Genotypic Diversity and Antimicrobial Resistance Profiles of Multidrug-Resistant Escherichia coli in Porcine Populations from Hubei, China
Xiaoyue Li, Zewen Liu, Ningning Wang, Rui Guo, Wenjie Chen, Wei Liu, Ting Gao, Keli Yang, Yongxiang Tian, Fangyan Yuan

TL;DR
This study examines drug-resistant E. coli in pigs from Hubei, China, finding high levels of antibiotic resistance and diverse genetic profiles.
Contribution
The study provides a comprehensive analysis of E. coli from pigs in Hubei, integrating genetic diversity, resistance, and virulence data.
Findings
E. coli isolates showed 100% resistance to lincosamides and sulfonamides, with all being multidrug-resistant.
ST46 was the most common sequence type, and isolates carried multiple resistance genes like blaTEM and mcr-1.
Abstract
The indiscriminate and excessive use of antimicrobial agents in livestock production is a major driver of antimicrobial resistance (AMR), thereby posing a grave threat to global public health. Although several surveillance studies have documented antimicrobial resistance patterns of swine-derived E. coli in different regions of China, comprehensive investigations integrating multilocus sequence typing (MLST), resistance determinants, and virulence gene profiles have remained scarce for central China, particularly Hubei province, since 2018. This study investigated the prevalence of antibiotic resistance, and molecular epidemiology of E. coli isolated from swine farms in Hubei province, China, while simultaneously analyzing their clonal and genetic diversity. A total of 148 E. coli isolates were collected from porcine sources in central China, revealing distinct regional variations in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1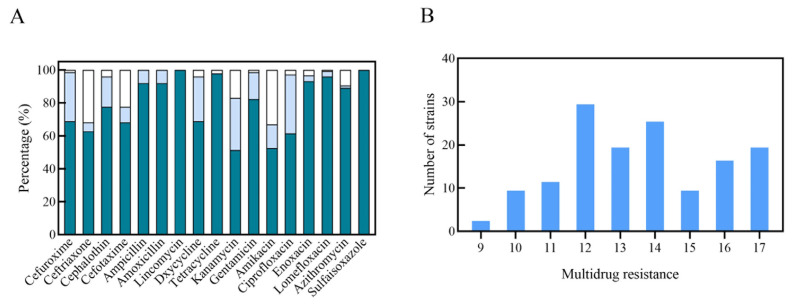 Figure 2
Figure 2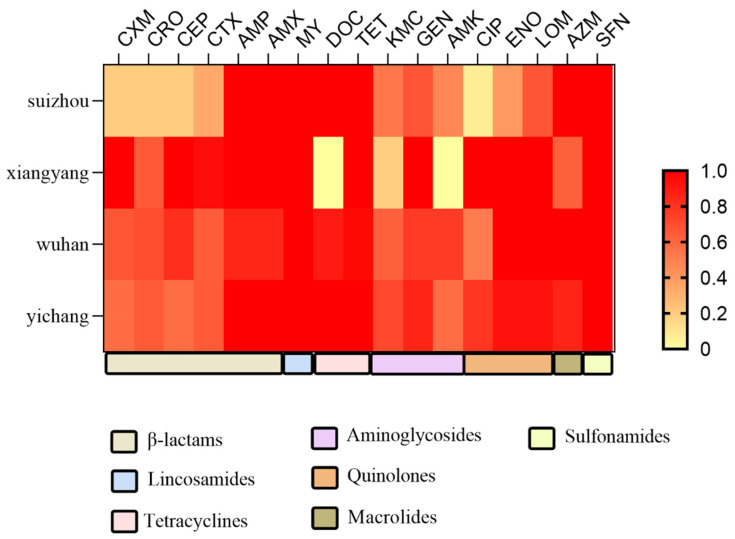 Figure 3
Figure 3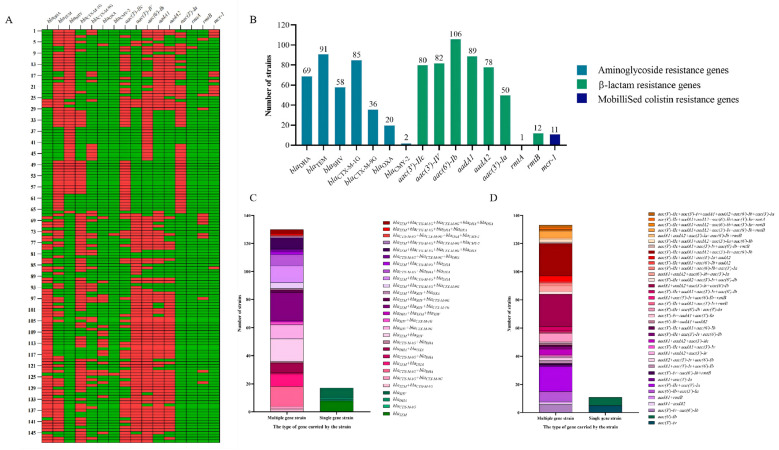 Figure 4
Figure 4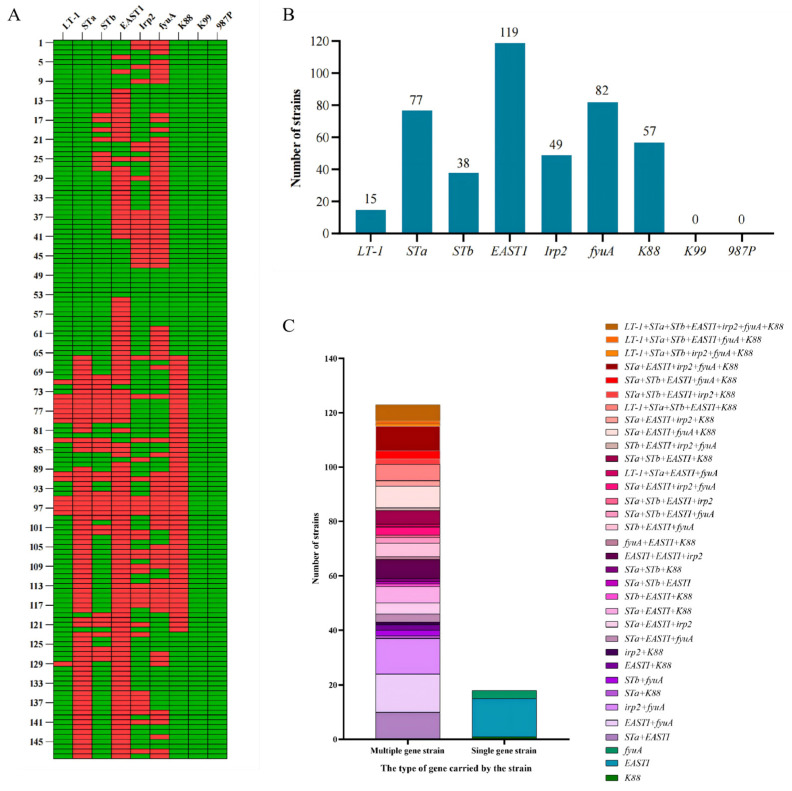 Figure 5
Figure 5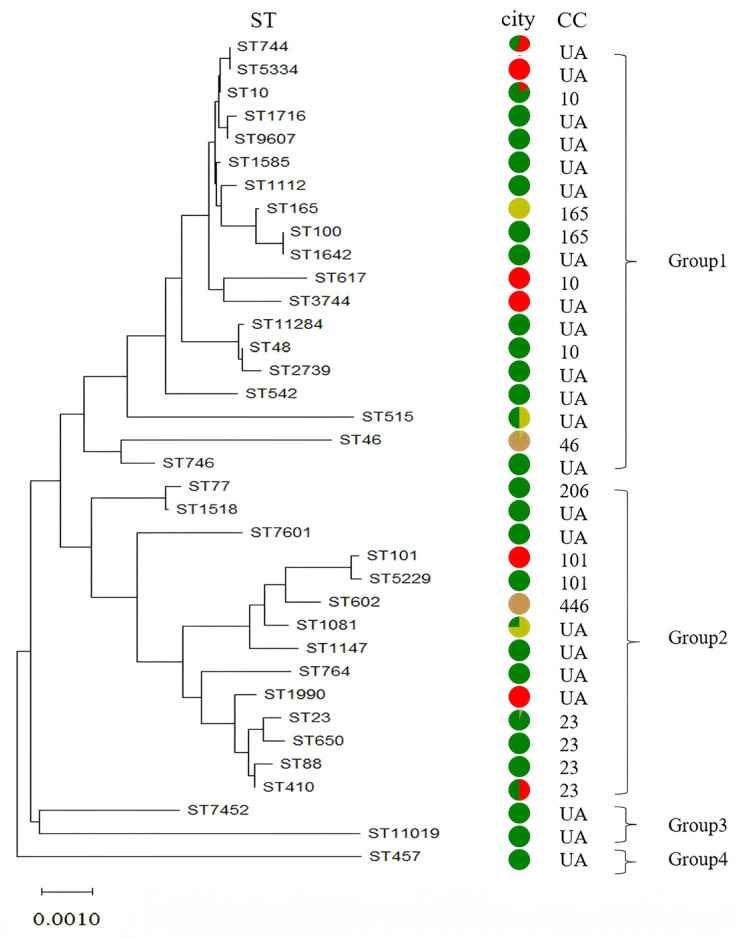 Figure 6
Figure 6 Figure 7
Figure 7 Figure 8
Figure 8 Figure 9
Figure 9- —the Technical Innovation Project of Hubei Province
- —the Hubei Provincial Major Science and Technology Innovation Plan
- —the Hubei Province Innovation Center of Agricultural Sciences and Technology
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEscherichia coli research studies · Antibiotic Resistance in Bacteria · Salmonella and Campylobacter epidemiology
1. Introduction
