Asymmetric Organocatalytic Addition of Malononitrile to Trifluoromethyl Arylketimines: A Viable Entry to Chiral α-CF3 Quaternary Aminoesters
Milena Ivkovic, Francesca Franco, Sergio Rossi, Sara Ferrario, Alessandra Puglisi, Maurizio Benaglia

TL;DR
This study presents a new method for creating chiral molecules with a CF3 group using asymmetric organocatalysis.
Contribution
A novel asymmetric organocatalytic method to synthesize chiral α-CF3 quaternary aminoesters is developed.
Findings
The reaction achieved up to 87/13 enantiomeric ratio using chiral iminophosphoranes.
The adduct was converted into enantioenriched aminoesters without stereochemical loss.
Computational studies helped explain the reaction mechanism and stereochemical outcome.
Abstract
In the present study the stereoselective addition of malononitrile to trifluoromethyl arylketimines promoted by chiral iminophosphoranes was investigated. A panel of structurally diverse enantiopure bifunctional superbases, which include thiourea or squaramide unit and a basic site connected by a chiral scaffold, was tested in the asymmetric organocatalytic reaction, to afford an adduct featuring a quaternary stereocenter, in up to a 87/13 enantiomeric ratio. The product was then converted in a single step transformation into the corresponding enantioenriched α-CF3 substituted quaternary aminoester, without any loss of stereochemical integrity. The absolute configuration of the final product was established by chemical correlation of the chiral compound with a known molecule. Preliminary computational studies were performed in order to elucidate the reaction mechanism and rationalize…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
