Kinetic and Potentiometric Characteristics of Ferredoxin: NADP+ Oxidoreductase from Chlorobaculum tepidum
Dominykas Laibakojis, Daisuke Seo, Narimantas Čėnas, Mindaugas Lesanavičius

TL;DR
This study investigates the redox properties and reaction mechanisms of a key enzyme in Chlorobaculum tepidum, revealing insights into its electron transfer behavior and potential applications in bioelectrochemical systems.
Contribution
The study provides new insights into the reoxidation mechanism of FAD in CtFNR and its electron transfer properties using various oxidants.
Findings
The reoxidation of FAD semiquinone is the rate-limiting step in CtFNR reactions.
The two-electron reduction midpoint potential of FAD at pH 7.0 is −0.282 V.
Electron transfer distances in reactions with Q and ArNO2 range from 2.6–3.4 Å.
Abstract
Chlorobaculum tepidum ferredoxin: NADP+ oxidoreductase (CtFNR) is a dimeric thioredoxin reductase (TrxR)-type FNR, whose mechanism and redox properties are poorly characterized. In this work, we focused on the reoxidation mechanisms of its flavin adenine dinucleotide (FAD) cofactor using quinones (Q), nitroaromatics (ArNO2), and other nonphysiological oxidants with different single-electron reduction midpoint potentials (E71) and electrostatic charge. Like in other FNRs, the rate-limiting step of the reaction is the reoxidation of FAD semiquinone (FADH•). However, only one FAD per dimer functions in CtFNR due to some nonequivalence of the NADP(H) binding domains in separate subunits. The reactivity of Q increases with increasing E71, while ArNO2 form another analogous series of lower reactivity. The compounds are reduced in a dominant single-electron way. These data are consistent with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
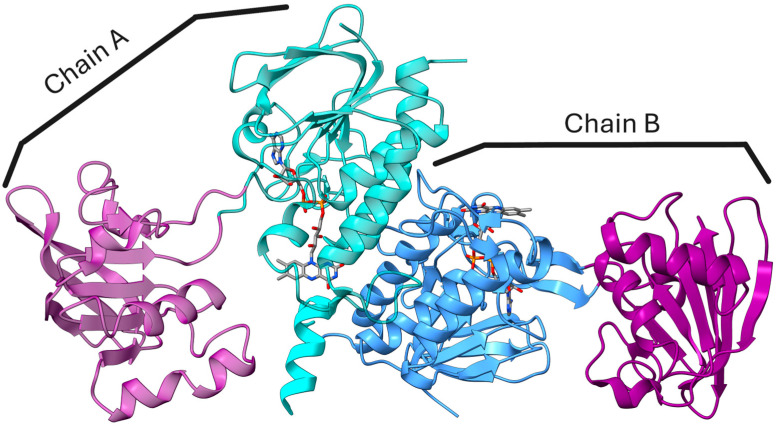 Figure 1
Figure 1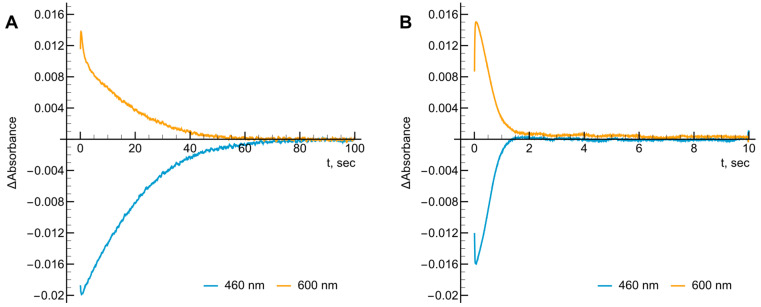 Figure 2
Figure 2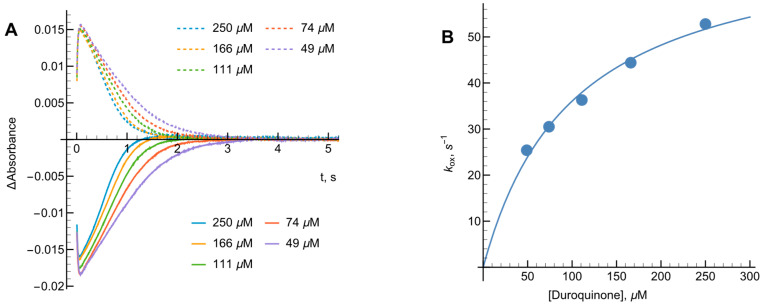 Figure 3
Figure 3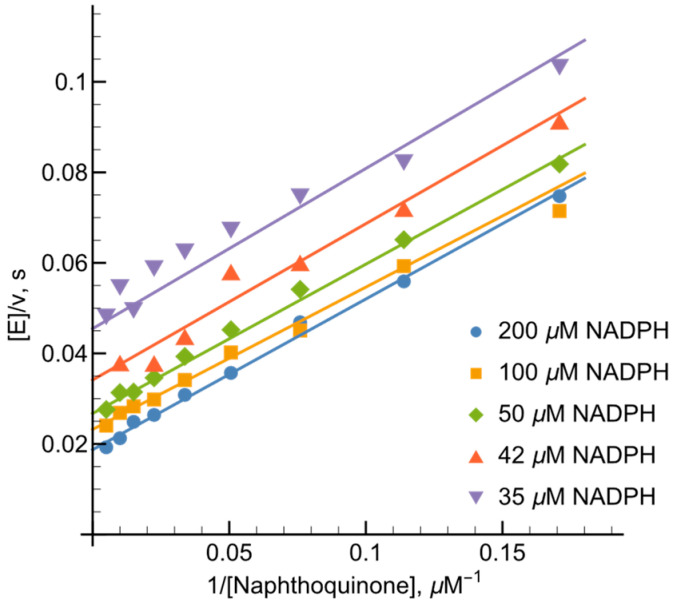 Figure 4
Figure 4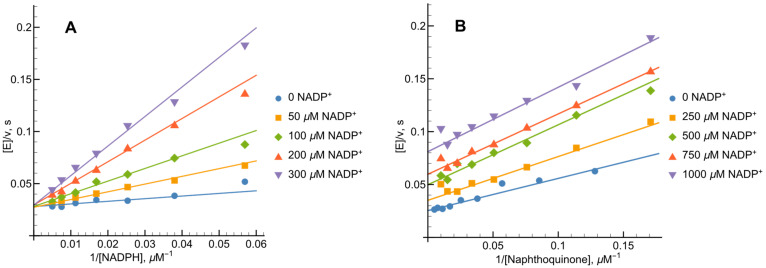 Figure 5
Figure 5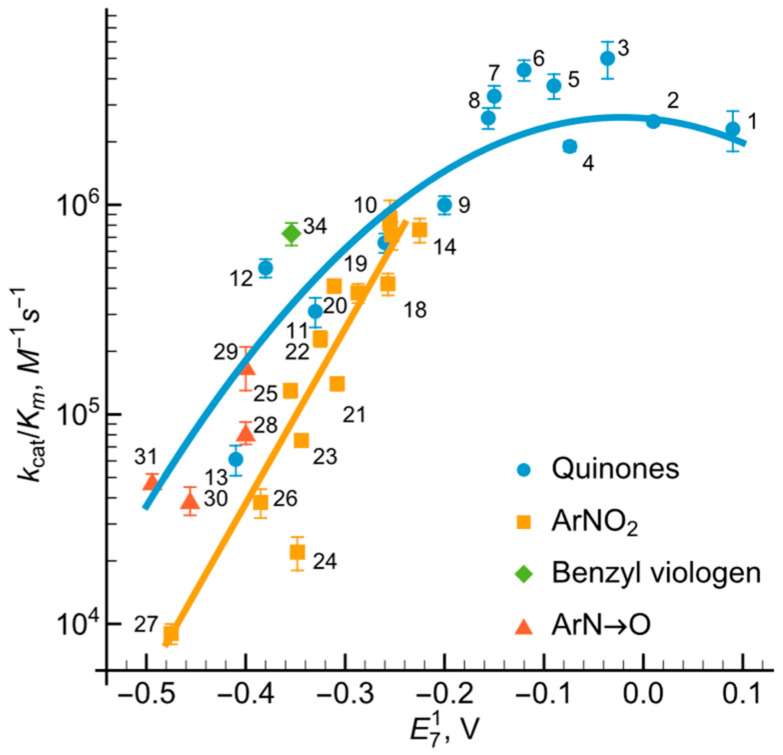 Figure 6
Figure 6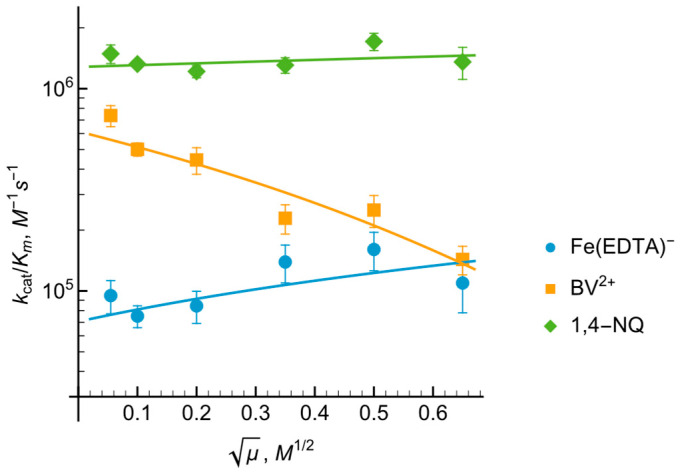 Figure 7