Escherichia coli is a predominant commensal bacterium residing as part of the natural gut microbiota in humans and warm-blooded animals [1,2,3,4]. However, pathogenic isolates of E. coli are a major cause of intestinal infections in neonatal piglets aged 1–10 days, primarily causing diarrhea and edema in this vulnerable population [5]. The lack of effective vaccines to prevent E. coli outbreaks attributable to its diverse serotypes and region-specific epidemiological variations has led to a heavy reliance on antimicrobial agents for disease management in most agricultural settings. The escalating use of antimicrobials in livestock production has precipitated the emergence of multidrug-resistant (MDR), extensively drug-resistant (XDR), and even pandrug-resistant (PDR) bacterial isolates, raising significant global health concerns. Of particular urgency is the rapid dissemination of antimicrobial resistance (AMR), a critical public health crisis that now permeates multiple sectors of society and imposes substantial economic burdens worldwide [6].
Antimicrobial resistance (AMR) has evolved into one of the most critical global health threats of the past three decades [7]. Of particular concern is the rising prevalence of drug-resistant Gram-negative bacteria in livestock populations, which can transmit to humans through environmental pathways and food chain contamination [8]. The intensive use of antibiotics in swine production has exacerbated this trend, with resistance profiles in pig-derived pathogens showing accelerated development when compared with other livestock sectors. Notably, surveillance data from Great Britain revealed higher AMR prevalence in porcine-derived E. coli isolates than those from cattle and sheep [9], underscoring the urgency to systematically characterize antibiotic resistance profiles of pathogenic E. coli in Chinese swine operations. Surveillance data from 2011–2012 revealed high resistance rates in swine E. coli isolates to tetracycline (79.57%), trimethoprim-sulfamethoxazole (73.12%), and kanamycin (55.91%) [10]. Subsequent monitoring in Guangdong province (2013–2016) detected multidrug resistance (MDR) in 100% of 333 E. coli isolates from commercial pig operations [11]. A surveillance of E. coli in Chinese pig farms from 2018 to 2019 indicated that 90.54% were multidrug-resistant (MDR) or had common resistance to tetracycline (96.26%) etc., and occasional resistance to last-resort drugs. Among 515 sequenced MDR isolates were 101 O-serogroups, 118 sequence types, 109 AMR genes, and 53 plasmid groups [12]. The results revealed that sequence types (ST88, ST100) and serotypes (O9:H19, O116:H11, O149:H10) of E. coli isolates exhibited enhanced virulence, while F18ab-fimbriated isolates carrying stx2A genes showed higher cytotoxicity, 95.8% were multidrug resistant, and 79% harbored oqxB-aac(3) gene clusters [13].