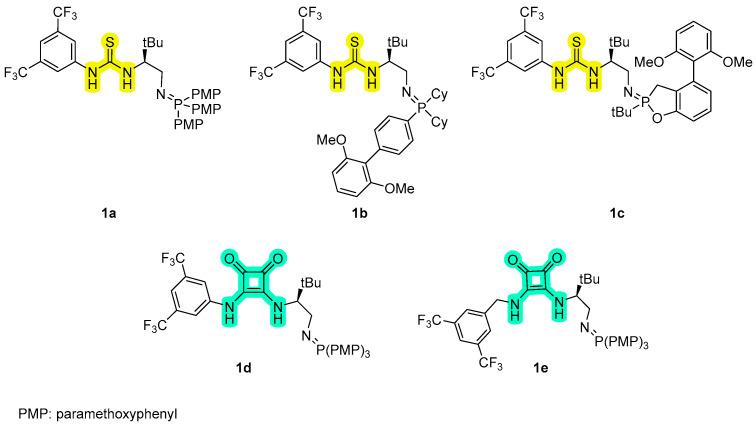 Figure 1
Figure 1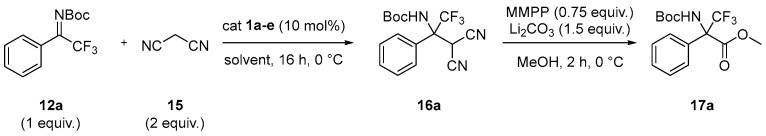 Figure 2
Figure 2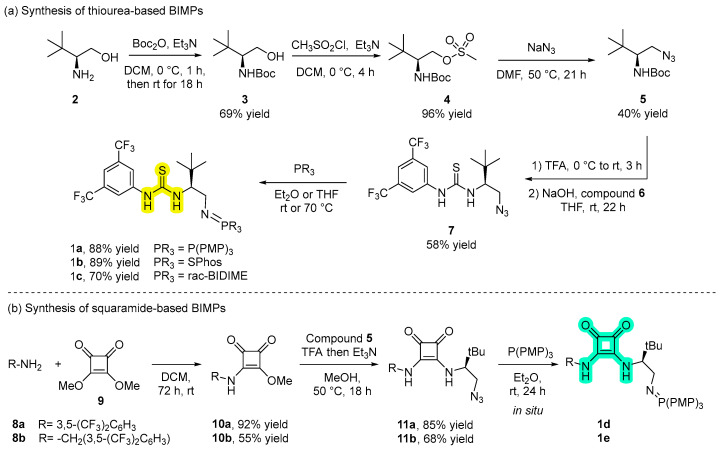 Figure 3
Figure 3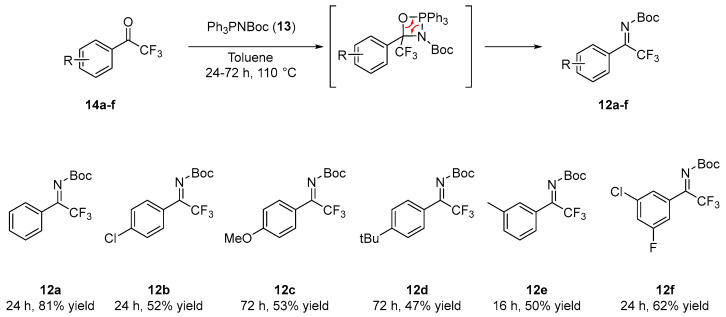 Figure 4
Figure 4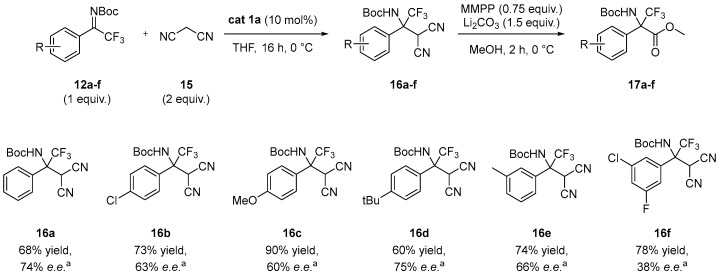 Figure 5
Figure 5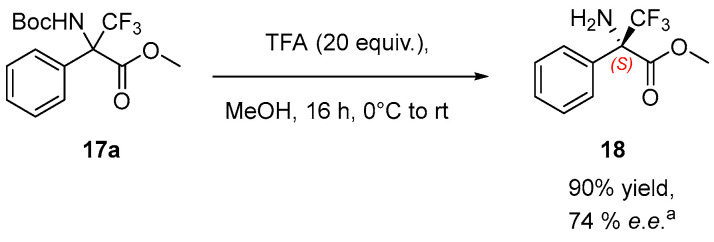 Figure 6
Figure 6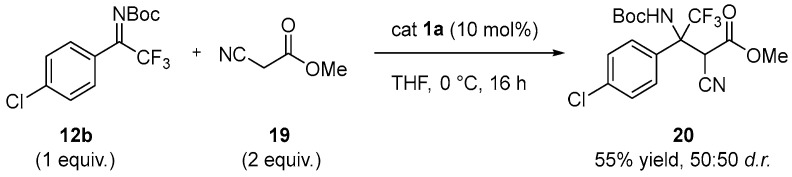 Figure 7
Figure 7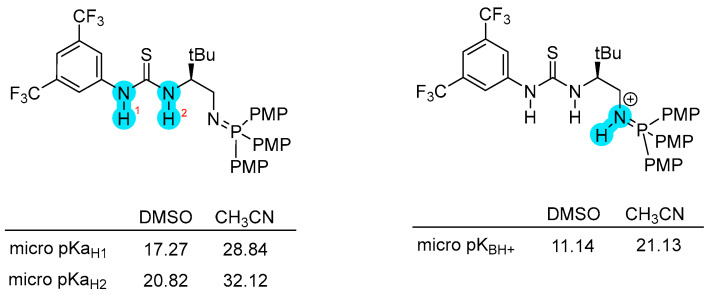 Figure 8
Figure 8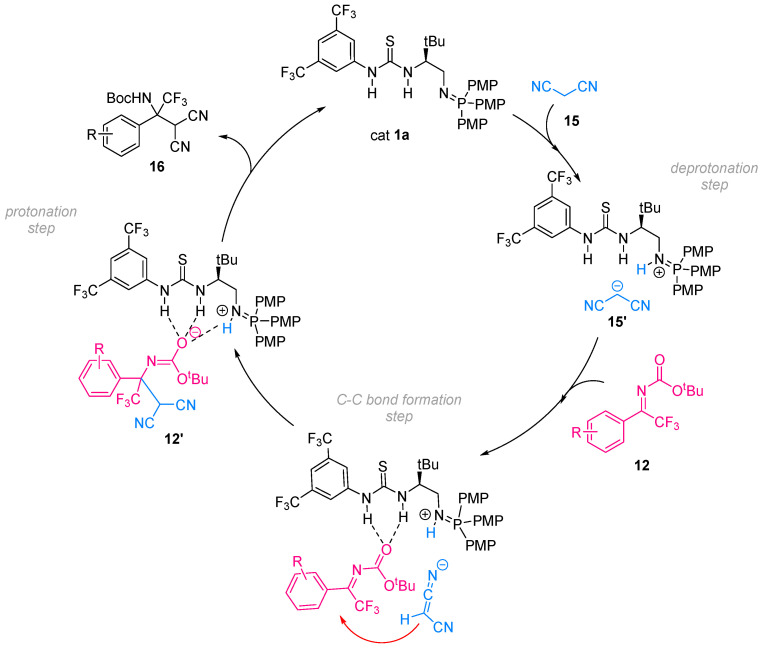 Figure 9
Figure 9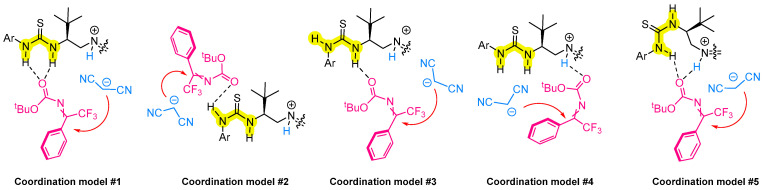 Figure 10
Figure 10- —ITN-EID project Marie Sklodowska-Curie Actions Innovative Training Network—TECHNOTRAIN H2020-MSCA-ITN-2018
- —European Union—NextGenerationEU
- —University of Milan
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Asymmetric Synthesis and Catalysis · Phosphorus compounds and reactions
1. Introduction
Chiral organosuperbases remove protons from acidic pronucleophiles to produce a reactive nucleophile. The relevant protonated catalysts may engage effectively with the reactants via hydrogen bonding or electrostatic interaction [1]. Organosuperbases are usually defined by their excellent solubility in organic solvents, notable tunability, and the ability to broaden the reaction scope to challenging substrates with elevated pKa values. Moreover, chiral organobase catalysts with an additional functional group alongside the basic site can exhibit dual functionalization, simultaneously activating both pronucleophiles and electrophiles [2].