Figure 7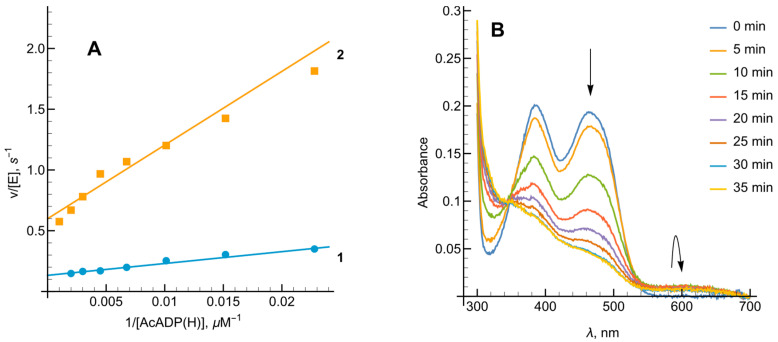 Figure 8
Figure 8 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMetal-Catalyzed Oxygenation Mechanisms · Photosynthetic Processes and Mechanisms · Metalloenzymes and iron-sulfur proteins
1. Introduction
Ferredoxin: oxidoreductases (FNRs, E.C. 1.18.1.2) transform a two-electron transfer into a single-electron one, and according to flavoenzyme classification belong to the class of dehydrogenases–electrontransferases [1,2]. A typical feature of FNRs is the relative high stability of a neutral flavin adenine dinucleotide semiquinone ( ) intermediate, which enables efficient redox reactions between two molecules of ferredoxin (Fd) and NADP(H). FNRs are found in all domains of life and function in versatile cellular metabolic processes. In oxygenic photosynthesis, plant-type FNRs reduce at the expense of two molecules of reduced Fd generated by an FeS-type photoreduction center photosystem I, thus providing NADPH for assimilation (Equation (1)) [3,4,5]. In nonphotosynthetic processes, FNRs reduce Fd by NADPH, with reduced Fd then being used for nitrate assimilation or the synthesis of cell membrane components [5,6,7]. In the case of iron deficiency low redox potential electron carrier flavodoxin (Fld) can function as the physiological partner of FNRs in place of Fd [8]. FNRs and their protein redox partners are characterized by electrostatic and hydrophobic interactions [9].
FNR from the thermophilic green sulfur bacterium Chlorobaculum tepidum (CtFNR) belongs to a relatively poorly studied group of thioredoxin reductase (TrxR)-type FNRs [10,11]. The distinguishing features of this type FNRs are a homodimeric structure and a FAD-binding domain comprising two discontinuous segments separated by an NADP(H)-binding domain (Figure 1) just as found in disulfide reductases [5]. In the case of CtFNR, the FAD-binding domain consists of amino acid residues 13–131 and 262–329, while residues 134–258 build up an NADP(H) binding domain [12]. The distance between the isoalloxazine ring of FAD and nicotinamide ring of is assumed to be approximately 15 Å in the crystal structure, too great for