Cyclic Amide-Linked Oxazolidinone Triazoles as Inhibitors of the T-Box Riboswitch
Eric Parsons, Ali H. Aldhumani, Emily A. Fairchild, Oluwaseun B. Adegbite, Jessica M. Roberts, Jennifer V. Hines, Stephen C. Bergmeier

TL;DR
This paper explores new antibiotic compounds that target a specific RNA structure in bacteria, offering a novel approach to combat antimicrobial resistance.
Contribution
The study introduces a macrocyclic oxazolidinone scaffold designed to improve RNA-binding affinity and inhibit T-box riboswitch function.
Findings
A macrocyclic oxazolidinone compound was identified that may interfere with tRNA-induced transcription through π–π stacking interactions.
Computational docking suggests the compound interacts with the antiterminator region of the T-box riboswitch.
The T-box riboswitch is a promising target for antibacterial agents due to its conserved nature and regulatory role in essential genes.
Abstract
Antimicrobial resistance remains a critical global health challenge, and was intensified by the COVID-19 pandemic. To address this growing threat, novel antibacterial agents targeting unconventional mechanisms are urgently needed. One promising strategy involves inhibiting bacterial riboswitches—RNA elements that regulate gene expression. Unlike most riboswitches that respond to small-molecule metabolites, the T-box riboswitch uniquely binds non-aminoacylated tRNA and is predominantly found in Gram-positive bacteria, making it an attractive target due to its conserved sequences and regulatory role over essential genes. This study explored oxazolidinone- and triazole-based compounds as potential inhibitors of the T-box riboswitch. Prior investigations into tricyclic oxazolidinones revealed an allosteric modulator that effectively inhibited T-box riboswitch transcriptional readthrough in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
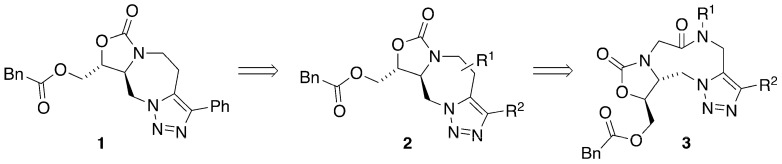 Figure 1
Figure 1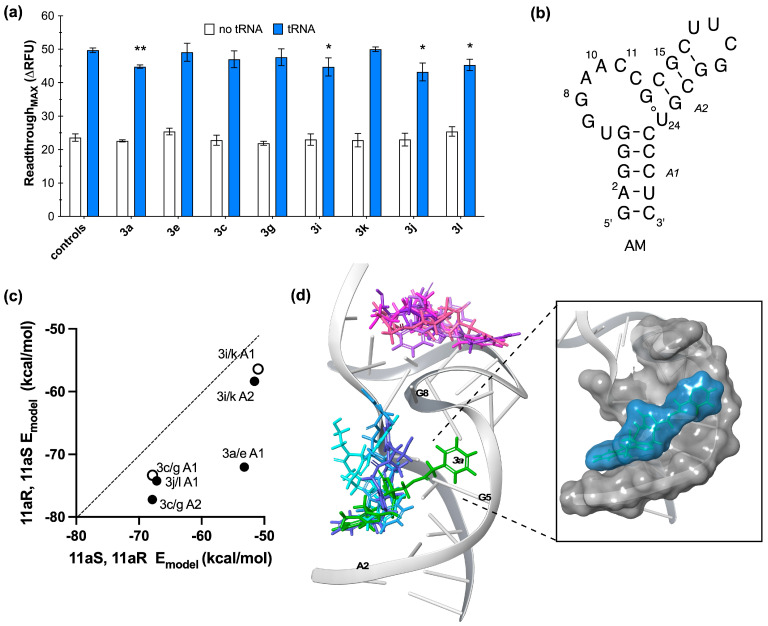 Figure 2
Figure 2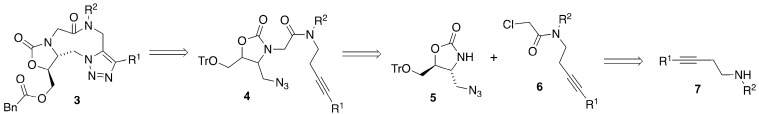 Figure 3
Figure 3 Figure 4
Figure 4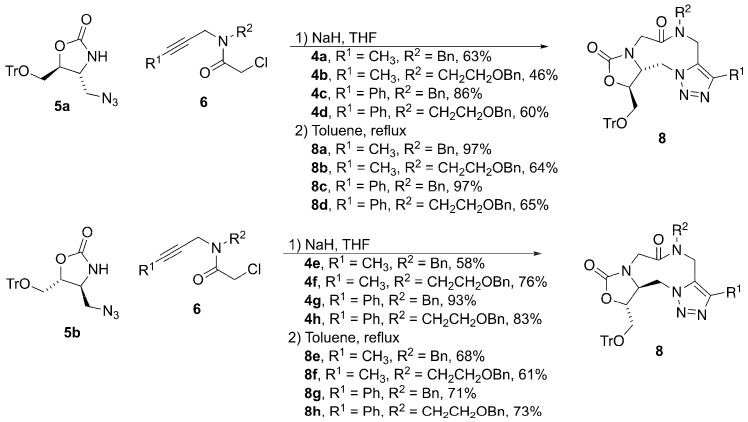 Figure 5
Figure 5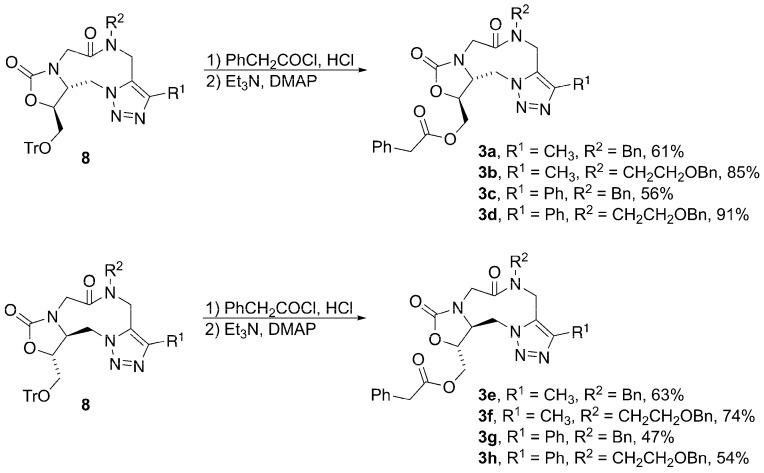 Figure 6
Figure 6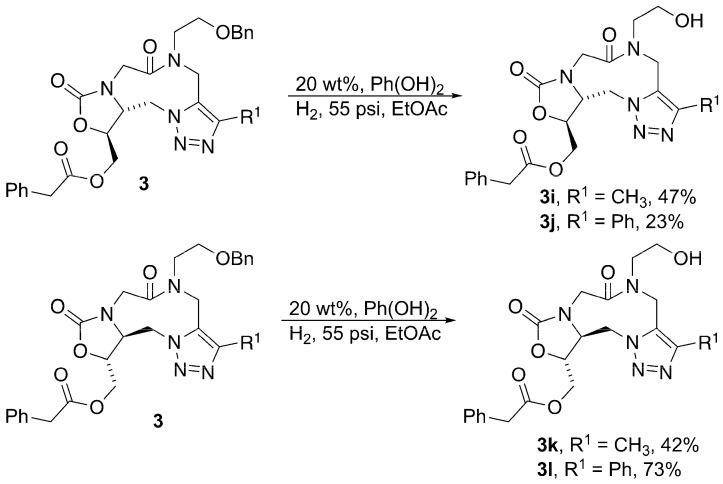 Figure 7
Figure 7- —National Institutes of Health
- —National Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRNA and protein synthesis mechanisms · RNA modifications and cancer · Bacterial Genetics and Biotechnology
1. Introduction
Antimicrobial resistance to current treatments continues to be a significant global health threat that was further exacerbated by the COVID pandemic [1,2]. The Centers for Disease Control and Prevention’s list of resistant threats include bacteria and fungi, with one-third (7/20) being Gram-positive bacteria [3]. To overcome this serious threat to human health, completely novel antibacterial agents are needed. Developing inhibitors that target the RNA of bacterial riboswitches is a novel area of antibacterial drug discovery [4,5,6]. Riboswitches are located in the 5′ untranslated region of mRNA and regulate gene expression at the level of transcription or translation by structurally responding to effector molecule binding or environmental conditions [7,8,9,10]. Most riboswitches respond to a small-molecule metabolite as their effector molecule, but the T-box riboswitch specifically binds and structurally responds to cognate non-aminoacylated tRNA. Found primarily in Gram-positive bacteria, the T-box riboswitch is an ideal target for antibacterial drug discovery due to regions of high sequence conservation and the many essential genes regulated [5,11,12].
Previous studies by our group have identified novel oxazolidinone ligands that bind the T-box riboswitch antiterminator element [13] and modulate tRNA-induced transcription readthrough [14,15]. T-box riboswitch inhibition by a small molecule has also been implicated in antibacterial activity specific to Gram-positive bacteria [16,17].