In this context, enantiopure bifunctional iminophosphoranes have found wide application in several transformations. In 2013 Dixon reported novel acyclic amino acid derived bifunctional iminophosphorane organocatalysts featuring a thiourea unit [3]. The high activity of the catalysts was demonstrated, among others, in the Mannich reaction of nitromethane to imines, direct aldol addition of aryl ketones to α-fluorinated ketones [4], 1,4-addition reactions (enantioselective sulfa-Michael to α-substituted acrylate esters [5,6], enantioselective cyclohexadienone desymmetrization [7], and synthesis of alkylidenecyclopropanes [8].
Following our interest in the synthesis of enantiopure fluorinated amines [9], we have recently reported a very efficient enantioselective organocatalytic addition of nitromethane to trifluoromethyl aryl ketimines promoted by electron-rich bifunctional iminophosphoranes, in up to 95% e.e. [10].
We wish to report here the stereoselective addition of malononitrile to trifluoromethyl arylketimines promoted by chiral iminophosphoranes. The addition products could be easily converted to enantioenriched α-CF_3_ substituted quaternary aminoesters, without any loss of stereochemical integrity [11].
2. Results and Discussion
2.1. Organocatalysts Synthesis
Taking advantage of their structural diversity and the tunability of both the basic and the hydrogen-bonding groups, a small library of bifunctional iminophosphorane (BIMPs) catalysts 1a–e were synthetized. The basicity of the iminophosphorane unit was systematically tuned by the choice of phosphines, using tris(4-methoxypenyl)phosphine (P(PMP)3), 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos) and 3-(tert-butyl)-4-(2,6-dimethoxyphenyl)-2,3-dihydrobenzo[d][1,3]oxaphosphole (rac-BIDIME), as representative partners. Thiourea- and squaramide-based H-bond donors were also explored as structural variations in the hydrogen-bond donor unit (Figure 1).