an efficient hydride transfer (Figure 1) [12]. The two nucleotide binding domains are connected by flexible hinge regions allowing for domain rotation during catalysis. As in other TrxR-type FNRs, there are also π–π stacking interactions between the isoalloxazine ring of FAD and Tyr57 on the si-side and Phe337 on its re-side [12]. Another notable peculiarity of CtFNR is the asymmetric arrangement of the homodimer, i.e., NADP(H) domain in one protomer is in “open” conformation, while the equivalent domain in the other protomer is “relatively closed” in the crystal structure. The asymmetry may be related to the presence of the distinct long hydrophilic C-terminal extension which acts as a subdomain to physically tether one protomer of the dimer while interacting with the other protomer [12].
As an obligate photoautotrophic anaerobe, C. tepidum utilizes the reverse tricarboxylic acid cycle for fixation and this pathway needs a continuous supply of low-potential reductants, e.g., reduced Fd [14,15,16]. C. tepidum utilizes an FeS-type photoreaction center as a sole photosystem for photosynthesis which reduces Fds directly as just the case of photosystem I in oxygenic photosynthesis [15,17,18]. Although C. tepidum FNR can catalyze the reduction of in the presence of photosynthetic reaction center and Fd from C. tepidum [11], kinetic details of the interactions of Fds with CtFNR have not been studied [17,19]. The positively charged domain of Lys60, Lys262 and Arg321 is suggested to be candidate for Fd binding based on the crystal structure [12].
While kinetic studies of CtFNR with electron donors/acceptors such as Fd are scarce, the reactions with NADPH and are studied with steady state and pre-steady state approaches [18,20]. At pH 7.0, the initial phase of reduction of the enzyme by excess NADPH is characterized by k ≥ 500 however, after 500 ms, the FAD absorbance decrease is about 50% of that expected during the complete CtFNR reduction. Similarly, reoxidation of dithionite-reduced CtFNR by restores only about 70% of the absorption of oxidized FAD. This is likely related to the above-mentioned structural nonequivalence of its subunits.