We have examined a number of oxazolidinone- and triazole-based compounds in order to understand how small molecules can interact with and inhibit the T-box riboswitch [14,15,18]. Most recently, we reported on the design, synthesis, and evaluation of a preliminary set of tricyclic oxazolidinones (Figure 1) [19]. Overall, the RNA-binding results for compound 1 suggest that these ligands act as allosteric modulators of T-box riboswitch function. While these ligands effectively inhibited transcriptional readthrough in the T-box riboswitch functional assay, there was modest to no inhibition in the formation of the tRNA–antiterminator complex observed in a fluorescence anisotropy assay and minimal stereoselectivity. Thus, rather than disrupting the complex formation via competitive inhibition, these ligands may bind to an allosteric site (a site other than where tRNA forms contacts with the antiterminator) to then cause a conformational change, which results in the observed disruption of the function of the T-box antiterminator [19].
Consequently, we set out to design an oxazolidinone-containing macrocycle that allowed for a larger RNA-binding footprint (to enhance stereoselective interactions) while retaining conformational restriction. We planned to introduce an additional substituent (e.g., R^1^) on the chain linking the triazole to the oxazolidinone, as shown in compound 2. Compound 3 appeared to be a synthetically tractable version of the proposed compound 2. Lactam 3 would allow for the introduction of a variety of substituents in the R^1^ position through substitution of the amide.
2. Results
For our initial studies, we planned a small selection of substituents for positions R^1^ and R^2^. Our previous studies [15,20], as well as other RNA/ligand studies [21], have indicated the importance of hydrophobic interactions and π–π stacking for conferring specificity, as opposed to electrostatic (ionic) interactions, which are much less specific. Consequently we looked at those analogs as well as H-bonding substituents.
The synthesis of triazole 3 is outlined in Scheme 1. Triazole 3 should be readily available through an intramolecular dipolar cycloaddition of azido alkyne 4. An alkylation of the known azido-oxazolidinone 5 with α-chloroamide 6 would provide azidoalkyne 4. The α-chloroamide 6 can be readily prepared by acylation of amine 7. This amine will provide a ready scaffold upon which to introduce both substituents R^1^ and R^2^.
As shown in Scheme 2, treatment of known (7a [22], 7c [23]) or commercially available (7b, 7d) amines with chloroacetyl chloride provided generally good yields of α-chloroamides 6a–6d.
Once the four required α-chloroamides 6a–6d were obtained, they were used for the alkylation of enantiopure oxazolidinone azides 5a and 5b (Scheme 3) [15]. Treatment of oxazolidinone 5 with NaH followed by the α-chloroamide provided intermediate alkynyl azides 4a–4h. These enantiopure azidoalkynes were then subjected to a thermal cycloaddition reaction to generate triazoles 8a–8h.
The trityl deprotection–acylation reaction follows a modified version of a one-pot procedure previously reported [24]. The original procedure uses non-distilled acid chloride to both deprotect and acylate a trityl-protected alcohol, as outlined in Scheme 4. This procedure, while effective, produced a byproduct that was difficult to remove. In order to avoid the formation of this byproduct, the phenylacetyl chloride was first distilled and a catalytic amount of anhydrous HCl added. In order to drive the reaction to completion, DMAP and Et_3_N were added to the reaction once it was shown by TLC that all of the trityl-protected triazole starting material had been consumed. This second step ensured that all of the primary alcohol that resulted from trityl group removal was acylated to form the desired ester product. This provided enantiopure final compounds 3a, 3c, 3e, and 3g.
The final step that was required in the synthesis of the desired enantiopure analogs was the reductive debenzylation of benzyl ethers 3b, 3d, 3f, and 3h to provide the primary alcohol (Scheme 5). Various reaction conditions were attempted, and eventually palladium hydroxide was determined to be the best choice as the catalyst to remove the benzyl group. With the purification and isolation of these last four analogs (3i–3l), the synthesis of the eight desired enantiopure analogs was complete.
To investigate the effect of the ligands on T-box riboswitch function, we used a fluorescence-monitored transcription of the glyQS riboswitch assay previously developed by our lab [25]. Four of the compounds (3a, 3i, 3j, and 3l) inhibited tRNA-induced glyQS transcription readthrough (Figure 2a). None of the ligands inhibited the basal level of transcription readthrough (p value > 0.5 in the absence of tRNA). For 3a, 3i, 3j, and 3l, this lack of basal level inhibition also indicates that the observed inhibition of tRNA-induced transcription readthrough is likely specific to the T-box riboswitch mechanism. Compound 3a showed the most significant inhibition of tRNA-induced transcription readthrough (14% inhibition, p value = 0.0015). The remaining compounds (3c, 3e, 3g, 3k) had no effect on riboswitch function under the conditions assayed (p value > 0.5 with and without tRNA).
Computational docking studies were conducted using Glide (Schrödinger) and our previously optimized protocol [15] to investigate possible ligand–antiterminator interactions. Previous studies with this protocol have demonstrated that the computationally derived E_model_ value for the most stable docking pose can correlate well with experimental binding data [19]. All of the ligands in this study were docked to the NMR-derived structure of antiterminator model RNA, AM [26], and poses selected based on lowest E_model_ value or docking location as indicated (Figure 2b–d).
Notably, 3a was the only ligand that docked entirely in the A1 helix. In the docking pose with the lowest E_model_ value, 3a aligned along the major groove of the A1 helix forming favorable van der Waals contacts with residues A2 to G4 and π–π stacking with the 3′ face of G5. Three of the compounds (3e, 3j, and 3l) docked partially in the A1 helix accompanied by significant interactions with the bulge nucleotides. The four remaining compounds had the lowest energy docked pose in the A2 helix (3c, 3g, 3k). Regarding stereospecific interactions, of the enantiomeric pairs, 3a compared to it’s enantiomer 3e had the greatest difference in E_model_ values (26%) and docking location (Figure 2c,d). Notably, 3a was the only ligand that docked entirely in the A1 helix.
For 3a, the combination of A1 helix docking location and riboswitch inhibition was consistent with ligand binding interfering with the antiterminator function. Previous studies in our lab identified that ligands that bind along the antiterminator A1 helix were antagonists, while ligands that docked only the bulge nucleotides in the antiterminator (U6-C12 of AM, Figure 2b) were agonists of T-box riboswitch function [15]. Given the potential significance of docking in the A1 helix, we also examined alternate docking poses for ligands 3c, 3g, 3i, and 3k other than their most stable A2 helix pose. While they all had a docking pose in the A1 helix with weaker E_model_ values (Figure 2c), each ligand’s pose also had significant contacts with the bulge nucleotides in addition to the A1 helix (Supplementary Material) and did not align along the groove of the A1 helix in the way that 3a did.
The consequences of a ligand binding along the A1 helix of the antiterminator can be interpreted in light of recent structural studies of the T-box riboswitch. In the cryo-EM structure of the glyQS discriminator region complexed with tRNA, the glyQS nucleotide U155 (equivalent to U6 in AM, Figure 2) forms a base pair with the 3′ terminal nucleotide of tRNA (A75) and is stacked on the 3′ face of glyQS G154 (G5 of AM) and subsequently stabilized by other long-range interactions [27]. Consequently, it is possible that 3a forming π–π stacking contacts with G5 in the antiterminator may disrupt formation of a tRNA–antiterminator conformer, which is required for full functioning of tRNA-induced transcription readthrough. The conformer of the tRNA–antiterminator complex needed for the subsequent stabilization could be sensitive to allosteric inhibition by a ligand binding along the A1 helix. This would be consistent with our previous identification of T-box riboswitch antagonists binding along the A1 helix [15] and identification of other likely allosteric inhibitors [19].
3. Materials and Methods
3.1. General Experimental
CH_2_Cl_2_ and THF were dried using a SOLV-TEK solvent purification system. All other dry solvents (acetone, DMF, toluene, Et_3_N, EtOAc) were purified by drying overnight with molecular sieves (4 Å) and distilling immediately before use. Unless otherwise noted, all reactions were conducted under an inert atmosphere (N_2_ or Argon). A DigiMelt MPA 160 melting point apparatus (SRS, Sunnyvale, CA, USA) was used to determine melting points, and all melting points are reported uncorrected. An AUTOPOL ^®^ IV (Rudolph Research Analytical, Hackettstown, NJ, USA) polarimeter with a sodium (λ = 589 nm) lamp was used to measure specific rotations, which are reported as follows: [α]λ ^T °C^ (c = g/100 mL, solvent). HPLC analysis for all tested compounds was performed using a Shimadzu LC-10AT instrument (Kyoto, Japan) equipped with a UV detector employing a Hypersil Gold C8 HPLC column (Waltham, MA, USA, 150 × 4.6 mm × 5 μm), with a flow rate of 1 mL/min (HPLC solvent systems described in General HPLC methods A and B). Tested compounds showed >95% purity unless otherwise stated. ^1^H NMR and ^13^C NMR spectra were recorded with either a Bruker Ascend 500 MHz spectrometer (Billerica, MA, USA) or a Bruker Avance 300 MHz spectrometer. Chemical shifts are reported in ppm on the δ scale relative to deuterated chloroform as an internal standard. NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant in Hz, integration. The capillary HRMS analyses were performed using a Thermo Scientific Q Exactive Plus Orbitrap mass spectrometer (Waltham, MA, USA). HPLC was performed using a 150 × 4.6 mm Hypersil GOLD C8 HPLC Column; Method A, 30% CH_3_CN in H_2_O (containing 1 mM HCO_2_H) going to 90% CH_3_CN over 14 min; Method B, 50% CH_3_CN in H_2_O (containing 1 mM HCO_2_H) going to 90% CH_3_CN over 14 min.