The synthetic routes to access the thiourea- and squaramide-based organocatalysts follow different pathways. In the case of thiourea based-BIMPs, catalysts 1a–c were prepared from commercially available l-tert-leucinol 2 according to the literature procedure [10]. As shown in Scheme 1a, the free amino group of l-tert-leucinol was Boc protected to give carbamate 3. Subsequent activation of the hydroxyl group with methanesulfonyl chloride results in the formation of mesylate 4, which then underwent nucleophilic substitution with NaN_3_ to give the corresponding azide 5. Deprotection of 5 with TFA, followed by treatment with aqueous NaOH and with 3,5-bistrifluoromethyl phenyl isothiocyanate 6, leads to the formation of organoazide 7, a common intermediate for the synthesis of catalysts 1a–c. Intermediate 7 was then reacted in the Staudinger reaction with the desired phosphine, to afford the corresponding thiourea-based BIMP catalysts in good yields.
In contrast, squaramide-based BIMPs were accessed by condensation of the corresponding amine intermediates with commercially available dimethyl squarate under mild conditions (Scheme 1b). Due to stability issues, iminophosphorane catalysts 1d–e were not isolated but generated in situ from stable precursors 11a–b during the asymmetric addition of malononitrile to trifluoromethyl aryl ketimines. 3,5-bis(trifluoromethyl)aniline 8a and 3,5-bis(trifluoromethyl)benzylamine 8b were reacted with 3,4-dimethoxy-3-cyclobutene-1,2-dione 9 to afford the corresponding derivatives 10a–b in good to excellent yields. At this point, a treatment with previously synthetized organoazide 5 afforded the desired stable precursors 11a–b which were converted in situ into the desired organocatalysts 1d–e by reaction with P(PMP)3 [12].
2.2. N-Boc Aryl Trifluoromethyl Ketimines Synthesis
N-Boc trifluoromethyl ketimines 12a–f where synthesized via a [2+2] cycloaddition of N-Boc-imino-(triphenyl)-phosphorane 13 with commercially available trifluoromethyl ketones 14a–f, performed in toluene under reflux. The resulting four-membered heterocyclic intermediate easily undergoes reverse [2+2] cycloaddition to yield the target ketimines in 24–72 h [13]. Six ketimines exhibiting different electronic and steric properties where then prepared in 47–81% yields (Scheme 2).
2.3. Enantioselective Mannich Addition of Malononitrile to N-Boc Aryl Trifluoromethyl Ketimine
Having synthesized the required precursors, we investigated the enantioselective addition of malononitrile 15 to N-Boc aryl trifluoromethyl ketimine 12a, selected as model compound, for the preparation of enantioenriched dicyano adduct 16a. The addition product 16a was then converted into the high-value α-trifluoromethyl α-amino esters 17a by reaction with magnesium bis(monoperoxyphthalate) hexahydrate (MMPP) [14], for which the enantiomeric excesses were determined. Reactions were performed on a 0.2 mmol scale of ketimine using 2 equiv. of malononitrile, 10 mol% of BIMP organocatalyst at 0 °C in 0.75 mL of the desired solvent. Selected results are reported in Table 1 (see Supporting Information for further details).
Using toluene as the solvent, we first evaluated the influence of the basic moiety of the bifunctional organocatalyst. Catalysts 1a, 1b, and 1c, bearing different iminophosphorane residues, afforded the desired product 16a in comparable yields (76–85%) and with enantioselectivities up to 56% e.e. (entries 1–3). In contrast, with catalysts 1d and 1e compound 16a was obtained in high yields but in racemic form, indicating that their structural features are not suitable for effective chiral discrimination in this transformation (entries 4–5). Since catalyst 1a provided the most promising results, it was selected as catalyst of choice for the following screening. Different solvents and temperatures were then investigated. Ethers such as THF, MTBE, and Me-THF (entries 6–8) lead to the formation of desired product with slightly reduced yields (63–68%) but significantly improved enantiomeric excesses (74% e.e. in THF and 72% e.e. in MTBE and Me-THF). In contrast, chlorinated and polar aprotic solvents such as DCM and CH_3_CN (entries 9–10) provide good yields but very low enantioselectivities. Reaction performed in dioxane at room temperature (entry 11) afforded compound 16a in moderate yield and enantioselectivity, whereas an attempt performed in THF at −20 °C for 96 h provided the desired product in 72% yield without improvement of the enantioselectivity (74% e.e. entry 12).
Having identified the best reaction conditions (Table 1, entry 7), the substrate scope of ketimines 12a–f in the organocatalytic enantioselective addition was investigated (Scheme 3).