These findings reflect global concerns where MDR Gram-negative pathogens contribute to 60–70% of antimicrobial treatment failures in veterinary practice, highlighting critical challenges for animal health management [14].
Extended-spectrum beta-lactamase (ESBL)-producing-E. coli is spreading worldwide and poses a public health issue [15]. Escherichia coli has evolved multiple β-lactamase-mediated resistance mechanisms, including extended-spectrum β-lactamases (ESBLs; SHV, TEM, OXA, and CTX-M types), plasmid-mediated AmpC β-lactamases (CMY-2, a plasmid-mediated AmpC-like gene) and carbapenemases (MBL, KPC, and class D oxacillinases) [16]. CTX-Ms formed the largest group of ESBLs, and they have become globally disseminated. Currently, CTX-M-15 (a part of the CTX-M-1 group) is the most frequent CTX-M worldwide. This is closely followed by CTX-M-14 (a member of the CTX-M-9 group) [17]. Given the limited data on the epidemiology of MDR E. coli from pigs in Hubei province in China, it is interesting to investigate the spread of these resistant bacteria, particularly the ESBL types, which may be the cause of the spread of resistance genes from pork to human.
Several epidemiological studies on E. coli have been initiated in parts of China. In northeastern China, a survey showed that the separation rate of E. coli isolated from pig fecal samples reached 88% [18]. An investigation on pig farms in Henan province found that the positive rate of E. coli was 70.74%, of which the commonly sequenced types (STs) 10 and 101 were identified [19]. However, data on the genetic diversity and antimicrobial resistance of E. coli is still restricted in the Hubei province of central China.
E. coli can induce porcine postweaning diarrhea (PWD) [20], and it may be associated with the expression of fimbriae to mediate adhesion and colonization of porcine enterocytes [21,22]. Most adhesins are related with fimbriae F18 and F4 (K88), F5 (K99), F6 (987P) and F41, with F18 and F4 (K88) being leading causes of PWD in piglets [23,24,25]. The predominant toxins of pathogenic E. coli include those that are heat-labile (LT-I and LT-II) [26,27] and heat-stable (STa and STb) [28], as well as enteroaggregative E. coli heat-stable enterotoxin 1 (EAST1) [29], and these produce other virulence factors, such as pathogenicity islands (PAI). The locus of enterocyte effacement (LEE) and high-pathogenicity islands (HPIs) are particularly significant PAIs. Irp2 and fyuA genes, located in high-pathogenicity islands (HPIs), encode a yesiniabactin-like iron-scavenging system [30]. However, E. coli isolated from pigs with diarrhea carry different types of virulence genes, which can enhance the pathogenicity of E. coli through synergistic effects. However, data on characteristics of virulence genes of E. coli is still restricted in the Hubei province of central China.
Multilocus sequence typing (MLST) has been used to study the evolution and epidemiology of a number of bacterial pathogens. It has become the method of choice for typing epidemiologically important isolates. MLST is a widely used and standardized molecular typing method that enables comparison of E. coli lineages across studies and geographic regions, and is particularly suitable for population structure and epidemiological investigations [31]. Investigating the trend characterization of epidemic isolates will help us to better understand epidemiology.
This present study was carried out to investigate the prevalence and characteristics of E. coli. The MLST genotypes, antibiotic resistance and virulence genes of E. coli isolates were examined, having been collected from 2019 to 2022 in Hubei province of China. These findings provide information and implications for safeguarding and commanding the occurrence of diseases in future studies.
2. Results
2.1. Isolation of E. coli Isolates and Antimicrobial Susceptibility Profile
E. coli isolates were obtained from the fecal matter of weaned piglets from Hubei province. A total of 148 isolates of E. coli were separated from samples and further identified by PCR tests as E. coli. There are 15 isolates of E. coli isolated from Suizhou city, 37 isolates of E. coli isolated from Xiangyang city, 82 isolates of E. coli isolated from Wuhan city and 14 isolates of E. coli isolated from Yichang city (Figure 1).
2.2. Antimicrobial Susceptibility Profile of E. coli Isolates
Antimicrobial susceptibility testing (AST) results showed that all of the E. coli isolates showed resistance to lincosamides and sulfonamide (Figure 2A). A high rate of resistance to sulfaisoxazole, tetracycline, lomefloxacin, enoxacin, ampicillin, amoxicillin, azithromycin, and gentamicin has also been observed for the isolates, among which 100% of the E. coli was resistant to sulfaisoxazole; 97.97% of the E. coli was resistant to tetracycline; 95.95% of the E. coli was resistant to lomefloxacin; 93.24% of the E. coli was resistant to enoxacin; 91.89% of the E. coli was resistant to ampicillin; 91.89% of the E. coli was resistant to amoxicillin; 89.19% of the E. coli was resistant to azithromycin; and 82.43% of the E. coli was resistant to gentamicin. The isolates demonstrated a relatively low rate of resistance to cephalothin (77.7%) cefuroxime (68.92%), doxycycline (68.92%), cefotaxime (68.24%), ceftriaxone (62.84%), ciprofloxacin (61.49%), amikacin (52.7%) and kanamycin (51.35%) (Figure 2A and Figure 3).