3.2. General Procedure A: Deprotection and Acylation Reaction
Triazole 9 was dissolved in dry CH_2_Cl_2_ (0.2 M) and cooled to 0 °C. Distilled phenylacetyl chloride (500 mol %) was added, followed by addition of 1.5 M anhydrous HCl in EtOAc (50 mol %). The reaction was stirred at 0 °C for 5 min, and was then warmed to room temperature. After approximately 18 h, at which point the trityl-protected triazole starting material was shown to be consumed by TLC, the reaction was cooled to 0 °C and DMAP (100 mol %) was added, followed by the addition of Et_3_N (700 mol %). The reaction was stirred for an additional 2 h, then methanol (4500 mol %) was added and stirred for 30 min. The reaction was concentrated and the resulting crude material was dissolved in EtOAc and washed with saturated aqueous NaHCO_3_ (2×) and water (2×). The combined aqueous phases were extracted twice with EtOAc. The combined organic phases were then washed with brine, dried over MgSO_4_, filtered, concentrated, and chromatographed.
((11S,11aR)-5-benzyl-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3a). Synthesized from triazole 9a (120 mg, 0.20 mmol) using general procedure A. The crude product was purified by column chromatography (55% EtOAc in hexanes) to afford 58 mg (61%) of 3a as a white solid, mp 115.0–116.0 °C; [α]D^25^ −73.714 (c = 0.175, CHCl_3_). ^1^H NMR (500 MHz, CDCl_3_) δ 7.42–7.24 (m, 8H), 7.15–7.09 (m, 2H), 5.00 (d, J = 14.2 Hz, 1H), 4.83 (d, J = 16.6 Hz, 1H), 4.67 (d, J = 17.7 Hz, 1H), 4.48 (d, J = 15.6 Hz, 1H), 4.38 (q, J = 3.8 Hz, 1H), 4.27 (d, J = 3.5 Hz, 2H), 4.25–4.21 (m, 1H), 4.22–4.17 (m, 1H), 3.87–3.78 (m, 2H), 3.67 (d, J = 6.4 Hz, 3H), 2.17 (s, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 168.1, 155.6, 142.9, 134.6, 133.3, 129.5, 129.4, 129.1, 128.9, 128.7, 128.1, 127.7, 72.8, 63.4, 57.7, 50.2, 49.0, 48.9, 41.4, 40.9, 10.3. HRMS: calculated for C_26_H_27_N_5_O_5_·H^+^, 490.2085; found, 490.2102. HPLC (254 nm, method B) 3.00 min, 98%.
((11S,11aR)-5-(2-(benzyloxy)ethyl)-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3b). Synthesized from triazole 9b (0.27 g, 0.41 mmol) using general procedure A. The crude product was purified by column chromatography (70% EtOAc in hexanes) to afford 180 mg (85%) of 3b as a white solid, mp 114.0–116.0 °C; [α]D^25^ −76.3 (c 0.35, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.41–7.21 (m, 10H), 4.80 (d, J = 7.8 Hz, 1H), 4.77 (d, J = 8.8 Hz, 1H), 4.65 (d, J = 17.7 Hz, 1H), 4.48–4.35 (m, 3H), 4.35 (d, J = 3.6 Hz, 2H), 4.27 (d, J = 3.4 Hz, 2H), 3.78–3.74 (m, 1H), 3.66 (s, 2H), 3.65–3.57 (m, 3H), 3.42 (d, J = 16.4 Hz, 1H), 3.33–3.24 (m, 1H), 2.21 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 168.4, 155.6, 142.7, 137.5, 133.3, 129.3, 129.1, 128.6, 128.1, 127.8, 127.7, 73.4, 72.8, 68.3, 63.4, 57.7, 57.6, 49.0, 48.9, 43.3, 41.3, 10.2. HRMS: calculated for C_28_H_31_N_5_O_6_·Na^+^, 556.21665; found, 556.21820. HPLC (254 nm, method B) 3.31 min, 100%.
((11S,11aR)-5-benzyl-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3c). Synthesized from triazole 9c (0.12 g, 0.18 mmol) using general procedure A. The crude product was purified by column chromatography (45% EtOAc in hexanes) to afford 0.056 g (56%) of 3c as a white solid, mp 122.0–124.0 °C; [α]D^25^ −65.4 (c 0.19, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.10 (m, 14H), 6.92 (d, J = 7.4 Hz, 2H), 4.86 (d, J = 16.7 Hz, 1H), 4.70 (d, J = 14.3 Hz, 1H), 4.53–4.45 (m, 2H), 4.41 (q, J = 3.9 Hz, 1H), 4.29–4.25 (m, 2H), 4.23 (dd, J = 15.6, 2.7 Hz, 1H), 4.06 (d, J = 14.2 Hz, 1H), 3.87–3.82 (m, 1H), 3.67 (s, 2H), 3.53 (d, J = 16.7 Hz, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 167.9, 155.4, 147.2, 134.1, 133.2, 129.5, 129.3, 129.0, 128.9, 128.8, 128.4, 128.3, 128.1, 127.6, 72.7, 63.4, 57.5, 50.6, 49.2, 48.8, 42.2, 41.2. HRMS: calculated for C_31_H_29_N_5_O_5_·H^+^, 552.2242; found, 552.2268; HPLC (254 nm, method B) 4.4 min, 99%.
((11S,11aR)-5-(2-(benzyloxy)ethyl)-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3d). Synthesized from triazole 9d (0.29 g, 0.40 mmol) using general procedure A. The crude product was purified by column chromatography (57.5% EtOAc in hexanes) to afford 0.22 g (91%) of 3d as a white solid, mp 127.0–128.3 °C; [α]D^25^ −48.219 (c 0.36, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.55–7.49 (m, 2H), 7.44–7.27 (m, 11H), 7.14–7.09 (m, 2H), 5.03 (d, J = 17.9 Hz, 1H), 4.85 (t, J = 17.3 Hz, 2H), 4.54 (s, 2H), 4.46 (q, J = 3.9 Hz, 1H), 4.29 (d, J = 3.5 Hz, 2H), 4.24 (s, 2H), 3.85–3.81 (m, 1H), 3.68 (s, 2H), 3.56–3.48 (m, 3H), 3.45 (d, J = 16.9 Hz, 1H), 3.43–3.36 (m, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 168.6, 155.6, 147.0, 137.4, 133.3, 129.8, 129.4, 129.1, 129.1, 128.9, 128.8, 128.6, 128.2, 127.9, 127.7, 73.2, 72.9, 67.9, 63.6, 57.6, 49.4, 48.9, 47.6, 44.3, 41.3. HRMS: calculated for C_33_H_33_N_5_O_6_·H^+^, 596.2504; found, 596.2526; HPLC (254 nm, method B) 5.1 min, 100%.
((11R,11aS)-5-benzyl-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3e). Synthesized from triazole 9e (120 mg, 0.20 mmol) using general procedure A. The crude product was purified by column chromatography (55% EtOAc in hexanes) to afford 61 mg (63%) of 3e as a white solid; mp 115.0–116.0 °C; [α]D^25^ +73.143 (c 0.18, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.42–7.24 (m, 8H), 7.15–7.09 (m, 2H), 5.00 (d, J = 14.2 Hz, 1H), 4.83 (d, J = 16.6 Hz, 1H), 4.67 (d, J = 17.7 Hz, 1H), 4.48 (d, J = 15.6 Hz, 1H), 4.38 (q, J = 3.8 Hz, 1H), 4.27 (d, J = 3.5 Hz, 2H), 4.25–4.21 (m, 1H), 4.22–4.17 (m, 1H), 3.87–3.78 (m, 2H), 3.67 (d, J = 6.4 Hz, 3H), 2.17 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 168.1, 155.6, 142.9, 134.6, 133.3, 129.5, 129.4, 129.1, 128.9, 128.7, 128.1, 127.7, 72.8, 63.4, 57.7, 50.2, 49.0, 48.9, 41.4, 40.9, 10.3. HRMS: calculated for C_26_H_27_N_5_O_5_·H^+^, 490.2085; found, 490.2102; HPLC (254 nm, method B) 3.0 min, 98%.