Para-substituted ketimines 16b–d bearing either electron-withdrawing or electron-donating substituents were well tolerated, providing the corresponding adducts 16a–d in yields of up to 90% (compound 16c) and enantioselectivities of up to 75% (compound 16d). When meta-substituted ketimine 12e was employed, product 16e was isolated in 74% yield and 66% e.e. Interestingly, electron poor disubstituted ketimine 12f was also successfully engaged in the enantioselective Mannich addition, affording the corresponding product 16f in 78% yield and 38% e.e.
To assign the absolute configuration of the reaction product, compound 17a was converted into compound 18 in 90% yield via amine deprotection without erosion of the enantiomeric excess (Scheme 4). According to the literature it was not possible to determine the enantiomeric excess of compound 18 by chiral GC or HPLC; therefore, the enantiomeric purity was assigned by comparison with a reported optical purity value [15]. The optical purity of compound 18 was determined by polarimetric analysis and compared with the reported literature values, confirming that the compound adopt the (S)-configuration [15].
To further evaluate the feasibility of the transformation and to access more synthetically demanding, highly functionalized compounds, the reaction was extended to the more challenging nucleophile methyl 2-cyanoacetate 19 in combination with ketimine 12b. Gratefully, under the optimized reaction conditions, the corresponding product 20 was obtained in 55% yield as a 1:1 mixture of diastereoisomers (Scheme 5). Further studies will be required to determine the enantiomeric excess.
2.4. Computational Analysis. pKa Values
In order to rationalize the stereochemical outcome of this transformation, computational analysis was performed. At first a DFT investigation of the acid/base properties of catalyst 1a was investigated. It is known that the pKa values of widely used chiral bifunctional dialkylamino and cinchona-derived (thio)urea organocatalysts fall within the range of 8.5–19.6 in DMSO [16], with the strongest acidic example being Schreiner’s thiourea [17,18], whereas one of the least acidic examples is the Jacobsen-List organocatalysts [19,20].
Although bifunctional iminophosphoranes are often described as superbases catalysts due to their pronounced reactivity in bifunctional activation [1,2], no experimental determinations of BIMPs intrinsic acid-base profile are currently available. Only a few examples related to the determination of iminophosphorane bases pKa have been reported [3,7]. For this reason, we computed the micro-pKa and pK_BH_^+^ values of catalyst 1a, considering both its neutral and protonated forms, in order to clarify its acid–base profile and its potential role in the catalytic activation of different substrates.
Quantum chemical methods have been widely employed for pKa predictions, using either direct methods based on Born–Haber cycles [21], or indirect approaches, commonly referred to as the “isodesmic method [22].” Direct methods provide results usually aligned with experimental measurements but require high computational costs, especially when explicit solvation models are employed, making them impractical for larger systems. Indirect isodesmic methods, on the other hand, are computationally less demanding but require the use of an experimental pKa value from a reference acid-base reaction involving species similar to those under investigation. In this approach, structural similarity is critical, since even small differences can lead to significant deviations in acidity, making the careful selection of an appropriately comparable reference a challenging task. Isodesmic method has been successfully applied to the determination of the acid-base profile of the iminophosphorane moiety of BIMPs (BH^+^ → B + H^+^ profile), using pKa of aniline as reference [23]; however, to the best of our knowledge, no pKa determination of the neutral structure has been computed.
To overcome the limitation of isodesmic approach, in 2022, Busch and co-workers reported an alternative and more general computational method able of predicting pKa values for a broad range of compounds across different solvents, which does not require being referred to a known reference species. This method is based on experimental aqueous pKa values of known molecules combined with absolute potentials of the standard hydrogen electrode (SHE) in non-aqueous media (LFESR approach) [24]. Inspired by these developments, we adopted this computational strategy to estimate the acidities of catalyst 1a in DMSO and CH_3_CN. Taking advantage of the LFESR approach, it was possible to estimate the proton solvation energy at a minimal computational cost. A summary of the pKa estimations is presented in Scheme 6. DFT calculations were performed using Gaussian 16, Revision C.01, the M06-2X functional and 6-311++G (d,p) basis set in combination with SMD solvation model since it offered the best combination of accuracy, reliability, and time consumption [24].