In our previous studies, we determined the mechanisms of reactions of homologous TrxR-type FNRs from Rhodopseudomonas palustris (RpFNR) and Bacillus subtilis (BsFNR) with nonphysiological oxidants quinones (Q), nitroaromatic compounds ( ), and inorganic complexes [21,22]. This type of reaction can reveal properties of redox proteins that are important for their reactions with physiological redox partners in solution, such as electrostatic or hydrophobic interaction, electron transfer mechanisms, etc., and is valuable for comparative analysis [23]. In many cases, especially when it comes to the reduction of quinones and nitroaromatics by flavoenzymes, this information can also elucidate the mechanisms of the therapeutic/toxic effects or biodegradation pathways of these compounds [24,25,26]. Moreover, FNRs in general have been recently investigated for various applications, e.g., for NADPH regeneration [27,28], and microbial production of ethanol or biofuels [29,30].
In this work we studied the reactions of CtFNR with a panel of nonphysiological oxidants, mainly quinones and nitroaromatics with a wide range of oxidative potency and different electrostatic charge. Taken together with the determination of the redox potentials of the first and second electron transfer of the FAD cofactor of this enzyme, these data provide valuable comparative information, further generalizing the catalytic mechanisms of TrxR-type FNRs.
2. Results
2.1. Presteady-State Kinetics Studies of CtFNR Reoxidation
Existing evidence suggests that FAD reduction in the dimeric CtFNR by NADPH may be partial [18,20]. Thus, we examined the possible manifestation of this phenomenon in the kinetics of CtFNR reoxidation under multiple turnover conditions by tetramethyl-1,4-benzoquinone (duroquinone, DQ). We utilized DQ as an electron acceptor due to its optical transparency above 460 nm and its semiquinone being rapidly reoxidized by oxygen. The control experiment performed without DQ shows very fast reduction of FAD by NADPH followed by its slow reoxidation by , evidenced by a slow reappearance of absorbance at 460 nm, and a corresponding initial absorbance increase with a slow decrease at 600 nm (Figure 2A). The addition of DQ increases the rate of reoxidation approximately 40-fold with spectral changes at both wavelengths being on the same timescale (Figure 2B). Most importantly, the maximum degree of FAD reduction determined from the difference in absorbance between oxidized and dithionite-reduced CtFNR ( [18]) was equal to 46.4% ( ), and 33.1% (DQ) which is 2–2.5 times less than in the case of R. palustris or B. subtillis FNR, when the studies were performed with the same NADPH and DQ concentrations [21,22]. These findings imply that only a fraction of the FAD cofactor participates in the catalytic turnover of CtFNR. However, for convenience, the rate constants presented later will be calculated based on the total enzyme concentration.
The rate constants of enzyme reoxidation ( ) under multiple turnover conditions were calculated using Equation (2) [31]:
In this expression, represents the starting NADPH concentration, while denotes the peak level of reduced enzyme observed during turnover. The term refers to the duration measured between the points where the reduced enzyme reaches half its maximal concentration during both the formation and decay phases. Based on the data in Figure 2A, the for oxygen was 1.8 . By varying DQ concentrations (Figure 3A,B) we calculated of 73.2 ± 4.6 at infinite oxidant concentration and the apparent bimolecular oxidation rate constant was determined to be 8.0 ± 1.4 × .
2.2. Steady-State Kinetics and Oxidant Substrate Specificity of CtFNR
At NADPH concentrations ranging from 20 to 200 µM, NADPH–oxidase activity of CtFNR was determined to be 2.1 , a value consistent with our presteady-state kinetic observations (Figure 2A). The values reported herein have been adjusted to account for this baseline oxidase activity. Previous research [21,22] has identified 1,4-naphthoquinone derivatives as effective nonphysiological oxidants for FNR enzymes. In our study, varying the concentrations of both NADPH and 1,4-naphthoquinone yielded a series of parallel lines in double-reciprocal (Lineweaver–Burk) plots (Figure 4). This kinetic pattern strongly suggests that CtFNR operates via a “ping-pong” catalytic mechanism, where the enzyme alternates between its oxidized and reduced forms without forming a ternary complex.
Calculated according to Equation (11) (see Section 4), the for 1,4-naphthoquinone reduction at saturating NADPH concentrations was 80.9 ± 10.3 . The bimolecular rate constants ( ) for 1,4-napthoquinone and NADPH were determined to be 3.3 ± 0.4 × and 9.3 ± 1.3 × , respectively.
We observed that acts as a product inhibitor of the CtFNR-catalyzed quinone reduction. Specifically, at a fixed 1,4-napthoquinone concentration (100 μM) exhibited competitive inhibition relative to NADPH (Figure 5A) with = 33.8 ± 4.0 μM (Equation (12)). Conversely, functioned as an uncompetitive inhibitor toward the oxidant (at 100 μM NADPH), characterized by parallel lines in the Lineweaver–Burk plot (Figure 5B). The resulting was to 447.4 ± 54.5 μM, as calculated according to Equation (13).
