droplet dense-phase compartments allowing liquid dilute-phase crowding environments to eliminate the kinetically trapped slow reaction.
At over 41% conversions, the K1 dispersion became bluish (Figure S22) suggesting the particle size increase. In contrast, the K2 and K3 dispersions remained transparent due to the “substantial DMA-dilution”. Furthermore, reaction resulted in K1 solution acidification but K2, K3 solution neutralization (Figure 7A). The K1 final product displays pKa of 1.76 (α-), 2.56 (β-) for COOH motifs, notably lower than the Asp100 homopolymer counterpart, with substantial pKa decrease (ΔpKa: α- > 2 pH units, β- > 4 units, Figure S23). Significant pKa perturbations are also observed in the K2 and K3 final products, suggesting substantial COO^−^•••imidazolium^+^ salt bridges in the final products. Furthermore, compared to the monomer/polymer coacervates (see Figure S17), the K1 final COOH motifs show negative pKa shift, whilst those in the K2, K3 products show a positive pKa shift (Figure 7B). The final imidazole buffer zooms re-emerged, with the pKa = 7.76 (K2 final) and 7.53 (K3 final) lower than that (8.38) of the K1 final due to the “substantial DMA-dilution”. The opposing trends of proton feedbacks (release vs. recruitment) led to K1 reaction solution acidification, but K2 and K3 reaction solution neutralization.
Figure 8A shows a decrease in ζ-potential for K1 droplets at 0–30% conversion, and then a gradual decrease due to the lamella formation discussed below. However, K2 and K3 droplets show a mild ζ-decrease owing to “substantial DMA-dilution” for the dilute-phase swelling and dense-phase sequestration. Figure 8B,C shows the intensity increase and size decrease of K1 coacervates caused by the droplets-to-lamellae transition discussed below. The low intensity but slight size increase of K2 and K3 droplets can be attributed to the considerable swelling caused by the “substantial DMA-dilution”.
Finally, we examined reaction coacervates using cryofixation TEM and XRD. Figure 9 indicates the “substantial DMA-dilution” liquefaction, as illustrated by the transitions from K1 droplets/lamellae A (17% conv.) to lamellae B–E (31–100%), from K2 droplets G (46%) to droplets/lamellae H (65%) to lamellae I, J (92–100%), and from K3 droplets K–M (26–71%) to droplets/lamellae N, O (92–100%). The kinetically trapped reaction observed in K1 was eliminated by liquefying the K2 and K3 coacervates. Additionally, Figure 9P reveals 0.87-nm d-spacing in K2 and K3 final products indicating sustainable dense-phase catalytic reaction sites. Another factor contributing to the reaction catalysis is the macromolecular crowding and confinement [37,39] by the DMA-rich dilute-phase crowding surroundings. The excluded volume effect [37,39] promotes growing chain compaction, facilitating chain propagation by reducing volume compared to monomers [14]. Consequently, the “substantial DMA-dilution” results in the sustainable catalytic sites in the apolar DMA-rich liquid-like dilute-phase crowding environments, as depicted in Figure 9Q.
In short, we developed the catalytic coacervates for photo-RAFT in dilution via site-specific monomer recruitment and localization, with the aid of macromolecular crowding and confinement. The “substantial DMA-dilution” ensured the sustainable catalytic sites within liquid-like apolar dilute-phase crowding environments, resulting in 97% conversion within 12 min. Sumerlin’s group [59] introduced an ultrafast UV-photoiniferter PISA; however, when targeting a DP of 50, the reaction slowed due to the lack of monomer compartmentalization in the micellar core [59]. Boyer and coworkers [60] achieved an ultrafast photoiniferter PISA under visible light, but at 50% v/v solids content. Shi [61] succeeded in an ultrafast ring-opening metathesis polymerization by means of monomer emulsification. An’s group [62,63,64] has demonstrated the remarkably high efficiencies of enzyme-catalyzed RAFT polymerization. Nevertheless, catalytic photo-RAFT through enzyme-like droplet compartmentalization for site-specific monomer recruitment/localization in dilution has hitherto been unreported. These findings underscore a key role of macromolecular crowding and confinement in the LLPS droplet compartmentalization and site-specific monomer recruitment/localization, and provide a practical guideline for rational design of catalytic coacervates toward lifelike reaction functionalities.