The computed acidity trends provide clear insight into the acid behavior of catalyst 1a in different solvents. In both DMSO and CH_3_CN media, the N–H group of the thiourea bound to the 3,5-bis(trifluoromethyl)phenyl ring (N–H1) is consistently more acidic than the N–H group located proximal to the iminophosphorane unit (N–H2), as evidenced by systematically lower micro-pKa values (pKa_H1_ < pKa_H2_). This identifies N-H1 group as the most acidic site of the catalyst. Although the overall acidity of the catalyst is formally determined by the macroscopic pKa, the two micro-pKa values differ by more than two orders of magnitude, making the contribution of the less acidic site negligible for all practical purposes. The absolute pKa values, however, exhibit a pronounced solvent dependence: all micro-pKa values are substantially higher in CH_3_CN than in DMSO, reflecting the reduced ability of acetonitrile to stabilize charged species.
Considering the acidity trends discussed above and based on previously reported stereoselective transformations promoted by this class of catalysts (ketimine nitro-Mannich reactions [10], sulfur–Michael additions [6], and conjugate additions to enone diesters [5]), a plausible reaction mechanism for the enantioselective addition of malononitrile 15 to ketimine 12a was proposed (Scheme 7).
Following the initial deprotonation of malononitrile 15 by the basic iminophosphorane moiety, the resulting malononitrile anion 15′ is engaged in a nucleophilic attack on the ketimine 12, which is simultaneously activated through dual hydrogen-bond coordination between the thiourea unit of the catalyst and the N-Boc protecting group of the substrate. This is consistent with the fact that the most acidic moiety of the catalyst is involved in hydrogen-bond formation with the ketimine, whereas the basic site, once protonated, interacts via electrostatic interaction with the malononitrile anion. It should be noted that the deprotonation step and nucleophilic attack could occur in a concerted manner or through discrete, sequential steps. However, according to this proposed catalytic cycle, the reactive partners are aligned within a well-defined chiral environment, favoring the formation of a new C–C bond in a stereoselective manner. After that, a proton transfer from the protonated iminophosphorane to the negatively charged N-Boc-protected intermediate 12′ occurs, leading to the formation of the desired N-Boc-protected fluorinated dicyano compound 16 and regenerating the free BIMP catalyst which is able to re-enter the catalytic cycle.
2.5. Computational Analysis
In order to confirm this hypothesis, preliminary computational studies were performed on the transition states involved in this transformation. For this purpose, calculations were performed considering the ketimine 12a and catalyst 1a, which has been shown to promote the formation of the desired compound (S)-16a with 74% e.e. To comprehensively investigate all relevant transition states, several key structural and mechanistic factors were considered. These included the E/Z configuration of ketimine 12a, the conformational flexibility of the thiourea unit (capable of adopting either s-cis or s-trans arrangements) and the hydrogen-bonding interactions within 12a and three distinct acidic sites of the catalyst (namely the N-H1 and N-H2 groups of the thiourea moiety and the P=NH^+^ group). Additionally, the approach of the nucleophilic malononitrile anion to either the re or si face of the imine becomes particularly relevant for the final configuration of the product.
Considering all these aspects, at least five different coordination models can be proposed, (Scheme 8) considering all of them and taking into account the E/Z configuration of the imine as well as the nucleophilic attack on the si/re face, four transition states can be identified for each model, resulting in a total of 20 transition states.
Initial conformational geometries were obtained by Monte Carlo conformational analysis performed with molecular mechanics calculations using the OPLS4 force field [25] of the Macromodel package in the Schrodinger suite [26]. These structures were then fully optimized in vacuo using the PM6 semiempirical method of the Gaussian package [27]. This level of theory was chosen as a compromise between computational cost and the need to explore the large number of transition states arising from the five proposed coordination models, enabling a rapid and systematic investigation of all relevant transition state geometries. Harmonic vibrational calculations were also performed to confirm that the optimized structures correspond to first-order saddle points, each exhibiting a single imaginary frequency. All five different coordination models where analyzed, and in the notation used for describing transition states, (s-cis)/(s-trans) refers to the conformation of the thiourea unit, E/Z indicates the configuration of the imine, and re/si denotes the face of the imine exposed to nucleophilic attack by malononitrile.