((11R,11aS)-5-(2-(benzyloxy)ethyl)-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3f). Synthesized from triazole 9f (0.26 g, 0.40 mmol) using general procedure A. The crude product was purified by column chromatography (70% EtOAc in hexanes) to afford 0.16 g (74%) of 3f as a white solid, mp 114.0–116.0 °C; [α]D^25^ +75.775 (c 0.355, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.41–7.21 (m, 10H), 4.80 (d, J = 7.8 Hz, 1H), 4.77 (d, J = 8.8 Hz, 1H), 4.65 (d, J = 17.7 Hz, 1H), 4.48–4.35 (m, 3H), 4.35 (d, J = 3.6 Hz, 2H), 4.27 (d, J = 3.4 Hz, 2H), 3.78–3.74 (m, 1H), 3.66 (s, 2H), 3.65–3.57 (m, 3H), 3.42 (d, J = 16.4 Hz, 1H), 3.33–3.24 (m, 1H), 2.21 (s, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 168.4, 155.6, 142.7, 137.5, 133.3, 129.3, 129.1, 128.6, 128.1, 127.8, 127.7, 73.4, 72.8, 68.3, 63.4, 57.7, 57.6, 49.0, 48.9, 43.3, 41.3, 10.2. HRMS: calculated for C_28_H_31_N_5_O_6_·Na^+^, 556.2167; found, 556.2182. HPLC (254 nm, method B) 3.3 min, 100%.
((11R,11aS)-5-benzyl-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3g). Synthesized from triazole 9g (0.10 g, 0.15 mmol) using general procedure A. The crude product was purified by column chromatography (40% EtOAc in toluene) to afford 0.039 g (47%) of 3g as a white solid, mp 122.0–124.0 °C; [α]D^25^ +64.571 (c 0.175, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.10 (m, 14H), 6.92 (d, J = 7.4 Hz, 2H), 4.86 (d, J = 16.7 Hz, 1H), 4.70 (d, J = 14.3 Hz, 1H), 4.53–4.45 (m, 2H), 4.41 (q, J = 3.9 Hz, 1H), 4.29–4.25 (m, 2H), 4.23 (dd, J = 15.6, 2.7 Hz, 1H), 4.06 (d, J = 14.2 Hz, 1H), 3.87–3.82 (m, 1H), 3.67 (s, 2H), 3.53 (d, J = 16.7 Hz, 1H). ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 167.9, 155.4, 147.2, 134.1, 133.2, 129.5, 129.3, 129.0, 128.9, 128.8, 128.4, 128.3, 128.2, 127.6, 72.7, 63.4, 57.5, 50.6, 49.2, 48.8, 42.2, 41.2. HRMS: calculated for C_31_H_29_N_5_O_5_·H^+^, 552.2242; found, 552.2269; HPLC (254 nm, method B) 4.4 min, 99%.
((11R,11aS)-5-(2-(benzyloxy)ethyl)-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3h). Synthesized from triazole 9h (0.25 g, 0.35 mmol) using general procedure A. The crude product was purified by column chromatography (52.5% EtOAc in hexanes) to afford 0.11 g (54%) of 3h as a white solid, mp 127.0–128.3 °C; [α]D^25^ +48.1 (c 0.36, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.55–7.49 (m, 2H), 7.44–7.27 (m, 11H), 7.14–7.09 (m, 2H), 5.03 (d, J = 17.9 Hz, 1H), 4.85 (t, J = 17.3 Hz, 2H), 4.54 (s, 2H), 4.46 (q, J = 3.9 Hz, 1H), 4.29 (d, J = 3.5 Hz, 2H), 4.24 (s, 2H), 3.85–3.81 (m, 1H), 3.68 (s, 2H), 3.56–3.48 (m, 3H), 3.45 (d, J = 16.9 Hz, 1H), 3.43–3.36 (m, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 168.6, 155.6, 147.0, 137.4, 133.3, 129.8, 129.4, 129.1, 129.0, 128.9, 128.8, 128.6, 128.2, 127.9, 127.7, 73.2, 72.9, 67.9, 63.6, 57.60, 49.4, 48.9, 47.6, 44.3, 41.3. HRMS: calculated for C_33_H_33_N_5_O_6_·H^+^, 596.2504; found, 596.2526. HPLC (254 nm, method B) 5.1 min, 100%.
3.3. General Procedure B: Debenzylation
To a 50 mL PARR reaction vessel was added 20 wt. % Pd(OH)2 on carbon (200 weight % added). EtOAc (2 mL) was then added to wet the catalyst. The requisite ester (100 weight %) was dissolved in EtOAc (4 mL) and then added to the PARR reaction flask, which was then sealed and filled with H_2_ at 55 psi. The PARR reaction vessel was evacuated with an aspirator and filled with H_2_ at 55 psi three times and was then left to shake/react overnight. After ~24 h (or until the reaction was shown to be complete), the flask was evacuated and the reaction mixture was filtered through a fritted glass funnel layered with sand, celite, and filter paper (in that order). The filtrate was then concentrated in vacuo and the crude product purified by column chromatography to afford the pure debenzylated product.
((11S,11aR)-5-(2-hydroxyethyl)-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3i). Synthesized from ester 3b (0.075 g, 0.14 mmol) using general procedure B. The crude product was purified by column chromatography (3% iPrOH in EtOAc) to afford 0.029 g (47%) of 3i as a white solid, mp 126.2–128.0 °C; [α]D^25^ −97.7 (c 0.13, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.41–7.23 (m, 5H), 4.89–4.80 (m, 2H), 4.60–4.45 (m, 3H), 4.45–4.40 (m, 1H), 4.29 (d, J = 3.5 Hz, 2H), 3.84–3.77 (m, 2H), 3.77–3.70 (m, 1H), 3.67 (s, 2H), 3.59 (dd, J = 14.2, 4.2 Hz, 1H), 3.44 (d, J = 16.2 Hz, 1H), 3.26–3.17 (m, 1H), 2.28 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 169.4, 155.7, 142.9, 133.3, 129.4, 129.1, 128.4, 127.7, 72.9, 63.4, 61.1, 57.7, 57.7, 50.2, 49.1, 48.9, 43.7, 41.4, 10.4. HRMS: calculated for C_21_H_25_N_5_O_6_·H^+^, 444.1877; found, 444.1891. HPLC (254 nm, method B) 2.4 min, 100%.
((11S,11aR)-5-(2-hydroxyethyl)-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3j). Synthesized from ester 3d (0.076 g, 0.127 mmol) using general procedure B. The crude product was purified by column chromatography (80:20:2, EtOAc:hexanes:Et_3_N) to afford 0.015 g (23%) of 3j as a white solid, mp (decomposed) 186.9–187.5 °C; [α]D^25^ −58.8 (c 0.08, CHCl_3_). ^1^H NMR (500 MHz, CDCl_3_) δ 7.54–7.26 (m, 10H), 5.11 (d, J = 17.8 Hz, 1H), 4.87 (d, J = 16.8 Hz, 1H), 4.71 (d, J = 18.0 Hz, 1H), 4.68–4.59 (m, 2H), 4.47 (q, J = 3.8 Hz, 1H), 4.30 (t, J = 3.2 Hz, 2H), 3.91–3.86 (m, 1H), 3.76–3.68 (m, 1H), 3.68 (s, 2H), 3.67–3.59 (m, 1H), 3.51–3.45 (m, 2H), 3.32–3.24 (m, 1H). ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 169.6, 155.5, 147.1, 133.2, 129.5, 129.3, 129.0, 128.8, 128.4, 128.2, 127.6, 72.8, 63.4, 60.7, 57.5, 50.3, 49.4, 48.7, 44.4, 41.2. HRMS: calculated for C_26_H_27_N_5_O_6_·H^+^, 506.2034; found, 506.2050. HPLC (254 nm, method B) 2.7 min, 100%.
((11R,11aS)-5-(2-hydroxyethyl)-3-methyl-6,9-dioxo-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3k). Synthesized from ester 3f (0.052 g, 0.098 mmol) using general procedure B. The crude product was purified by column chromatography (3% iPrOH in EtOAc) to afford 0.018 g (42%) of 3k as a white solid, mp 126.2–128.0 °C; [α]D^25^ +99.2 (c 0.12, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.41–7.23 (m, 5H), 4.89–4.80 (m, 2H), 4.60–4.45 (m, 3H), 4.45–4.40 (m, 1H), 4.29 (d, J = 3.5 Hz, 2H), 3.84–3.77 (m, 2H), 3.77–3.70 (m, 1H), 3.67 (s, 2H), 3.59 (dd, J = 14.2, 4.2 Hz, 1H), 3.44 (d, J = 16.2 Hz, 1H), 3.26–3.17 (m, 1H), 2.28 (s, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 170.9, 169.4, 155.6, 142.9, 133.3, 129.4, 129.1, 128.4, 127.7, 72.9, 63.4, 61.1, 57.7, 57.7, 50.2, 49.1, 48.9, 43.7, 41.4, 10.4. HRMS: calculated for C_21_H_25_N_5_O_6_·H^+^, 444.1877; found, 444.1891. HPLC (254 nm, method B) 2.4 min, 100%.