To evaluate the oxidant specificity of CtFNR, we characterized a diverse array of nonphysiological electron acceptors. This includes quinones (Q), nitroaromatics ( ), aromatic N-oxides (ArN→O) with single-electron reduction midpoint potentials ( ) spanning 0.09 V to −0.494 V. Additionally, single-electron acceptors such as ferricyanide, benzyl viologen ( ) and were included. The calculated and values at 100 µM NADPH are summarized in Table 1, derived from non-linear regression using Equation (9) or Equation (10). Notably, the for DQ closely aligns with the apparent bimolecular reaction rate constant observed in our presteady-state analysis (Table 1, Figure 3B), validating the consistency of our kinetic model.
The reactivity of the examined nitroaromatics, expressed as log , demonstrated a linear correlation with their values (Figure 6), where nifuroxime and nitrofurantoin emerged as the most efficient oxidants. In contrast, the reactivity profiles for quinones and aromatic N-oxides followed a parabolic dependence on with both chemical groups displaying comparable log magnitudes. Notably, the single-electron acceptor benzyl viologen (compound 34, Figure 6) showed a reactivity level consistent with quinones of similar reduction potentials. Collectively, these data suggest that the redox reactions are governed primarily by thermodynamic driving force ( ) rather than specific structural recognition of the oxidant’s scaffold.
Our results demonstrate that CtFNR primarily reduces quinones and nitroaromatics to their radical forms. To quantify this, we measured the single-electron flux using the cytochrome c reduction assay at pH < 7.2 [34]. For CtFNR, the ratio of cytochrome c reduction to the doubled rate of NADPH oxidation was 155%, indicating a 77% single-electron flux. When 4-nitroacetophenone was employed as the oxidant, we observed a 90% single-electron flux. Interestingly, the addition of superoxide dismutase (SOD) reduced the cytochrome c reduction rate by 25%. This suggests that the nitroaromatic radical reacts with oxygen to generate superoxide, which subsequently contributes to cytochrome c reduction [35,36].
To investigate the role of electrostatic forces in CtFNR reoxidation, we examined the impact of ionic strength on oxidant reactivity (Figure 7). The log for the cationic benzyl viologen ( ) decreased as ionic strength increased, whereas the reactivity of anionic showed the opposite trend. The reactivity of neutral 1,4-naphthoquinone was largely independent of ionic strength. These observations suggest that these electorn acceptors interact with a negatively charged region of the CtFNR protein surface.
2.3. Redox Potential Determination
Building on our previous characterization of FNRs from R. palustris and B. subtilis [21,22], we utilized the Haldane relationship to determine the standard redox potential ( ) of the couple in CtFNR. For two-electron hydride transfer, the potential difference between reactants is described by Equation (3):
To circumvent the kinetic complexities of reduction, we employed the analog 3-acetylpyridine adenine dinucleotide phosphate (AcADP(H), = −0.258 V [37]). We monitored the forward reaction (AcADPH oxidation) by observing 1 mM ferricyanide reduction, with AcADPH generated in situ using a glucose-6-phosphate dehydrogenase system. The rate constants obtained were = 1.63 ± 0.12 and = 1.6 ± 0.3 × (on a two-electron basis) (Figure 8A). For the reverse reaction (reduction of by 200 µM NADPH) the constants were = 7.54 ± 0.24 and = 1.0 ± 0.1 × . No inhibition was observed when performing the transhydrogenase reaction with constant NADPH concentrations in the range of 25–200 µM. These data yielded an equilibrium constant K = 0.16 ± 0.04, corresponding to a calculated of −0.282 ± 0.003 V.
To evaluate the concentration of at equilibrium and determine the individual redox potentials for the two single-electron transfers, we initially followed the photoreduction methodology used for RpFNR and BsFNR using 5-deazaFMN and EDTA [21,22]. However, this approach was ineffective for CtFNR, even at high reagent concentrations. As an alternative, we achieved enzyme-bound FAD reduction by employing a catalytic amount of NADPH (1 μM) and an enzymatic regeneration system (glucose-6-phosphate/glucose-6-phosphate dehydrogenase). Strictly anaerobic conditions were maintained using an excess of glucose, glucose oxidase and catalase to scavenge residual and . After establishing anaerobiosis, the enzyme reduction begins immediately upon the introduction of NADPH, marked by an absorbance decrease at 460 nm and increase at 600 nm, characteristic of the neutral FAD semiquinone ( ) (Figure 8B). Upon reintroducing oxygen to the sample, the absorbance at 460 nm and 600 nm rapidly returned to the initial values.
Based on established extinction coefficients ( ) [38], the maximum semiquinone yield was calculated to be at 11%. By utilizing sub-stoichiometric concentration of NADPH, we minimized spectral interference from enzyme-NADP(H) complexes [18,20]. The separation of the two single-electron transfer potentials can then be calculated based on the semiquinone formation constant (Equations (4) and (5)):
where is the maximum concentration of the semiquinone, and is the total concentration of the enzyme [39]. According to our data, = 0.061 and = −0.070 V, which corresponds to = −0.317 V and = −0.247 V. In this case, questions may arise about the actual percentage of in equilibrium, since only one of the two subunits is involved in catalysis (Figure 2A,B). However, it can be argued that the different activities of the subunits are associated with the nonequivalence of the NADP(H) binding domains, since the FAD isoalloxazine environment is the same in both subunits [12]. Moreover, the reduction of CtFNR in our experiment proceeds quite slowly, and its degree of reduction significantly exceeds 50% (Figure 8B). Therefore, we believe that under these conditions a redox equilibrium is established involving both FAD-binding domains, and the resulting percentage reflects identical redox properties of both subunits. On the other hand, the possibility that the obtained values reflect the average of the redox potentials of both FADs cannot be excluded; however a more detailed study of these details is a subject of our further studies.