A detailed analysis of the possible 20 transition states is available in the Supporting Information. Although additional computational studies at higher level are necessary, from the preliminary investigation, considering that imine Z is known to be more stable, it resulted that low energy TS could be found with both coordination modes one and five, where a clear preference for nucleophilic attack on the si face of the Z-configured imine is present, in line with the experimental observations. While the results provide a useful preliminary overview of the relative stabilities of the 20 transition states across the five proposed coordination models, they should be interpreted with caution. The absolute values and relative ordering may change at a higher level of theory and when solvent effects are considered, which are known to influence both the stabilization of transition states and the stereochemical outcome. Therefore, the present data are most useful for identifying trends and for selecting the most relevant transition states to be further optimized and evaluated using more accurate computational methods.
3. Material and Methods
Reactions were carried out under a positive pressure of nitrogen and dry solvents were used. Reactions were monitored by thin layer chromatography (TLC) on Macherey-Nagel (Düren, Germany) pre-coated silica gel plates (0.25 mm) and visualized by UV irradiation at 254 nm. Whenever necessary, a ninhydrin solution or a permanganic solution was used as stains for developing TLC plates. Flash chromatography was performed on standard flash column chromatography on Merck (Darmstadt, Germany). silica gel 60 (particle size: 0.04–0.063 mm). Hexane, pentane, ethyl acetate (EtOAc), dichloromethane (DCM), methanol (MeOH), and diethyl ether (Et_2_O) were used as standard eluent solvents.
^1^H NMR, ^13^C NMR and ^19^F NMR spectra were recorded at 25 °C on Bruker Avance spectrometers (300 MHz, 75 MHz, 282 MHz) or NEO 400 MHz (Bruker, Billerica, MA, USA) (400 MHz for ^1^H NMR, 101 MHz for ^13^C NMR). Deuterated solvents acquired from Sigma-Aldrich (Steinheim, Germany). were used as supplied. The spectra were recorded in ppm using the solvent peak as a reference for ^1^H and ^13^C NMR spectra (7.26, 77.16 for CDCl_3_).
High-resolution mass spectra (HRMS) were obtained from the Unitech COSPECT center, University of Milan and performed on a Q-TOF Synapt G2-Si (Waters, Milford, MA, USA) using an Acquity UPLC I-Class photodiode array (PDA) detector (Waters, Milford, MA, USA).
Enantiomeric excess determinations were performed with an Agilent (Santa Clara, CA, USA) Instrument Series 1100, using a Chiralpak AD or a Lux Phenomenex 3 µm Amylose-1 as column (eluent: n-hexane/isopropanol 95:5 according to the sample, flow rate as specified).
XYZ geometries of transition states are available as a separate supporting material on a Dataverse repository.
3.1. General Procedure for the Synthesis of N-Boc Trifluoromethyl Aryl Ketimines (12a–f)
According to the literature procedure [13], N-Boc-imino-(triphenyl)-phosphorane 13 (2 equiv.) was added to a solution of 300 mg (1 equiv.) of the corresponding commercially available trifluoroacetophenone 14a–f in dry toluene (5 mL). The reaction mixture was heated and stirred for 24–72 h (depending on starting ketone) at 110 °C. The reaction was monitored by TLC and/or ^1^H NMR in CDCl_3_. After the consumption of starting ketone, the reaction was cooled down to room temperature and toluene was removed under reduced pressure. The residue was purified by silica gel column chromatography (n-Hexane/AcOEt from 98:2 to 90:101) to afford ketimines 12a–f in modest to good yields. All the analytical data are in agreement with the literature [10,28].
3.2. General Procedure for the Enantioselective Synthesis of 15a–f
A 10 mL Schlenk tube under nitrogen was charged with ketimine 12a–f (0.2 mmol, 1 equiv., 0.27 M in THF) and 14.6 mg of iminophosphorane catalyst 1a (0.02 mmol, 0.1 equiv.). The reaction mixture was cooled down to 0 °C, and 26.4 mg of malononitrile 15 (0.4 mmol, 2 equiv.) were added. The reaction mixture was stirred for 16 h at 0 °C, then THF was removed under reduced pressure and the crude was purified by column chromatography on silica gel (n-Hexane/AcOEt from 100:0 to 90:10) to afford compounds 16a–f.