(11R,11aS)-5-(2-hydroxyethyl)-6,9-dioxo-3-phenyl-4,5,6,7,9,11,11a,12-octahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonin-11-yl)methyl 2-phenylacetate (3l). Synthesized from ester 3h (0.052 g, 0.084 mmol) using the general procedure for debenzylation. The crude product was purified by column chromatography (85% EtOAc in hexanes) to afford 0.032 g (73%) of 3l as a white solid, mp (decomposed) 186.9–187.5 °C; [α]D^25^ +58.8 (c 0.09, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.54–7.26 (m, 10H), 5.11 (d, J = 17.8 Hz, 1H), 4.87 (d, J = 16.8 Hz, 1H), 4.71 (d, J = 18.0 Hz, 1H), 4.68–4.59 (m, 2H), 4.47 (q, J = 3.8 Hz, 1H), 4.30 (t, J = 3.2 Hz, 2H), 3.91–3.86 (m, 1H), 3.76–3.68 (m, 1H), 3.68 (s, 2H), 3.67–3.59 (m, 1H), 3.51–3.45 (m, 2H), 3.32–3.24 (m, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 170.8, 169.6, 155.5, 147.1, 133.2, 129.5, 129.3, 129.0, 128.8, 128.4, 128.2, 127.6, 72.9, 63.4, 60.7, 57.5, 50.3, 49.4, 48.7, 44.4, 41.2. HRMS: calculated for C_26_H_27_N_5_O_6_·H^+^, 506.2034; found, 506.2050. HPLC (254 nm, method B) 2.7 min, 100%.
3.4. General Procedure C: Alkylation of Oxazolidinone
Enantiopure azide 5 [15] (100 mol %) was dissolved in dry THF (0.55 M), cooled to 0 °C, and stirred under argon. In one portion, 60% NaH (150 mol %) was added and the solution was warmed to room temperature and stirred for one hour. After one hour, the solution was cooled back to 0 °C, and α-chloroamide 6 (120 mol %) dissolved in dry THF (1.0 M) was added. The solution was once again warmed to room temperature and stirred until the reaction was shown to have reached completion by TLC (2–4 h is typical). The reaction was quenched with saturated aqueous NH_4_Cl, and the THF was removed in vacuo. The mixture was then diluted with water, and extracted thrice with EtOAc. The combined organic phases were washed twice with brine, dried with MgSO_4_, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to afford the pure azidoalkyne product (4) as a white solid.
2-((4R,5S)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-benzyl-N-(but-2-yn-1-yl)acetamide (4a). Synthesized from enantiopure azide 5a (0.51 g, 1.23 mmol) and 6a (0.35 g, 1.48 mmol) using general procedure C. The crude product was purified by column chromatography (22.5% EtOAc in hexanes) to afford 0.47 g (63%) of 4a as a white solid, mp 52.4–56.5 °C; [α]D^25^ +4.5 (c 0.36, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.47–7.22 (m, 20H), 4.79–4.52 (m, 2H), 4.33 (dd, J = 62.4, 16.9 Hz, 1H), 4.22–3.82 (m, 5H), 3.64–3.35 (m, 4H), 1.80 (d, J = 12.0 Hz, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.1, 166.9, 157.8, 143.4, 136.3, 135.7, 129.1, 128.7, 128.6, 128.4, 128.0, 127.7, 127.3, 126.6, 87.3, 81.3, 80.5, 75.7, 73.3, 72.8, 64.1, 57.6, 52.2, 52.1, 49.4, 48.9, 44.5, 44.3, 36.2, 35.4, 3.5, 3.5. HRMS: calculated for C_37_H_35_N_5_O_4_·Na^+^, 636.2581; found, 636.2587; HPLC (254 nm, method B) 8.1 min, 99%.
2-((4R,5S)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-(2-(benzyloxy)ethyl)-N-(but-2-yn-1-yl)acetamide (4b). Synthesized from enantiopure azide 5a (0.75 g, 1.81 mmol) and 6b (0.61 g, 2.17 mmol) using general procedure C. The crude product was purified by column chromatography (25% EtOAc in hexanes) to afford 0.55 g (46%) of 4b as a white solid, mp 45.9–48.3 °C; [α]D^25^ +10.5 (c 0.32, CHCl_3_). ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.40 (m, 7H), 7.34–7.29 (m, 11H), 7.24 (d, J = 7.3 Hz, 2H), 4.55–4.44 (m, 2H), 4.38–4.28 (m, 1H), 4.27–4.11 (m, 2H), 4.13–4.02 (m, 2H), 3.85 (dd, J = 57.6, 5.3 Hz, 1H), 3.77–3.52 (m, 4H), 3.38 (ddd, J = 27.8, 10.1, 5.3 Hz, 3H), 3.06 (ddd, J = 67.2, 13.2, 4.1 Hz, 1H), 1.83–1.76 (m, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 167.5, 158.0, 143.6, 143.4, 137.8, 128.8, 128.7, 128.2, 128.1, 127.5, 127.4, 87.4, 80.3, 78.0, 75.8, 73.8, 67.7, 64.4, 57.6, 54.0, 51.0, 46.4, 35.2, 3.6. HRMS: calculated for C_39_H_39_N_5_O_5_·Na^+^, 680.2843; found, 680.2843. HPLC (254 nm, method B) 9.9 min, 93%.
2-((4R,5S)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-benzyl-N-(3-phenylprop-2-yn-1-yl)acetamide (4c). Synthesized from enantiopure azide 5a (0.45 g, 1.09 mmol) and 6c (0.39 g, 1.3 mmol) using general procedure C. The crude product was purified by column chromatography (20% EtOAc in hexanes) to afford 0.63 g (86%) of 4c as a white solid, mp 55.6–66.0 °C; [α]D^25^ +2.9 (c 0.39, EtOAc); ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.21 (m, 25H), 4.84–4.63 (m, 2H), 4.58–4.44 (m, 1H), 4.44–4.14 (m, 3H), 4.04–3.89 (m, 2H), 3.54 (ddd, J = 46.9, 12.8, 3.7 Hz, 1H), 3.48–3.37 (m, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.3, 157.9, 143.4, 133.2, 131.7, 130.0, 129.1, 128.7, 128.6, 128.3, 127.9, 127.3, 126.7, 87.3, 85.2, 84.6, 83.6, 82.9, 75.8, 64.1, 57.7, 52.2, 49.5, 44.5, 36.9, 35.9. HRMS: calculated for C_42_H_37_N_5_O_4_·Na^+^, 698.2739; found, 698.2749; HPLC (254 nm, method B) 10.0 min, 95%.
2-((4R,5S)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-(2-(benzyloxy)ethyl)-N-(3-phenylprop-2-yn-1-yl)acetamide (4d). Synthesized from enantiopure azide 5a (0.66 g, 1.59 mmol) and 6d (0.65 g, 1.9 mmol) using general procedure C. The crude product was purified by column chromatography (30% EtOAc in hexanes) to afford 0.40 g (60%) of 4d as a white solid, mp 50.2–52.9 °C; [α]D^25^ +7.4 (c 0.29, CHCl_3_). ^1^H NMR (500 MHz, CDCl_3_) δ 7.47–7.37 (m, 8H), 7.31 (dq, J = 7.5, 4.7 Hz, 14H), 7.26–7.21 (m, 3H), 4.56–4.44 (m, 3H), 4.44–4.32 (m, 2H), 4.26–4.16 (m, 1H), 4.16–4.06 (m, 1H), 3.92–3.62 (m, 5H), 3.58–3.34 (m, 3H), 3.07 (ddd, J = 64.1, 13.2, 4.2 Hz, 1H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.5, 157.9, 143.6, 137.7, 131.8, 128.7, 128.6, 128.5, 128.5, 128.4, 128.1, 128.0, 127.7, 127.3, 87.3, 84.3, 84.1, 75.7, 73.8, 67.7, 64.3, 57.6, 50.9, 46.6, 43.9, 35.6. HRMS: calculated for C_44_H_41_N_5_O_5_·Na^+^, 742.2999; found, 742.3037. HPLC (254 nm, method B) 10.4 min, 93%.
2-((4S,5R)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-benzyl-N-(but-2-yn-1-yl)acetamide (4e). Synthesized from enantiopure azide 5b (0.51 g, 1.22 mmol) and 6a (0.35 g, 1.47 mmol) using general procedure C. The crude product was purified by column chromatography (22.5% EtOAc in hexanes) to afford 0.43 g (58%) of 4e as a white solid, mp 52.4–56.5 °C; [α]D^25^ −4.6 (c 0.35, CHCl_3_). ^1^H NMR (500 MHz, CDCl_3_) δ 7.47–7.22 (m, 20H), 4.79–4.52 (m, 2H), 4.33 (dd, J = 62.4, 16.9 Hz, 1H), 4.22–3.82 (m, 5H), 3.64–3.35 (m, 4H), 1.80 (d, J = 12.0 Hz, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.1, 166.9, 157.8, 143.4, 136.3, 135.7, 129.1, 128.7, 128.6, 128.4, 128.0, 127.7, 127.3, 126.6, 87.3, 81.3, 80.5, 75.7, 73.3, 72.8, 64.1, 57.6, 52.2, 52.1, 49.4, 48.9, 44.5, 44.3, 36.2, 35.4, 3.5, 3.5. HRMS: calculated for C_37_H_35_N_5_O_4_·Na^+^, 636.2581; found, 636.2587; HPLC (254 nm, method B) 8.1 min, 99%.