3. Discussion
The results of this work, complementing our previous studies on TrxR-type RpFNR and BsFNR [21,22], allow us to draw more general conclusions about the catalytic mechanisms and thermodynamic properties of this relatively poorly studied group of enzymes. Although the mechanisms of reactions with nonphysiological oxidants are not necessarily analogous to reactions with physiological redox partners, they provide important information that may be valuable in the application of these enzymes.
First, as in the case of RpFNR and BsFNR and other types of FNRs, reoxidation of CtFNR with duroquinone results in a transient 600 nm absorption (Figure 2 and Figure 3) [21,22,23]. It is consistent with a two-step reoxidation process with oxidation being rate limiting. Next, CtFNR catalysis proceeds according to a “ping-pong” mechanism (Figure 4) [21,22]. Thus, it can be stated that the reduction and oxidation half-reactions occur independently, but at the same or significantly overlapping binding sites. Given that at a fixed NADPH concentration, the apparent of the reactions vary significantly with different oxidants (Table 1), this indicates that the oxidative half-reaction is rate-limiting. As in the case of RpFNR and BsFNR [21,22], is a competitive inhibitor for NADPH and an uncompetitive inhibitor for the oxidant (Figure 5A,B). This indicates that binds much more efficiently to the oxidized form of the enzyme than to the reduced form, in this case its semiquinone form. Since oxidants are likely to interact with the negatively charged domain near the isoalloxazine (Figure 7), a possible candidate is the conserved Asp64 [11]. Similar patterns have been observed in the reactions of oxidants with BsFNR containing the Asp57 residue, which in turn corresponds to Asp56 in RpFNR [22].
One of the most important features that unites CtFNR with RpFNR, BsFNR and other types of FNRs is that the log values for quinones, aromatic nitrocompounds and *N-*oxides increase with an increase in their (Figure 6) [21,22,23], i.e., the reactivity is governed by the single-electron accepting potency of oxidants rather than by their structural features. Taken together with the dominant single-electron flux during Q and reduction, this is consistent with the “outer sphere” single electron transfer model [40,41]. In this case, the rate constant for the electron transfer between two reactants ( ) is dependent on the electron self-exchange rate constants of those reactants ( and ) and the reaction equilibrium constant (K):
where log K is expressed as in Equation (5), and
and Z is the frequency factor, . According to Equations (5)–(7), log for a reaction of electron donor with a series of oxidants with similar values will exhibit a parabolic dependence on , or a linear one, if = ±0.15 V. It has been established that the of , ~ , is 100 times lower than that of Q and ArN→O, ~ [42,43,44], which would lead to a 10-fold lower reactivity of when compared to quinones of similar values. A similar difference in their reactivity is observed experimentally (Figure 6).
Focusing on specific properties of CtFNR, we have for the first time determined its standard redox potential of −0.282 V, which is more negative than that of BsFNR (−0.240 V) and similar to that of R. palustris (−0.276 V) [21,22]. The similarities of the amino acids in the FAD isoalloxazine ring environment of these TrxR-type FNRs have been discussed in previous papers [21,22], therefore only the differences that may influence the standard redox potentials of the enzymes and their stability will be discussed. The *si-*side of the FAD isoalloxazine of CtFNR is shielded by Tyr57, along with that RpFNR and BsFNR have homologous Tyr49 and Tyr50, respectively [45]. In contrast, the re-side of the isoalloxazine of CtFNR is stacked with Phe337, which corresponds toTyr328 in RpFNR and His324 in BsFNR [45]. The effect of the presence of His324 in BsFNR is similar to the case for flavodoxin from Desulfovibrio vulgaris, where Tyr98His substitution increased the of FMN by 0.07 V [46]. The presence of the imidazole group of histidine is thought to stabilize the anionic form of the reduced flavin, i.e., to make its oxidation more difficult. On the other hand, the Tyr98Phe substitution has almost no effect on of flavodoxin [46]. These differences in match those observed in the aforementioned TrxR-type FNRs. It should also be noted that in the case of CtFNR, the 11% stability of at equilibrium is much lower than that of RpFNR, 26.5%, or BsFNR, 44% [21,22]. In this context, BsFNR and RpFNR have Thr326 and Thr330, respectively, which form H-bonds with N5 of isoalloxazine, while in CtFNR these functions are performed by Ser339 [12,18,20]. It is possible that the substitution of Thr for Ser destabilizes , as is known in the case of neuronal NO synthase, where the Ser1176Thr mutation significantly stabilizes FAD semiquinone [47].