3.3. General Procedure for Oxidative Decyanation of 16a–f
According to the literature [14], under nitrogen atmosphere, a flask was charged with compounds 16a–f (1 equiv., 0.1 M in dry methanol) and dry methanol at 0 °C. Magnesium monoperoxyphthalate hexahydrate (MMPP, 0.75 equiv.), and Li_2_CO_3_ (1.5 equiv.) were added to the stirring solution. The reaction mixture was stirred for 2 h at 0 °C. The reaction was stopped and quenched by water and extracted with DCM (3 × 5 mL). The combined organic layers were dried over anhydrous Na_2_SO_4_, filtered, and concentrated under reduced pressure to afford quaternary amino esters 17a–f.
Determination of enantiomeric excesses was conducted by chiral HPLC (column: Chiralpak AD, Lux Phenomenex 3 µm Amylose-1; eluent: n-hexane/isopropanol 95:5, flow rate 1 mL/min).
4. Conclusions
In conclusion, a new mild and efficient methodology for the synthesis of α-trifluoromethyl α-aminoesters was disclosed. Good yields, up to 90%, and enantioselectivities up to 75% were achieved. The methodology was applied to a small library of ketimines and a very preliminary attempt of using methyl 2-cyanoacetate as nucleophile is reported. The absolute configuration of the reaction product was assigned as (S) enantiomer, based on literature data. Preliminary computational investigation on the pKa values of the bifunctional iminophosphorane [29,30] catalysts and studies of the possible transition states of the reaction were also performed, thus opening the way to a better understanding of the mode of action of those bifunctional organosuperbases and to a rationalization of the observed stereochemical outcome of the organocatalytic transformation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Wang Y. Cao Z. Li Q. Lin G. Zhou J. Tian P. Activating Pronucleophiles with High p Ka Values: Chiral Organo-Superbases Angew. Chem. Int. Ed.2020598004801410.1002/anie.20191348431788933 · doi ↗ · pubmed ↗
- 2Formica M. Rozsar D. Su G. Farley A.J.M. Dixon D.J. Bifunctional Iminophosphorane Superbase Catalysis: Applications in Organic Synthesis Acc. Chem. Res.2020532235224710.1021/acs.accounts.0c 0036932886474 · doi ↗ · pubmed ↗
- 3Núñez M.G. Farley A.J.M. Dixon D.J. Bifunctional Iminophosphorane Organocatalysts for Enantioselective Synthesis: Application to the Ketimine Nitro-Mannich Reaction J. Am. Chem. Soc.2013135163481635110.1021/ja 409121 s 24107070 PMC 3931333 · doi ↗ · pubmed ↗
- 4Thomson C.J. Barber D.M. Dixon D.J. Catalytic Enantioselective Direct Aldol Addition of Aryl Ketones to α-Fluorinated Ketones Angew. Chem. Int. Ed.2020595359536410.1002/anie.20191612931961990 · doi ↗ · pubmed ↗
- 5Farley A.J. Sandford C. Dixon D.J. Bifunctional Iminophosphorane Catalyzed Enantioselective Sulfa-Michael Addition to Unactivated α-Substituted Acrylate Esters J. Am. Chem. Soc.2015137159921599510.1021/jacs.5b 1022626679772 · doi ↗ · pubmed ↗
- 6Yuan K. Ristache A.J. Kosc S.M. Crumpton A. Dixon D.J. Bifunctional Iminophosphorane Superbases Enable the Highly Enantioselective Sulfa-Michael Addition to Fully Substituted Cyclopropene Carboxylic Acid Derivatives J. Am. Chem. Soc.2025147400454005010.1021/jacs.5c 0784941123596 PMC 12593379 · doi ↗ · pubmed ↗
- 7Poh C.Y.X. Rozsar D. Yang J. Christensen K.E. Dixon D.J. Bifunctional Iminophosphorane Catalyzed Amide Enolization for Enantioselective Cyclohexadienone Desymmetrization Angew. Chem. Int. Ed.202463 e 20231540110.1002/anie.20231540138055190 · doi ↗ · pubmed ↗
- 8Golec J.C. Tan D.H. Yamazaki K. Hiekink E.H. Christensen K.E. Hamlin T.A. Dixon D.J. Catalytic enantioselective synthesis of alkylidenecyclopropanes Nature 202564593293810.1038/s 41586-025-09485-y 40789330 PMC 12460155 · doi ↗ · pubmed ↗