2-((4S,5R)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-(2-(benzyloxy)ethyl)-N-(but-2-yn-1-yl)acetamide (4f). Synthesized from enantiopure azide 5b (0.75 g, 1.81 mmol) and 6c (0.61 g, 2.17 mmol) using the general procedure for making azidoalkynes. The crude product was purified by column chromatography (30% EtOAc in hexanes) to afford 0.91 g (76%) of 4f as a white solid, mp 45.9–48.3 °C; [α]D^25^ −10.2 (c 0.33, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.40 (m, 7H), 7.34–7.29 (m, 11H), 7.24 (d, J = 7.3 Hz, 2H), 4.55–4.44 (m, 2H), 4.38–4.28 (m, 1H), 4.27–4.11 (m, 2H), 4.13–4.02 (m, 2H), 3.85 (dd, J = 57.6, 5.3 Hz, 1H), 3.77–3.52 (m, 4H), 3.38 (ddd, J = 27.8, 10.1, 5.3 Hz, 3H), 3.06 (ddd, J = 67.2, 13.2, 4.1 Hz, 1H), 1.83–1.76 (m, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.5, 158.0, 143.6, 143.4, 137.8, 128.8, 128.7, 128.2, 128.1, 127.5, 127.4, 87.4, 80.3, 78.0, 75.8, 73.8, 67.7, 64.4, 57.6, 54.1, 51.0, 46.4, 35.2, 3.6. HRMS: calculated for C_39_H_39_N_5_O_5_·Na^+^, 680.2843; found, 680.2843; HPLC (254 nm, method B) 9.9 min, 93%.
2-((4S,5R)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-benzyl-N-(3-phenylprop-2-yn-1-yl)acetamide (4g). Synthesized from enantiopure azide 5b (0.45 g, 1.09 mmol) and 6c (0.39 g, 1.3 mmol) using general procedure C. The crude product was purified by column chromatography (5% EtOAc in toluene) to afford 0.69 g (93%) of 4g as a white solid, mp 55.6–66.0 °C; [α]D^25^ −2.9 (c 0.38, EtOAc); ^1^H NMR (500 MHz, CDCl_3_) δ 7.46–7.21 (m, 25H), 4.84–4.63 (m, 2H), 4.58–4.44 (m, 1H), 4.44–4.14 (m, 3H), 4.04–3.89 (m, 2H), 3.54 (ddd, J = 46.9, 12.8, 3.7 Hz, 1H), 3.48–3.37 (m, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.3, 157.9, 143.4, 133.2, 131.7, 130.0, 129.1, 128.8, 128.6, 128.3, 127.9, 127.3, 126.7, 87.3, 85.2, 84.6, 83.6, 82.9, 75.8, 64.1, 57.7, 52.2, 49.5, 44.5, 36.9, 35.9. HRMS: calculated for C_42_H_37_N_5_O_4_·Na^+^, 698.2739; found, 698.2749; HPLC (254 nm, method B) 10.0 min, 95%.
2-((4S,5R)-4-(azidomethyl)-2-oxo-5-((trityloxy)methyl)oxazolidin-3-yl)-N-(2-(benzyloxy)ethyl)-N-(3-phenylprop-2-yn-1-yl)acetamide (4h). Synthesized from enantiopure azide 5b (0.70 g, 1.69 mmol) and 6d (0.70 g, 2.03 mmol) using general procedure C. The crude product was purified by column chromatography (30% EtOAc in hexanes) to afford 1.01 g (83%) of 4h as a white solid, mp 50.2–52.9 °C; [α]D^25^ −7.6 (c 0.29, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.47–7.37 (m, 8H), 7.31 (dq, J = 7.5, 4.7 Hz, 14H), 7.26–7.21 (m, 3H), 4.56–4.44 (m, 3H), 4.44–4.32 (m, 2H), 4.26–4.16 (m, 1H), 4.16–4.06 (m, 1H), 3.92–3.62 (m, 5H), 3.58–3.34 (m, 3H), 3.07 (ddd, J = 64.1, 13.2, 4.2 Hz, 1H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.5, 157.9, 143.6, 137.7, 131.8, 128.7, 128.6, 128.5, 128.5, 128.4, 128.1, 128.0, 127.7, 127.3, 87.3, 84.3, 84.1, 75.7, 73.8, 67.7, 64.3, 57.6, 50.9, 46.6, 43.9, 35.6. HRMS: calculated for C_44_H_41_N_5_O_5_·Na^+^, 742.2999; found, 742.3037; HPLC (254 nm, method B) 10.4 min, 93%.
3.5. General Procedure C: Preparation of α-Chloroamides
Secondary amine 7 (100 mol %) was dissolved in anhydrous CH_2_Cl_2_ (0.6 M). Freshly distilled Et_3_N (300 mol %) was added to the solution, which was then cooled to 0 °C. Freshly distilled chloroacetyl chloride (200 mol %) dissolved in anhydrous CH_2_Cl_2_ (5.5 M) was then added dropwise over 20 min. The solution was stirred for an additional 5 min at 0 °C, and was then warmed to room temperature. When the reaction was shown to be complete by TLC (typically 1–2 h), it was quenched with water and diluted with CH_2_Cl_2_. The layers were separated and the organic phase was then washed twice with water. The combined aqueous layers were then extracted twice with CH_2_Cl_2_. The combined organic layers were washed with brine, dried with MgSO_4_, filtered, and concentrated in vacuo. The crude product was then purified by column chromatography to afford the α-chloroamide product (6) as a dark-brown oil.
N-benzyl-N-(but-2-yn-1-yl)-2-chloroacetamide (6a). Synthesized from amine 7a (0.30 g, 1.9 mmol) using general procedure C. The crude product was purified by column chromatography (10% EtOAc in hexanes) to afford 0.36 g (80%) of 6a. ^1^H NMR (300 MHz, CDCl_3_) δ 7.42–7.20 (m, 5H), 4.72 (s, 1H), 4.69 (s, 1H), 4.24 (s, 1H), 4.18 (s, 1H), 4.10 (s, 1H), 3.94 (d, J = 2.6 Hz, 1H), 1.86–1.76 (m, 3H). ^13^C NMR (125 MHz, CDCl_3_) δ 167.0, 136.6, 129.3, 129.1, 129.0, 128.9, 128.9, 128.6, 128.4, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.1, 81.7, 80.9, 73.4, 50.7, 49.2, 44.1, 42.9, 41.7, 37.4, 35.8, 3.7. HRMS: calculated for C_13_H_14_ClNO·H^+^, 236.0837; found, 236.0837. HPLC (254 nm, method B) 3.4 min, 100%.
N-(2-(benzyloxy)ethyl)-N-(but-2-yn-1-yl)-2-chloroacetamide (6b). Synthesized from 7b (0.45 g, 2.21 mmol) using general procedure C. The crude product was purified by column chromatography (15% EtOAc in hexanes) to afford 0.37 g (59%) of 6b. ^1^H NMR (500 MHz, CDCl_3_) δ 7.39–7.27 (m, 5H), 4.57 (d, J = 2.5 Hz, 2H), 4.28 (d, J = 2.0 Hz, 1H), 4.17 (d, J = 3.9 Hz, 4H), 4.11 (s, 1H), 3.29 (dd, J = 19.0, 7.6 Hz, 2H), 2.12–1.94 (m, 1H), 0.94 (dd, J = 21.5, 6.7 Hz, 6H); ^13^C NMR (125 MHz, CDCl_3_) δ 166.7, 166.3, 137.2, 136.9, 128.3, 128.2, 127.8, 127.8, 127.7, 80.8, 80.8, 80.5, 79.7, 71.7, 71.3, 57.2, 57.1, 54.8, 53.7, 41.3, 40.9, 38.1, 35.3, 26.9, 26.6, 19.8, 19.7; HRMS: calculated for C_15_H_18_ClNO_2_·H^+^, 280.1098; found, 280.1099; HPLC (254 nm, method B) 3.5 min, 100%.
N-benzyl-2-chloro-N-(3-phenylprop-2-yn-1-yl)acetamide (6c). Synthesized from 7c (0.35 g, 1.59 mmol) using general procedure C. The crude product was purified by column chromatography (80% CH_2_Cl_2_ in hexanes) to afford 0.28 g (60%) of 6c. ^1^H NMR (300 MHz, CDCl_3_) δ 7.45–7.25 (m, 10H), 4.78 (d, J = 7.1 Hz, 2H), 4.48 (s, 1H), 4.27 (d, J = 13.8 Hz, 2H), 4.14 (s, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 166.8, 136.0, 135.5, 131.7, 130.1, 129.1, 128.8, 128.8, 128.5, 128.4, 128.4, 128.3, 128.3, 128.1, 127.8, 126.8, 122.4, 121.9, 85.2, 84.6, 83.2, 82.6, 50.6, 49.1, 41.3, 37.5, 35.9; HRMS: calculated for C_18_H_16_ClNO·H^+^, 298.0993; found, 298.0993; HPLC (254 nm, method B) 4.9 min, 95%.