As shown in Figure 2B, all of the isolates were multidrug-resistant (MDR) isolates, and most of the isolates were resistant to more than three drug classes. AST revealed that the 20 isolates showed severe resistance profiles that were resistant to the seventeen antimicrobial agents tested. Overall, most of the isolates were conferring resistance to 12–14 of the antimicrobial agents tested. Among the antibiotics tested, high resistance rates were observed for sulfaisoxazole, azithromycin, lomefloxacin, enoxacin, amikacin, tetracycline, doxycycline, lincomycin, cefuroxime, ceftriaxone, cephalothin, and cefotaxime.
E. coli in different regions has separate antibiotic resistance. The AST results revealed that resistance to lincomycin, and sulfaisoxazole was a common phenotype of the isolates from the pig farms in four cities, including Xiangyang, Yichang, Suizhou, and Wuhan in Hubei province (Figure 3). E. coli isolates resistant to doxycycline and amikacin were isolated, including in Yichang, Suizhou, and Wuhan in Hubei province; Xiangyang was the only region from which no isolates from pig farms with the abovementioned resistant phenotypes were detected (Figure 3). Notably, MDR isolates were identified on pig farms in Hubei province, but a relatively high proportion of the MDR isolates were identified on farms in Wuhan and Yichang relative to those from the other city.
2.3. Detection of Antimicrobial Resistance Genes
16 antibiotic resistance genes (ARGs) in 148 isolates of E. coli were detected (Figure 4A) using PCR-based assays. Of these, aac(6′)-Ib (71.6%, 106/148), blaTEM (61.5%, 91/148), aadA1 (60.1%, 89/148), blaCTX-M-1G (57.4%, 85/148), aac(3′)-Iv (55.4%, 82/148), aac(3′)-IIc (54.1%, 80/148), aadA2 (52.7%, 78/148), and blaDHA (46.9%, 69/148) were prevalent with higher detection rates (Figure 4B). Among the 148 E. coli isolates, 147 isolates carried β-lactamase genes with a detection rate of 99.3%, of which, the detection rates of blaSHV, blaTEM, blaOXA, blaCMY-2 and blaDHA were 39.2% (58/148), 61.5% (91/148), 13.5% (20/148),1.4% (2/148) and 46.6% (69/148), respectively. The detection rate of blaCTX-M-1G in CTX-M was 57.4% (highest) and the detection rate of blaCTX-M-9G was 24.3%.
Among the 148 E. coli isolates, 144 isolates carried aminoglycoside-modifying enzyme genes with a detection rate of 97.3%, of which the detection rates of rmtB (8.1%) and aac(3′)-Ia (33.8%) were prevalent with lower detection rates. Amongst the isolates positive for aminoglycoside-modifying enzyme genes, only one strain was positive for rmtA. In addition, the detection rate of the mobilized colistin resistance gene mcr-1 was 7.4%.
Antibiotic resistance genes were detected in all 147 isolates, with only one strain having no detected antibiotic genes. Of the 147 isolates, 91 (61.5%) had more than 6 ARGs, 4 (2.7%) isolates had more than 10 ARGs, and 2 isolates had 11 ARGs (Supplementary Table S1).
2.4. Various Extended-Spectrum β-Lactamase Genes Were Present in the Isolates
Among all of the isolates tested, 147 (99.3%) carried ESBLs and 130 of them harbored various number of ESBLs. The most frequent was blaTEM + blaSHV + blaCTX-M-1G (13.5%, 20/148); 64 isolates were found to have two different ESBLs genes; 52 isolates were found to have three different ESBLs genes; 11 isolates were found to have four different ESBLs genes; and 3 isolates were found to have five ESBLs genes—blaTEM, blaCTX-M-1G, blaCTX-M-9G, blaDHA and blaOXA (Figure 4C, Supplementary Table S1).
2.5. Various Extended-Spectrum Aminoglycoside-Modifying Enzyme Genes Were Present in the Isolates
Amongst the 148 isolates tested, 144 (97.3%) isolates carried aminoglycoside-modifying enzyme genes and 133 of them harbored various numbers of aminoglycoside-modifying enzyme genes. The most frequent were aadA1 + aadA2 + aac(3′)-Iv + aac(6′)-Ib and aac(3′)-IIc + aadA1 + aadA2 + aac(3′)-Iv + aac(6′)-Ib (15.5%, 23/148); 34 isolates were found to have two different ESBLs genes; 22 isolates were found to have three different ESBLs genes; 41 isolates were found to have four different ESBLs genes; 27 isolates were found to have five different ESBLs genes; and 9 isolates were found to have six different ESBLs genes (Figure 4D, Supplementary Table S1).
2.6. Prevalence of Virulence Genes in E. coli Isolates from Pigs
The distribution of eight virulence genes in 148 isolates of E. coli has been examined in this study. As shown in Figure 5A,B, the virulence gene EASTI (80.4%, 119/148) was found in most of the E. coli isolates, followed by fyuA (55.4%, 82/148), STa (52%, 77/148), K88 (38.5%, 57/148) and irp2 (3.1%, 49/148). While K99 and 987P were not detected in all isolates.
Among the 148 isolates tested, 141 (95.3%) carried virulence genes and 123 of these harbored a various number of virulence genes. The most frequent were EASTI+ and EASTI + fyuA+ (9.9%, 14/141), followed by irp2 + fyuA+ (9.2%, 13/141), STa + EASTI (7.1%, 10/141), STa + EASTI + irp2 + fyuA + K88+ (6.4%, 9/141), STa + EASTI + fyuA + K88+ (5.7%, 8/141), EASTI + EASTI + irp2+ (5.0%, 7/141), LT-1 + STa + STb + EASTI + irp2 + fyuA + K88+ (4.3%, 6/141), LT-1 + STa + STb + EASTI + K88+ (4.3%, 6/141), and STa + EASTI + K88+ (4.3%, 6/141) (Figure 5C, Supplementary Table S1).