Some mechanistic details of the CtFNR-catalyzed electron transfer can be quantitatively assessed using the data in Figure 6. According to Mauk et al., the electron transfer distance ( ) in the reactions of metalloproteins with inorganic complexes at infinite ionic strength, i.e., in the absence of electrostatic effects, can be related to the electron self-exchange rate constant ( ) of metalloproteins [48]:
Based on the fact that when = 0, we have used this approach for the comparative purposes to estimate the values of various flavoenzymes dehydrogenases–electrontransferases, including the Trx-type FNRs [21,22,23]. Assuming that oxidation is the rate-limiting step, we obtained that at = −0.317 V of the oxidant, the log values for CtFNR are 3.6 ± 0.3 (Q), and 4.6 ± 0.4 ( ). This gives the values of 3.4 ± 0.3 Å and 2.6 ± 0.3 Å, respectively. Since it is possible that only one CtFNR subunit can participate in the reaction, the value of can be multiplied by 2, and accordingly the value of can be multiplied by 4. According to Equation (8), this would further reduce the value of by 0.5 Å. One may note that these values are lower than those estimated in the cases of RpFNR (5.2–5.4 Å) or BsFNR (3.8 Å) [21,22]. This can be attributed either to the insufficient accuracy of Equation (8), or to the specific structural features CtFNR. Recent studies have shown that the reduced FAD of CtFNR is in a more viscous environment than in RpFNR and BsFNR, and, correspondingly, contains less bound molecules close to the active center [49]. This can decrease the local dielectric constant at the environment of isoalloxazine, which in turn may decrease the solvent reorganization energy, thus increasing an intrinsic reactivity of flavin cofactor [50,51,52]. Taken together, these data indicate that the intrinsic activity of CtFNR towards nonphysiological oxidants is similar to that of RpFNR and BsFNR, thus ruling out the effects of possible shielding of isoalloxazine by C-terminal extension [12].
In conclusion, the studies of the reactions of CtFNR and other TrxR-type FNRs with nonphysiological oxidants can provide valuable information on their structural and catalytic properties, which, however, may not entirely clarify their physiologically relevant functions. On the other hand, this information may be valuable from an application perspective, including the use of anoxygenic phototrophic bacteria in photobioelectrochemical systems and microbial fuel cells [53,54,55]. In this case, a very important aspect is the ensuring of efficient electron transfer between the cellular redox enzymes and the electrode, which is carried out by redox-active mediators [56]. Our data on the efficiency of reduction of the redox mediators by flavoenzymes are directly related to this problem.
4. Materials and Methods
C. tepidum ferredoxin:NADP^+^ oxidoreductase was purified as previously described and its concentration was determined spectrophotometrically according to [12]. NADP(H), 3-acetylpyridineadenine dinucleotide phosphate ( ), horse heart cytochrome c, superoxide dismutase, glucose oxidase, glucose 6-phosphate, glucose 6-phosphate dehydrogenase, 5-deazaFMN and other commercially available reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) and used as received. 5-(Aziridin-1-yl)-2,4-dinitrobenzamid synthesized as described in [57], was generous gift of Dr. Vanda Miškinienė (Institute of Biochemistry, Vilnius University). 2,4,6-Trinitrotoluene and N-methylpicramide synthesized as described in [58,59] and tirapazamine derivatives synthesized as described in [60,61,62] were a generous gift of Dr. Jonas Šarlauskas (Institute of Biochemistry, Vilnius University). The structural formulae of nonphysiological electron acceptors are given in Figure 9:
The steady-state kinetics experiments were performed using a Cary60 UV/Vis spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). All experiments were performed in 0.02 M Hepes/NaOH + 1 mM EDTA buffer solution (pH 7.0) at 25 °C. The kinetic data were fitted to the Michaelis–Menten equation in Mathematica (Wolfram Research, Inc., Mathematica, Version 14.0, Champaign, IL, USA (2024)) (Equations (9) and 10) to yield the steady-state parameters of the reactions, namely the catalytic constant , bimolecular reaction rate constants (or catalytic efficiency constants) and substrate inhibitions constants (where applicable) of the oxidants under a constant concentration of NADPH:
where v is the reaction rate, [E] is the CtFNR concentration, [S] is the concentration of the oxidant and represents the number of molecules of NADPH oxidized by a single native molecule of the enzyme per second at saturated concentrations of both substrates. The fitted parameters correspond to the reciprocal intercepts and slopes of Lineweaver–Burk plots, [E]/v vs. 1/[S], respectively. The concentrations of the enzyme used in the experiments were 5–50 nM. The kinetic parameters of the steady-state reactions according to the ‘ping-pong’ mechanism were calculated according to Equation (11):
where Q is the electron acceptor.