N-(2-(benzyloxy)ethyl)-2-chloro-N-(3-phenylprop-2-yn-1-yl)acetamide (6d). Synthesized from 7d (0.70 g, 2.64 mmol) using general procedure C. The crude product was purified by column chromatography (15% EtOAc in hexanes) to afford 0.61 g (67%) of 6d. ^1^H NMR (500 MHz, CDCl_3_) δ 7.40–7.27 (m, 10H), 4.53 (s, 2H), 4.49 (s, 2H), 4.26 (d, J = 11.1 Hz, 2H), 3.80–3.75 (m, 2H), 3.73–3.70 (m, 2H); ^13^C NMR (125 MHz, CDCl_3_) δ 166.9, 166.5, 137.9, 137.5, 131.6, 128.6, 128.4, 128.2, 127.7, 127.4, 122.4, 121.9, 84.1, 83.8, 83.3, 73.3, 73.1, 68.5, 67.3, 47.3, 46.7, 41.4, 39.8, 35.9; HRMS: calculated for C_20_H_20_ClNO_2_·H^+^, 342.1255; found, 342.1255; HPLC (254 nm, method B) 5.0, 87%.
3.6. General Procedure E: Intramolecular Dipolar Cyclization (9)
A solution of azidoalkyne (4) was dissolved in distilled toluene (0.008 M) was heated to reflux until TLC showed the reaction to have reached completion (18–48 h). The reaction was then concentrated in vacuo, and the crude product was purified by column chromatography to afford the pure triazole product (9) as a white solid.
(11S,11aR)-5-benzyl-3-methyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo [3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8a). Synthesized from azidoalkyne 4a (0.29 g, 0.48 mmol) using general procedure E. The crude product was purified by column chromatography (40% EtOAc in hexanes) to afford 0.28 g (97%) of 8a as a white solid, mp 168.0–169.0 °C; [α]D^25^ −32.0 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.38 (d, J = 7.8 Hz, 6H), 7.35–7.29 (m, 9H), 7.29–7.24 (m, 3H), 7.19–7.11 (m, 2H), 5.08 (dd, J = 25.7, 15.3 Hz, 2H), 4.78 (d, J = 17.7 Hz, 1H), 4.49 (d, J = 15.4 Hz, 1H), 4.36 (q, J = 3.6 Hz, 1H), 4.30–4.21 (m, 2H), 4.01–3.96 (m, 1H), 3.94 (d, J = 16.5 Hz, 1H), 3.82 (d, J = 14.3 Hz, 1H), 3.49 (dd, J = 10.6, 3.7 Hz, 1H), 3.08 (dd, J = 10.6, 2.8 Hz, 1H), 2.17 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.5, 156.4, 143.1, 134.6, 129.1, 128.8, 128.6, 128.5, 128.2, 128.0, 127.9, 127.5, 87.2, 74.6, 63.2, 57.9, 50.0, 49.5, 49.1, 41.1, 10.2. HRMS: calculated for C_37_H_35_N_5_O_4_·Na^+^, 636.2581; found, 636.2589; HPLC (254 nm, method B) 5.8 min, 94%.
(11S,11aR)-5-(2-(benzyloxy)ethyl)-3-methyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8b). Synthesized from azidoalkyne 4b (0.50 g, 0.76 mmol) using general procedure E. The crude product was purified by column chromatography (60% EtOAc in hexanes) to afford 0.32 g (64%) of 8b as a white solid, mp 209.0–211.0 °C; [α]D^25^ −25.0 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.38–7.24 (m, 24H), 5.06 (d, J = 16.8 Hz, 1H), 4.89 (d, J = 17.7 Hz, 1H), 4.69 (d, J = 17.7 Hz, 1H), 4.49–4.37 (m, 2H), 4.36 (s, 3H), 3.95–3.91 (m, 1H), 3.85 (d, J = 16.2 Hz, 1H), 3.70–3.63 (m, 1H), 3.64–3.59 (m, 2H), 3.48 (dd, J = 10.7, 3.8 Hz, 1H), 3.28 (dt, J = 13.2, 6.2 Hz, 1H), 3.07 (dd, J = 10.8, 2.8 Hz, 1H), 2.22 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.8, 156.4, 143.1, 142.6, 137.5, 128.6, 128.6, 128.5, 128.2, 128.1, 127.8, 127.5, 87.2, 74.6, 73.4, 68.4, 63.2, 57.9, 49.5, 49.2, 47.3, 43.5, 10.2; HRMS: calculated for C_39_H_39_N_5_O_5_·H^+^, 658.3024; found, 658.3050; HPLC (254 nm, method B) 8.6 min, 98%.
(11R,11aS)-5-benzyl-3-phenyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo [3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8c). Synthesized from azidoalkyne 4c (0.29 g, 0.43 mmol) using general procedure E. The crude product was purified by column chromatography (30% EtOAc in hexanes) to afford 0.28 g (97%) of 8c as a white solid, mp (decomposed) 241 °C; [α]D^25^ +3.1 (c 0.4, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.45–7.12 (m, 23H), 6.97–6.92 (m, 2H), 5.15 (d, J = 16.6 Hz, 1H), 5.05 (d, J = 17.9 Hz, 1H), 4.77 (d, J = 14.2 Hz, 1H), 4.57–4.49 (m, 2H), 4.37 (q, J = 3.5 Hz, 1H), 4.30 (dd, J = 15.4, 2.6 Hz, 1H), 4.06 (d, J = 14.2 Hz, 1H), 4.04–4.02 (m, 1H), 3.98 (d, J = 16.6 Hz, 1H), 3.50 (dd, J = 10.6, 3.8 Hz, 1H), 3.08 (dd, J = 10.7, 2.8 Hz, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.4, 156.4, 143.1, 134.2, 129.7, 128.9, 128.8, 128.8, 128.5, 128.5, 128.3, 127.6, 87.3, 74.6, 63.3, 57.8, 50.6, 49.7, 49.1, 42.4; HRMS: calculated for C_42_H_37_N_5_O_4_·Na^+^, 698.2738; found, 698.2752; HPLC (254 nm, method B) 8.0 min, 98%.
(11R,11aS)-5-(2-(benzyloxy)ethyl)-3-phenyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8d). Synthesized from azidoalkyne 4d (0.53 g, 0.73 mmol) using general procedure E. The crude product was purified by column chromatography (35% EtOAc in hexanes) to afford 0.34 g (65%) of 8d as a white solid, mp 211.0–213.0 °C; [α]D^25^ +16.7 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.54 (dd, J = 7.5, 2.2 Hz, 2H), 7.44–7.36 (m, 9H), 7.35–7.23 (m, 12H), 7.11 (dd, J = 7.1, 2.5 Hz, 2H), 5.12 (t, J = 17.3 Hz, 2H), 4.90 (d, J = 17.9 Hz, 1H), 4.60–4.49 (m, 2H), 4.41 (q, J = 3.5 Hz, 1H), 4.24 (s, 2H), 3.99 (dt, J = 4.1, 2.0 Hz, 1H), 3.89 (d, J = 16.6 Hz, 1H), 3.56–3.46 (m, 4H), 3.45–3.35 (m, 1H), 3.09 (dd, J = 10.7, 2.8 Hz, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 169.0, 156.4, 146.9, 143.1, 137.4, 129.9, 129.0, 128.8, 128.7, 128.6, 128.5, 128.3, 128.2, 127.9, 127.7, 127.6, 87.3, 74.7, 73.3, 68.0, 63.3, 57.9, 49.8, 49.1, 47.6, 44.5; HRMS: calculated for C_44_H_41_N_5_O_5_·H^+^, 720.3180; found, 720.3161; HPLC (254 nm, method B) 8.7 min, 100%.
(11R,11aS)-5-benzyl-3-methyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo [3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8e). Synthesized from azidoalkyne 4a (0.35 g, 0.58 mmol) using general procedure E. The crude product was purified by column chromatography (40% EtOAc in hexanes) to afford 0.24 g (68%) of 8e as a white solid, mp 168.0–169.0 °C; [α]D^25^ +32.6 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.38 (d, J = 7.8 Hz, 6H), 7.35–7.29 (m, 9H), 7.29–7.24 (m, 3H), 7.19–7.11 (m, 2H), 5.08 (dd, J = 25.7, 15.3 Hz, 2H), 4.78 (d, J = 17.7 Hz, 1H), 4.49 (d, J = 15.4 Hz, 1H), 4.36 (q, J = 3.6 Hz, 1H), 4.30–4.21 (m, 2H), 4.01–3.96 (m, 1H), 3.94 (d, J = 16.5 Hz, 1H), 3.82 (d, J = 14.3 Hz, 1H), 3.49 (dd, J = 10.6, 3.7 Hz, 1H), 3.08 (dd, J = 10.6, 2.8 Hz, 1H), 2.17 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.5, 156.4, 143.1, 134.6, 129.1, 128.8, 128.6, 128.5, 128.2, 128.0, 127.9, 127.5, 87.2, 74.6, 63.2, 57.9, 50.0, 49.5, 49.1, 41.1, 10.2; HRMS: calculated for C_37_H_35_N_5_O_4_·Na^+^, 636.2581; found, 636.2589; HPLC (254 nm, method B) 5.8 min, 94%.