2.7. Multi-Locus Sequence Typing (MLST)
The genetic diversity of these E. coli isolates was analyzed with MLST. A total of 148 isolates were analyzed utilizing MLST, the identification of 38 sequence types (STs). Among these isolates, 101 out of the 148 isolates possessed 14 different STs which belonged to 7 CCs. The remaining 46 isolates belonged to 23 different unassigned STs. The presence of new STs was due to new combinations of previously known alleles in adk (allele 7), fumC (allele 7), gyrB (allele 193), icd (allele 1), mdh (allele 986), purA (allele 159) and recA (allele 139). The predominant STs were ST46, ST23 and ST10 containing 29 (19.59%), 20 (13.51%), 12 (8.11%) isolates respectively. Ten STs contained three or more isolates with ST602, ST165, ST100, ST744, ST7452, ST1642, ST1081, ST515, ST48 and ST101 comprising nine (6.08%), eight (5.41%), eight (5,41%), seven (4.73%), five (3.38%), four (2.70%), four (2.70%), four (2.70%), three (2.03%) and three (2.03%) isolates, respectively. Seven STs (ST410, ST650, ST764, ST1147, ST1518, ST2739 and ST5229) contained two isolates each. Eight STs (ST77, ST88, ST457, ST542, ST617, ST710, ST746, ST1112, ST1585, ST1716, ST1990, ST3744, ST5334, ST7601, ST9607, ST11019 and ST11284) had only one isolate each (Figure 6).
Minimum-spanning trees showed that the tested E. coli were mainly classed into seven clonal complexes and other unassigned clone complexes. CC-46 was the most frequently isolated clonal complex, containing 29 isolates belonging to one ST, and accounted for 19.59% (29/148) of all the isolates. The major clonal complexes also included CC-23 (25/148, 16.89%), CC-10 (16/148, 10.81%) and CC-165 (16/148, 10.81%). The other isolates covered 28 STs belonging to three CCs and were unassigned.
To further analyze sequence types (STs), a neighbor-joining phylogenetic analysis was performed based on concatenated MLST sequences (Figure 6). The NJ phylogram classified the identified STs into four major phylogenetic groups. Group 4 comprised a single isolate belonging to an unassigned ST, whereas Group 3 included six isolates representing two different unassigned STs. In contrast, Groups 1 and 2 encompassed the majority of isolates (n = 140) and contained most of the dominant clonal complexes, including CC-10, CC-23, CC-46, CC-101, CC-165, CC-206, CC-446, as well as several unassigned STs. Specifically, CC-10 included ST10, ST48, and ST617; CC-23 included ST23, ST88, ST410, and ST650; CC-101 included ST101 and ST5229; and CC-165 included ST100 and ST165 (Figure 6).
3. Discussion
E. coli is one of the main pathogenic bacteria that impact the production and growth of pigs on pig farms. It is associated with gastrointestinal diseases such as diarrhea, edema disease, and systemic infections such as septicemia and polyserositis [2]. These diseases caused by E. coli can increase mortality, morbidity and growth delays of piglets, which are responsible for economic losses. This study analyzed the prevalence, genetic diversity and antibiotic resistance of disease, which may help us to improve methods of prevention and treatment.
E. coli isolates exhibit high AMR with widespread multidrug resistance and extensive carriage of resistance genes, showing regional differences in resistance phenotypes.
From 2002 to 2008, the prevalence of E. coli isolated from pork chop samples was 44% in the United States [32]. However, various incidence rates have also been reported in different regions of China. From 2003 to 2009, the prevalence of E. coli isolates from pig farms was 77.78% in China [33]. From 2013 to 2016, the positive rates of E. coli between farm 1 and farm 2 were 40.25% and 59.75%. respectively, in Guangdong province [11]. Between 2016 and 2017, a survey indicated that the separation rate of E. coli isolated from pig fecal swabs reached 88% in northeastern China, including Heilongjiang, Jilin and Liaoning [18].
Diarrhea in weaned piglets driven by E. coli remains a principal cause of economic losses for the pig industry. This commonly requires antimicrobial drug treatment, which is of considerable expense when used to cure piglets infected with this pathogen. In 2011, the study of Agersө et al. found that 32% of isolates have multi-drug resistance, mainly concentrated on ampicillin (27%) and tetracycline (29%) [34]. In 2012, Tadesse et al. tested 1729 isolates of E. coli antibiotic susceptibility varied from different sources, the resistance rate of E. coli increased from 7.2% to 63.6% but the most common resistance was to tetracycline, streptomycin and sulfonamides [35]. A total of 131 E. coli isolates were obtained from the pigs presenting from diarrhea in Switzerland from 2014 to 2015, the isolates exhibited resistance to tetracycline (50%), sulfamethoxazole (49%), ampicillin (26%), gentamicin (17%), and ciprofloxacin (8%) [36]. However, this caused a rise in the employment of various antimicrobial agents, such as lincosamides, tetracyclines and sulfonamides, which may expand antimicrobial resistance.
In this study, antimicrobial susceptibility testing of the E. coli isolates revealed the highest prevalence of resistance to three classes of antimicrobials: lincosamides, tetracyclines, and sulfonamides. This observation is likely associated with the long-term irrational use of these three antibiotics for controlling bacterial diarrhea in piglets within Hubei province.
Notably, over 80% of the E. coli isolates exhibited high resistance rates to seven antimicrobials, specifically tetracycline, lomefloxacin, enoxacin, ampicillin, amoxicillin, azithromycin, and gentamicin. Resistance was also detected against β-lactam drugs: the resistance rate to cephalothin (a first-generation cephalosporin) reached 77.7%, which was higher than that to cefuroxime (a second-generation cephalosporin, 68.9%), the third-generation cephalosporins cefotaxime (68.2%) and ceftriaxone (62.8%).
All isolates displayed varying degrees of resistance to the tested antimicrobials, and most E. coli isolates exhibited a high prevalence of multidrug resistance. Among them, 20 isolates were resistant to all 17 tested antimicrobials. Overall, more than half of the isolates showed resistance to 9–17 antimicrobial agents. The most common MDR pattern was resistance to 12 antimicrobials (cefuroxime, amikacin, ampicillin, amoxicillin, lincomycin, doxycycline, tetracycline, gentamicin, enoxacin, lomefloxacin, azithromycin, and sulfaisoxazole), which was observed in 30 isolates.