The competitive inhibition constant of vs. NADPH was calculated according to Equation (12):
The uncompetitive inhibition constant of vs. electron acceptor (Q) was calculated according to Equation (13):
The rates of enzymatic NADPH oxidation in the presence of quinones, nitroaromatics, aromatic N-oxides or single electron acceptors were determined according to and they were corrected for the intrinsic NADPH oxidase activity of CtFNR (2.1 ) and/or nonenzymatic NADPH oxidation by high-potential quinones. When 50 μM of cytochrome c was added to the reaction mixture, its quinone- and nitroaromatic-mediated reduction was assessed according to . The ferricyanide reduction rate was measured according to . The rate of CtFNR-catalyzed reduction of by NADPH was determined according to and AcADPH was prepared in situ by reducing with 10 mM glucose 6-phosphate and 0.01 mg/mL glucose 6-phosphate dehydrogenase with its concentration determined according to [37]. NaCl was used to vary the ionic strength of the buffer solution. The stock solutions of organic compounds were prepared in DMSO. The final concentration of DMSO in reaction mixtures was 1% (v/v). The starting concentrations for the oxidants ranged from 100 to 1000 μM, and every compound was measured in a series of measurements with 1.5× serial dilutions for 7–10 different concentrations. The reactions were initiated by adding CtFNR to the mixture of the buffer solution, NADPH and electron acceptor in the cuvette.
Presteady-state kinetics assays were performed under aerobic conditions using SX20 stopped-flow system (Applied Photophysics, Leatherhead, UK). The reduction of CtFNR by NADPH and its reoxidation by a quinone or oxygen was evaluated at 460 nm and 600 nm. The CtFNR in syringe 1 (~10 μM) was mixed with the contents of syringe 2 (100 μM NADPH or 100 μM NADPH + 100–500 μM of duroquinone).
The reduction of CtFNR under anaerobic conditions was performed using an NADPH regeneration system with a catalytic amount of NADPH. The anaerobic cuvette contained 10 mM glucose, 50 nM glucose oxidase, 50 nM catalase, 10 mM glucose 6-phosphate, 0.01 mg/mL glucose 6-phosphate dehydrogenase and 1 μM NADPH. CtFNR (~200 μM) was placed in a side arm and the cuvette was flushed with oxygen free argon for 30 min. The reaction was initiated by mixing CtFNR with the cuvette contents for a final CtFNR concentration of ~20 μM. The progress of the reaction was followed spectrophotometrically for 3 h.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hemmerich P. Massey V. The Role of the Apoprotein in Directing Pathways of Flavin Catalysis Proceedings of the Third International Symposium on Oxidases and Related Redox Systems, State University of New York at Albany Albany, NY, USA 3–7 July 1979379405
- 2Massey V. Hemmerich P. Active-site probes of flavoproteins Biochem. Soc. Trans.1980824625710.1042/bst 00802467399046 · doi ↗ · pubmed ↗
- 3Shin M. Arnon D.I. Enzymic Mechanisms of Pyridine Nucleotide Reduction in Chloroplasts J. Biol. Chem.19652401405141110.1016/S 0021-9258(18)97591-514284756 · doi ↗ · pubmed ↗
- 4Ceccarelli E.A. Arakaki A.K. Cortez N. Carrillo N. Functional plasticity and catalytic efficiency in plant and bacterial ferredoxin-NADP(H) reductases Biochim. Biophys. Acta Proteins Proteom.2004169815516510.1016/j.bbapap.2003.12.00515134648 · doi ↗ · pubmed ↗
- 5Aliverti A. Pandini V. Pennati A. de Rosa M. Zanetti G. Structural and functional diversity of ferredoxin-NADP+ reductases Arch. Biochem. Biophys.200847428329110.1016/j.abb.2008.02.01418307973 · doi ↗ · pubmed ↗
- 6Nogués I. Tejero J. Hurley J.K. Paladini D. Frago S. Tollin G. Mayhew S.G. Gómez-Moreno C. Ceccarelli E.A. Carrillo N. Role of the C-Terminal Tyrosine of Ferredoxin-Nicotinamide Adenine Dinucleotide Phosphate Reductase in the Electron Transfer Processes with Its Protein Partners Ferredoxin and Flavodoxin Biochemistry 2004436127613710.1021/bi 049858 h 15147197 · doi ↗ · pubmed ↗
- 7Röhrich R.C. Englert N. Troschke K. Reichenberg A. Hintz M. Seeber F. Balconi E. Aliverti A. Zanetti G. Köhler U. Reconstitution of an apicoplast-localised electron transfer pathway involved in the isoprenoid biosynthesis of Plasmodium falciparum FEBS Lett.20055796433643810.1016/j.febslet.2005.10.03716289098 · doi ↗ · pubmed ↗
- 8Goñi G. Herguedas B. Hervás M. Peregrina J.R. De la Rosa M.A. Gómez-Moreno C. Navarro J.A. Hermoso J.A. Martínez-Júlvez M. Medina M. Flavodoxin: A compromise between efficiency and versatility in the electron transfer from Photosystem I to Ferredoxin-NADP+ reductase Biochim. Biophys. Acta Bioenerg.2009178714415410.1016/j.bbabio.2008.12.00619150326 · doi ↗ · pubmed ↗