(11R,11aS)-5-(2-(benzyloxy)ethyl)-3-methyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8f). Synthesized from azidoalkyne 4b (0.51 g, 0.78 mmol) using general procedure E. The crude product was purified by column chromatography (60% EtOAc in hexanes) to afford 0.32 g (61%) of 8f as a white solid, mp 209.0–211.0 °C; [α]D^25^ +24.6 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.38–7.24 (m, 24H), 5.06 (d, J = 16.8 Hz, 1H), 4.89 (d, J = 17.7 Hz, 1H), 4.69 (d, J = 17.7 Hz, 1H), 4.49–4.37 (m, 2H), 4.36 (s, 3H), 3.95–3.91 (m, 1H), 3.85 (d, J = 16.2 Hz, 1H), 3.70–3.63 (m, 1H), 3.64–3.59 (m, 2H), 3.48 (dd, J = 10.7, 3.8 Hz, 1H), 3.28 (dt, J = 13.2, 6.2 Hz, 1H), 3.07 (dd, J = 10.8, 2.8 Hz, 1H), 2.22 (s, 3H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.8, 156.4, 143.1, 142.6, 137.5, 128.6, 128.6, 128.5, 128.2, 128.1, 127.8, 127.5, 87.2, 74.6, 73.4, 68.4, 63.2, 58.0, 49.5, 49.2, 47.3, 43.5, 10.21; HRMS: calculated for C_39_H_39_N_5_O_5_·H^+^, 658.3024; found, 658.3050; HPLC (254 nm, method B) 8.6 min, 98%.
(11R,11aS)-5-benzyl-3-phenyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo [3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8g). Synthesized from azidoalkyne 4c (0.34 g, 0.51 mmol) using general procedure E. The crude product was purified by column chromatography (30% EtOAc in hexanes) to afford 0.25 g (71%) of 8g as a white solid, mp (decomposed) 241 °C; [α]D^25^ −3.4 (c 0.4, CHCl_3_;); ^1^H NMR (500 MHz, CDCl_3_) δ 7.45–7.12 (m, 23H), 6.97–6.92 (m, 2H), 5.15 (d, J = 16.6 Hz, 1H), 5.05 (d, J = 17.9 Hz, 1H), 4.77 (d, J = 14.2 Hz, 1H), 4.57–4.49 (m, 2H), 4.37 (q, J = 3.5 Hz, 1H), 4.30 (dd, J = 15.4, 2.6 Hz, 1H), 4.06 (d, J = 14.2 Hz, 1H), 4.04–4.02 (m, 1H), 3.98 (d, J = 16.6 Hz, 1H), 3.50 (dd, J = 10.6, 3.8 Hz, 1H), 3.08 (dd, J = 10.7, 2.8 Hz, 1H); ^13^C NMR (125 MHz, CDCl_3_) δ 168.4, 156.4, 143.1, 134.2, 129.7, 128.9, 128.9, 128.8, 128.5, 128.5, 128.3, 127.6, 87.3, 74.6, 63.3, 57.8, 50.6, 49.7, 49.1, 42.4; HRMS: calculated for C_42_H_37_N_5_O_4_·Na^+^, 698.2738; found, 698.2752; HPLC (254 nm, method B) 8.0 min, 98%.
(11R,11aS)-5-(2-(benzyloxy)ethyl)-3-phenyl-11-((trityloxy)methyl)-4,5,11a,12-tetrahydrooxazolo[3,4-a][1,2,3]triazolo[1,5-d][1,4,7]triazonine-6,9(7H,11H)-dione (8h). Synthesized from azidoalkyne 4d (0.50 g, 0.70 mmol) using general procedure E. The crude product was purified by column chromatography (40% EtOAc in hexanes) to afford 0.37 g (73%) of 8h as a white solid, mp 211.0–213.0 °C; [α]D^25^ −16.3 (c 0.3, CHCl_3_); ^1^H NMR (500 MHz, CDCl_3_) δ 7.54 (dd, J = 7.5, 2.2 Hz, 2H), 7.44–7.36 (m, 9H), 7.35–7.23 (m, 12H), 7.11 (dd, J = 7.1, 2.5 Hz, 2H), 5.12 (t, J = 17.3 Hz, 2H), 4.90 (d, J = 17.9 Hz, 1H), 4.60–4.49 (m, 2H), 4.41 (q, J = 3.5 Hz, 1H), 4.24 (s, 2H), 3.99 (dt, J = 4.1, 2.0 Hz, 1H), 3.89 (d, J = 16.6 Hz, 1H), 3.56–3.46 (m, 4H), 3.45–3.35 (m, 1H), 3.09 (dd, J = 10.7, 2.8 Hz, 1H). ^13^C NMR (125 MHz, CDCl_3_) δ 169.0, 156.4, 146.9, 143.1, 137.4, 129.9, 129.0, 128.8, 128.7, 128.6, 128.5, 128.3, 128.2, 127.9, 127.7, 127.6, 87.3, 74.7, 73.3, 68.0, 63.3, 57.9, 49.8, 49.1, 47.6, 44.5. HRMS: calculated for C_44_H_41_N_5_O_5_·H^+^, 720.3180; found, 720.3161; HPLC (254 nm, method B) 8.7 min, 100%.
3.7. Transcription Assays
The T-box riboswitch in vitro transcription readthrough assay was conducted as previously described [25]. Triplicate reactions of 0.1 mM ligand, 30 nM tRNA, 5 mM spermidine, 0.05 U/uL RNAP, and 2.5% DMSO were run for 6 h and the data analyzed as previously described [25] to determine the Readthrough_max_. Unpaired, two-tailed t tests were conducted using Prism 9.0 (GraphPad) of each ligand reaction compared to the respective control in the absence of ligand. Percentage inhibition of tRNA-induced transcription readthrough was calculated as [1 − [(ΔRFU_tRNA+L_ − ΔRFU_L_)/(ΔRFU_tRNA+0_ − ΔRFU_0_)]] × 100, where ΔRFU_tRNA+0_ and ΔRFU_0_ are the average Readthrough_max_ values in the absence of ligand for the control reactions with or without tRNA, respectively, and ΔRFU_tRNA+L_ and ΔRFU_L_ are the respective average values in the presence of ligand.
3.8. Computational Docking
The computational docking was conducted similarly to that previously described using the Glide module of the Schrödinger software suite (version 2022-2, Schrödinger, New York, NY, USA) [15]. Ligands were initially built in ChemDraw 22.0.0.22, exported as .sdf files and then prepared for docking using Ligprep (Schrödinger) with the OPLS4 forcefield. The docking grid was generated from 1N53 PDB [26] using Glide with the OPLS2005 forcefield (non-default settings were: grid-centered on residues 2–15, 22–28 (numbering as in Figure 2); dock ligands ≤ 20 Å; ligand midpoint box 25 Å in x,y,z) and the ligands docked using Glide with the OPLS2005 forcefield (non-default settings were: standard precision docking; include aromatic H and halogens as hydrogen bond acceptors; keep at most 5 poses, no minimization).
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Antimicrobial Resistance Collaborators Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis Lancet 202239962965510.1016/S 0140-6736(21)02724-035065702 PMC 8841637 · doi ↗ · pubmed ↗
- 2U.S. Department of Health and Human Services, CDC COVID-19: U.S. Impact on Antimicrobial Resistance, Special Report U.S. Department of Health and Human Services, CDC Atlanta, GA, USA 202210.15620/cdc:117915 · doi ↗
- 3U.S. Department of Health and Human Services, CDC Antibiotic Resistance Threats in the United States U.S. Department of Health and Human Services, CDC Atlanta, GA, USA 201910.15620/cdc:82532 · doi ↗
- 4Panchal V. Brenk R. Riboswitches as Drug Targets for Antibiotics Antibiotics 2021104510.3390/antibiotics 1001004533466288 PMC 7824784 · doi ↗ · pubmed ↗
- 5Giarimoglou N. Kouvela A. Maniatis A. Papakyriakou A. Zhang J. Stamatopoulou V. Stathopoulos C. A Riboswitch-Driven Era of New Antibacterials Antibiotics 202211124310.3390/antibiotics 1109124336140022 PMC 9495366 · doi ↗ · pubmed ↗
- 6Motika S.E. Ulrich R.J. Geddes E.J. Lee H.Y. Lau G.W. Hergenrother P.J. Gram-Negative Antibiotic Active Through Inhibition of an Essential Riboswitch J. Am. Chem. Soc.2020142108561086210.1021/jacs.0c 0442732432858 PMC 7405991 · doi ↗ · pubmed ↗
- 7Fukunaga K. Dhamodharan V. Miyahira N. Nomura Y. Mustafina K. Oosumi Y. Takayama K. Kanai A. Yokobayashi Y. Small-Molecule Aptamer for Regulating RNA Functions in Mammalian Cells and Animals J. Am. Chem. Soc.20231457820782810.1021/jacs.2c 1233236991533 PMC 10103173 · doi ↗ · pubmed ↗
- 8Crielaard S. Maassen R. Vosman T. Rempkens I. Velema W.A. Affinity-Based Profiling of the Flavin Mononucleotide Riboswitch J. Am. Chem. Soc.2022144104621047010.1021/jacs.2c 0268535666649 PMC 9204756 · doi ↗ · pubmed ↗