For context, relevant studies conducted in other regions of China have reported comparable trends. Jiang et al. found that E. coli isolates had high resistance rates to ampicillin (99.5%), tetracycline (93.4%), and amoxicillin (65.1%), with prevalent resistance also detected against cephalosporins, quinolones, and aminoglycosides [37]. Meng et al. similarly noted that the majority of E. coli isolates were resistant to tetracycline (79.57%), trimethoprim-sulfamethoxazole (73.12%), and kanamycin (55.91%) [10]. In contrast, a study in Sichuan province (2012–2013) reported that E. coli isolates had the highest resistance to sulfamethoxazole (61.6%), followed by tetracycline (61.2%), ampicillin (48.2%), and kanamycin (22.4%) [38]. Additionally, a multi-provincial survey (covering seven provinces) showed that E. coli isolates from pig farms had resistance rates of 81.44% to ampicillin, 94.37% to tetracycline, and 88.36% to sulfaisoxazole [39]. Collectively, these findings provide critical insights and implications for guiding the rational application of antibiotics in swine production in future research and practice.
It has been documented that E. coli has a great ability to accumulate ARGs, mainly through horizontal gene transfer [40]. In this study, a large number of ARGs are detected in various isolates and it was found that a higher proportion contained six or more resistance genes. One of the most common antibiotic resistance mechanisms in E. coli is mediated by the production of β-lactamase, ESBLs are an increasing cause of resistance to third-generation cephalosporins, including the fourth-generation cephalosporins in Enterobacteriaceae [41,42]. The blaTEM, blaCTX-M and blaSHV types have been recognized as the most prevalent ESBL genes that confer antibiotic resistance among pathogens [43,44]. In this study, five ESBL genes and two AmpC enzyme genes (blaDHA, blaCMY-2) were detected. Among the five ESBL genes, blaTEM (61.5%) was the most prevalent gene, similar to the ESBL epidemic in Guizhou in 2021 [45]; followed by blaCTX-M-1 at 57.4%, blaSHV at 39.2%, blaCTX-M-9G at 24.3% and blaOXA at 13.5%. ESBLs are paradigmatic of resistance, are usually encoded on mobile genetic elements that accelerate their dissemination, and have become another challenge for drug resistance control in pig farms [46].
It is inevitable for bacteria to develop resistance to aminoglycosides due to their widespread use. There have been reports of aminoglycoside resistance in both Gram-negative and Gram-positive bacteria [47]. In this study, the detection rate of five aminoglycoside-modifying enzyme genes (aac(6′)-Ib, aac(3′)-IV, aac(3′)-IIc, aadA1, aadA2) exceeds 50%, and it is possible to carry multiple aminoglycoside modifying enzyme genes in a single strain, leading to high levels of resistance to aminoglycosides. In 2015, the plasmid-mediated colistin resistance gene, mcr-1, was reported for the first time in E. coli isolate from the animals and their food in China [48], it has quickly spread to human pathogens [49]. The transfer of colistin resistance by plasmid has been ascribed to the mcr-1 gene, which is the most predominant type of mcr gene [50]. The low detection rate (7.4%) of mcr-1 in this study likely results from the low use of polymyxins in pig feed in Hubei. It is important to monitor such isolates closely to prevent their spread. The concern of antibiotic resistance in these categories is serious and needs urgent attention. Additionally, it demands urgent attention in terms of the regularization monitoring of antimicrobial susceptibilities and the efficient administration of bacterial infections to restrict the further spread of multidrug resistance in Hubei province.
In past decades, the occurrence and spread of PWD in piglets has caused massive economic losses to the development of the pig farming industry in China [51]. The highest detection rate of virulence factor has been found for EAST1 (80.4%), followed by fyuA (55.4%). Fimbriae adhesins are necessary in the pathogenetic mechanism, the most common adhesins on E. coli from PWD in pigs are fimbriae F4 and F18 [52]. F4 (K88) (38.5%) was identified in this study, which indicates that it is closely related with pathogenic E. coli. For enterotoxins detected in this study, the gene encoding the heat-stable enterotoxin STa was frequently detected (52.0%), which is similar to findings reported in Korea and other countries [53,54], followed by the heat-stable enterotoxin STb (25.7%). The detection of heat-labile enterotoxin (LT-1) (10.1%) was less than that of the STa and STb in this study. It is possible that the comparative identification of enterotoxins seems to vary from one geographic area to another. This study finds the distribution and characteristics of virulence factors in E. coli in Hubei province, and the data may be useful for establishing preventive measures for PWD.
The application of MLST in E. coli isolates facilitates a better understanding of the genetic diversity of these E. coli isolates. In this study, a total of 148 isolates were analyzed utilizing MLST, leading to the identification of 38 sequence types (STs), the most frequent ST was ST46, followed by ST23, ST10, ST602, ST165, ST100 and ST744. These isolates belonged to seven CCs and other unassigned clone complexes, including CC46, CC23, CC10 and so on. This reveals that there is abundant diversity in ST types among porcine E. coli isolates in Hubei province. Notably, the ST distribution profile in our study exhibits distinct discrepancies compared with previously reported data, both from the same region (Hubei province) and other regions of China. First, from the city-level perspective, no city-exclusive STs were identified in this study: dominant sequence types (e.g., ST46, ST23, ST10) were detected across multiple cities in Hubei province, indicating regional circulation of these lineages rather than strict geographic restriction. This pattern may reflect potential dissemination via animal movement, shared farming practices, or environmental transmission routes between farms in different cities. Second, our study identified ST46 as the most prevalent sequence type, which differs from findings reported in other regions of China. For instance, Yang et al. [55] found that ST10 was the most common sequence type (22/171, 12.9%), followed by ST48 (16/171, 9.4%) and ST744 (8/171, 4.7%). Additionally, the prevalence of ST10 in our study (12/148, 8.11%) was lower than that documented in previous research. Zhang et al. [11] have reported that the proportion of ST10 reached 50% (16/32). The potential reasons are the limitation of the data size, regional epidemiological characteristics of E. coli clones, and differences in sample collection strategies (e.g., variations in farm scale, feeding patterns, and disease prevalence periods between studies). Recently, E. coli ST10 has not only been detected in animals in China but also isolated from human infections in China [56], indicating the potential zoonotic risk of this ST clone, which warrants further surveillance. These findings highlight the necessity of the continuous regional epidemiological surveillance of E. coli ST types, as the distribution of dominant clones may vary geographically, which is crucial for guiding local disease prevention and control strategies.