3. Conclusions
In summary, we developed catalytic coacervates for aqueous photo-RAFT in dilution, focusing on the site-specific monomer recruitment/localization through macromolecular crowding and confinement. Our results revealed that both the block and statistical block copolymers underwent hydrogen-bonded interpolymer complexes, resulting in complex ion-clusters. Intercluster complexation led to LLPS and droplet compartmentalization into charged dense-phase and neutral dilute-phase compartments. These droplets exhibited the capacity of site-specific monomer recruitment and localization via salt-bridging molecular recognition. The “substantial DMA-dilution” offered liquid-like apolar dilute-phase crowding surroundings for the sustainable dense-phase catalytic reaction sites, enabling the reaction catalysis to 97% conversion in 12 min. Notably, the strategies used for catalytic coacervates mirror those seen in enzyme active sites, such as creating apolar microenvironments, perturbing pKa of ionizable groups, and involving proton feedback mechanisms. Given that these active coacervates originated from an amino acid monomer, we propose that the active coacervates could represent rudimentary precursors of synthetic enzymes. These findings underscored the key role of macromolecular crowding and confinement in the LLPS droplet compartmentalization and site-specific monomer recruitment/localization that are essential for enzyme reaction catalysis, and provided practical guidelines for creating the catalytic coacervates toward lifelike reaction functionalities.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Shin Y. Brangwynne C.P. Liquid phase condensation in cell physiology and disease Science 2017357 eaaf 438210.1126/science.aaf 438228935776 · doi ↗ · pubmed ↗
- 2Dignon G.L. Best R.B. Mittal J. Biomolecular phase separation: From molecular driving forces to macroscopic properties Annu. Rev. Phys. Chem.202071537510.1146/annurev-physchem-071819-11355332312191 PMC 7469089 · doi ↗ · pubmed ↗
- 3Alberti S. Gladfelter A. Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates Cell 201917641943410.1016/j.cell.2018.12.03530682370 PMC 6445271 · doi ↗ · pubmed ↗
- 4Galvanetto N. IvanovićM.T. Chowdhury A. Sottini A. Nüesch M.F. Nettels D. Best R.B. Schuler B. Extreme dynamics in a biomolecular condensate Nature 202361987688310.1038/s 41586-023-06329-537468629 PMC 11508043 · doi ↗ · pubmed ↗
- 5Hyman A.A. Weber C.A. Juelicher F. Liquid-liquid phase separation in biology Annu. Rev. Cell Dev. Biol.201430395810.1146/annurev-cellbio-100913-01332525288112 · doi ↗ · pubmed ↗
- 6Banani S.F. Lee H.O. Hyman A.A. Rosen M.K. Biomolecular condensates: Organizers of cellular biochemistry Nat. Rev. Mol. Cell Biol.20171828529810.1038/nrm.2017.728225081 PMC 7434221 · doi ↗ · pubmed ↗
- 7Hanopolskyi A.I. Mikhnevich T.A. Paikar A. Nutkovich B. Pinkas I. Dadosh T. Smith B.S. Orekhov N. Skorb E.V. Semenov S.N. Interplay between autocatalysis and liquid-liquid phase separation produces hierarchical microcompartments Chem 202393666368410.1016/j.chempr.2023.08.029 · doi ↗
- 8The Nobel Prize in Chemistry 2009 (Information for the Public)—The Key to Life at the Atomic Level Available online: https://www.nobelprize.org/uploads/2018/06/popular-chemistryprize 2009.pdf(accessed on 24 December 2025)