Based on the detected virulence gene combinations, most isolates could be classified as enterotoxigenic E. coli (ETEC), which is consistent with their isolation from diarrheic piglets. Several dominant STs, including ST46, ST23, and ST10, were detected across multiple cities, suggesting regional dissemination rather than city-specific circulation. Notably, some of these STs have also been reported in human infections, indicating potential zoonotic relevance. Although this study was not designed to establish causal relationships between virulence and antimicrobial resistance, an isolate-level integrative analysis revealed that several multidrug-resistant isolates simultaneously harbored multiple virulence-associated genes. However, no clear linear correlation was observed between the number of resistance genes and virulence genes across isolates. This finding suggests that antimicrobial resistance and virulence traits are not directly proportional but may be co-selected under similar selective pressures in intensive swine production systems.
Dominant STs exhibited heterogeneous resistance and virulence profiles, indicating that clonal background alone does not determine pathogenic potential. The coexistence of multidrug resistance and enterotoxigenic virulence factors in several isolates underscores their epidemiological relevance. The detection of similar STs across multiple cities supports the hypothesis of regional dissemination and highlights the importance of continuous surveillance to monitor the spread of multidrug-resistant E. coli within and beyond swine populations.
Several limitations should be acknowledged in this study. First, although phenotypic resistance to sulfonamides was universally observed, sulfonamide resistance genes were not investigated, which limits mechanistic interpretation and warrants further molecular characterization in future studies. Second, while multiple antimicrobial resistance genes and virulence-associated genes were detected, a systematic one-to-one correlation between resistance genotype and phenotypic susceptibility, as well as between virulence gene content and clinical severity, could not be established. This was partly due to the absence of detailed clinical outcome data and individual antimicrobial treatment histories for the sampled piglets. Third, although transmission patterns and horizontal gene transfer were discussed, the lack of whole-genome sequencing data constrained comprehensive analyses of mobile genetic elements and resistance gene dissemination. Future studies integrating genomic, clinical, and antimicrobial usage data will be essential to identify high-risk lineages and better understand the evolution and spread of multidrug-resistant E. coli.
In this study, a high antimicrobial resistance and the genotypic diversity of E. coli were isolated from swine in Hubei province and observed. From the results obtained it can be concluded that these isolates present highly prevalent multi-drug resistance. These data provide a greater understanding of the genetic diversity and antimicrobial resistance of E. coli. From a One Health perspective, the “risk” associated with individual isolates should be interpreted as a composite characteristic arising from the combined presence of multidrug resistance and key virulence determinants rather than from sequence type alone. The presence of multidrug-resistant diarrheagenic E. coli in swine populations represents a potential reservoir for resistance determinants that may disseminate across animals, humans, and the environment, emphasizing the need for integrated antimicrobial stewardship strategies.
4. Materials and Methods
4.1. Bacterial Isolates and Identification
From 2019 to 2022, a total of 148 fecal swab samples were collected from pig farms in 4 cities: Xiangyang, Yichang, Suizhou, and Wuhan in Hubei province. The samples were taken from diseased piglets with diarrhea symptoms. These isolates were obtained using MacConkey agar, which was incubated at 37 °C for 24 h. Pink colonies were picked and purified again, then bacterial isolates cultured in Luria–Bertani (LB) broth at 37 °C for 24 h. And further confirmed by the PCR amplification of the 16S rRNA gene with primers (16S-F: 5′-AGAGTTTGATCATGGCTCAG-3′; 16S-R: 5′-TAGGGTTACCTTGTTACGACTT-3′) as previously described [55]. PCR amplification was performed as previously described, with minor modifications to the annealing temperature and extension time to optimize amplification efficiency. Primer sequences remained unchanged.
4.2. Antibiotic Resistance Profiles
According to the guidelines of the Clinical and Laboratory Standards Institute [57], the confirmed E. coli were assayed for antimicrobial susceptibility testing. E. coli isolates were examined for susceptibility to antimicrobial drugs utilizing a disk diffusion assay. All samples were analyzed for the presence of resistant bacteria. A total of 17 antimicrobials were tested, comprising cefuroxime (CXM), ceftriaxone (CRO), cephalothin (CEP), cefotaxime (CTX), ampicillin (AMP), amoxicillin (AMX), lincomycin (MY), doxycycline (DOC), tetracycline (TET), kanamycin (KMC), gentamicin (GEN), amikacin (AMK), ciprofloxacin (CIP), enoxacin (ENO), lomefloxacin (LOM), azithromycin (AZM), and sulfaisoxazole (SFN). Inoculated plates were incubated at 37 °C for 24 h and the diameters of the inhibition zone were subsequently measured (in mm). Antimicrobial susceptibility testing was performed using the disk diffusion method. Isolates were categorized as susceptible, intermediate, or resistant according to the Clinical and Laboratory Standards Institute (CLSI) M100 guidelines based on inhibition zone diameters. The E. coli strain ATCC 25922 was utilized for quality control. Multidrug resistance (MDR) was defined as resistance to at least one antimicrobial agent in three or more different antimicrobial classes, in accordance with established criteria [58]. The antimicrobial agents tested in this study represented eight antimicrobial classes, including cephalosporins, penicillins, lincosamides, tetracyclines, aminoglycosides, fluoroquinolones, macrolides, and sulfonamides.
4.3. Detection of Antibiotic Resistance Genes
To extract the total DNA of each E. coli strain, we boiled the lysis of the isolated colonies. PCR was used to identify genes responsible for resistance to β-lactamase genes (blaDHA, blaCMY-2, blaTEM, blaSHV, blaCTX-M-1G, blaCTX-M-9G, blaOXA), aminoglycoside-modifying enzyme genes (aac(3′)-Ia, aac(3′)-IIc, aac(3′)-IV, aac(6′)-Ib, aadA1, aadA2, rmtA, rmtB), and polypeptide resistance gene mcr-1. The primers for PCR are listed in Table 1 and were synthesized at Sangon Biotech, Shanghai, China.
4.4. Detection of Virulence-Associated Genes
Considering the contribution of virulence genes to the invasiveness and pathogenicity of E. coli, DNA was isolated and we identified nine virulence-associated genes of each strain, including LT-1, STa, STb, EAST1, irp2, fyuA, K88, K99, and 987P, which were amplified by PCR, as previously described [51,68,69,70]. These virulence genes were selected because they represent major adhesins, enterotoxins, and pathogenicity island-associated factors commonly implicated in porcine enterotoxigenic E. coli (ETEC) infections. The primer sequences of virulence genes are shown in Table 2. PCR reactions were carried out in a final volume of 25 μL containing 12.5 μL mix (Vazyme Biotech, Nanjing, China), 1 μL each of primers, and 2 μL bacterial DNA. PCR was performed in a GeneAmp PCR System 9700 (Applied Bio-systems, Darmstadt, Germany), under the following conditions: initial denaturation at 95 °C for 5 min; cycling consisted of 35 cycles of 30 s at 94 °C, 30 s at 58 °C, and 1 min at 72 °C, with a final extension at 72 °C for 10 min. The resulting amplification products were separated by electrophoresis in 1% agarose gel, stained with ethidium bromide and visualized using a GelDoc XR system (Bio-Rad, Shanghai, China).
4.5. MLST and Phylogenetic Tree
The multilocus sequence typing (MLST) was executed on E. coli isolates according to the E. coli MLST database guidelines (https://enterobase.warwick.ac.uk/species/ecoli/allele_st_search, accessed on 9 January 2023), according to the protocols published on the web site. Briefly, the seven house-keeping genes adk, fumC, gyrB, icd, mdh, purA and recA were amplified employing a PCR protocol, and the amplicons sequenced utilizing the amplification primers. We investigated individual gene sequences and allocated an allelic profile number in line with the MLST database. Sequence type (ST) and clone complex (CC) designations of each strain were composed of seven alleles. Multilocus sequence typing (MLST)-based phylogenetic analysis was performed using concatenated sequences of seven housekeeping genes. Sequence alignment was conducted in MEGA version 11.0, and a neighbor-joining (NJ) phylogenetic tree was constructed using the Kimura 2-parameter (K2P) substitution model. The robustness of the tree topology was assessed by 1000 bootstrap replicates.
4.6. Statistical Analysis
The Student’s t-test was employed to analyze the data. When p < 0.05, difference was considered statistically significant. The analysis was performed using GraphPad Prism version 8 (GraphPad Software, Inc., San Diego, CA, USA).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marques C. Belas A. Franco A. Aboim C. Gama L.T. Pomba C. Increase in antimicrobial resistance and emergence of major international high-risk clonal lineages in dogs and cats with urinary tract infection: 16 year retrospective study J. Antimicrob. Chemother.20187337738410.1093/jac/dkx 40129136156 PMC 5890753 · doi ↗ · pubmed ↗
- 2Fairbrother J.M. NadeauÉ. Gyles C.L. Escherichia coli in postweaning diarrhea in pigs: An update on bacterial types, pathogenesis, and prevention strategies Anim. Health Res. Rev.20056173910.1079/AHR 200510516164007 · doi ↗ · pubmed ↗
- 3Suryadevara M. Clark A.E. Wolk D.M. Carman A. Rosenbaum P.F. Shaw J. Molecular Characterization of Invasive Staphylococcus aureus Infection in Central New York Children: Importance of Two Clonal Groups and Inconsistent Presence of Selected Virulence Determinants J. Pediatr. Infect. Dis. Soc.20132303910.1093/jpids/pis 08726619440 · doi ↗ · pubmed ↗
- 4Moore J.E. Watabe M. Millar B.C. Rooney P.J. Loughrey A. Goldsmith C.E. Direct molecular (PCR) detection of verocytotoxigenic and related virulence determinants (eae, hyl, stx) in E. coli O 157:H 7 from fresh faecal material Br. J. Biomed. Sci.20086516316510.1080/09674845.2008.1197812118986109 · doi ↗ · pubmed ↗
- 5Casey W.T. Mc Clean S. Exploiting molecular virulence determinants in Burkholderia to develop vaccine antigens Curr. Med. Chem.2015221719173310.2174/092986732266615040811130425850766 · doi ↗ · pubmed ↗
- 6Yang S.C. Lin C.H. Aljuffali I.A. Fang J.Y. Current pathogenic Escherichia coli foodborne outbreak cases and therapy development Arch. Microbiol.201719981182510.1007/s 00203-017-1393-y 28597303 · doi ↗ · pubmed ↗
- 7Beasley D.W. Davis C.T. Whiteman M. Granwehr B. Kinney R.M. Barrett A.D. Molecular determinants of virulence of West Nile virus in North America Arch. Virol. Suppl.200418354110.1007/978-3-7091-0572-6_415119761 · doi ↗ · pubmed ↗
- 8Umpierrez A. Bado I. Oliver M. Acquistapace S. Etcheverría A. Padola N.L. Vignoli R. Zunino P. Zoonotic Potential and Antibiotic Resistance of Escherichia coli in Neonatal Calves in Uruguay Microbes Environ.20173227528210.1264/jsme 2.ME 1704628904264 PMC 5606698 · doi ↗ · pubmed ↗