4,5-Dihydro-2H-pyridazin-3-ones as a Platform for the Construction of Chiral 4,4-Disubstituted-dihydropyridazin-3-ones
Paul Joël Henry, Gabriel Burel, William Nzegge, Mario Waser, Jean-François Brière

TL;DR
This paper presents a new method to synthesize chiral dihydropyridazin-3-ones using 4,5-dihydro-2H-pyridazin-3-ones as a starting platform.
Contribution
A novel asymmetric synthesis pathway for 4,4-disubstituted dihydropyridazin-3-ones using chiral catalysts is introduced.
Findings
Chiral ammonium salt phase-transfer catalysts enable enantioselective conjugate additions to DHPDOs.
The method allows for the formation of all-carbon stereocenters in dihydropyridazin-3-one derivatives.
Moderate to good enantioselectivities and reasonable yields are achieved with acrylate and quinone methide substrates.
Abstract
4,5-Dihydro-2H-pyridazin-3-ones (DHPDOs) are important synthetic as well as naturally occurring heterocycles. We herein report the synthesis of various 4-monofunctionalized 4,5-dihydro-2H-pyridazin-3-ones and their use as starting materials to access 4,4-disubstituted dihydropyridazin-3-ones in an asymmetric fashion. By using chiral ammonium salt phase-transfer catalysts, conjugate additions of these scaffolds to classical acrylate-based Michael acceptors, as well as quinone methides, can be carried out with moderate to good enantioselectivities and in reasonable yields, affording a new pathway to dihydropyridazin-3-one derivatives with an all-carbon stereocenter.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
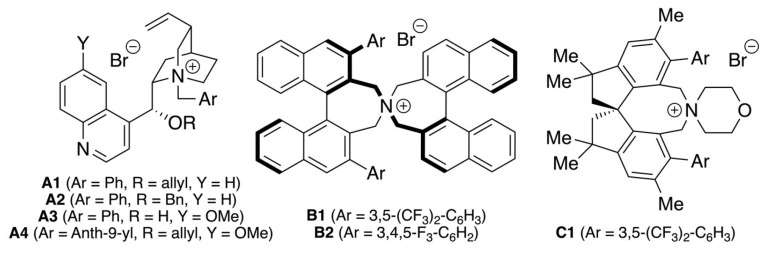 Figure 1
Figure 1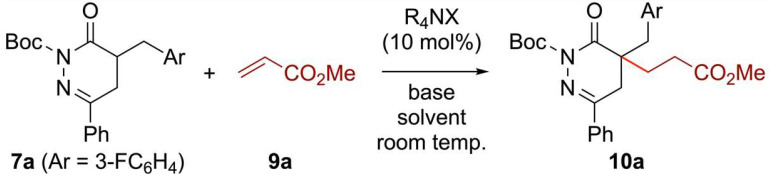 Figure 2
Figure 2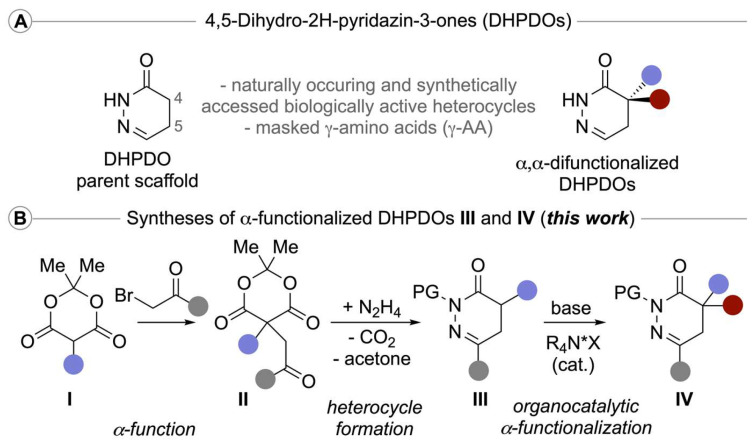 Figure 3
Figure 3 Figure 4
Figure 4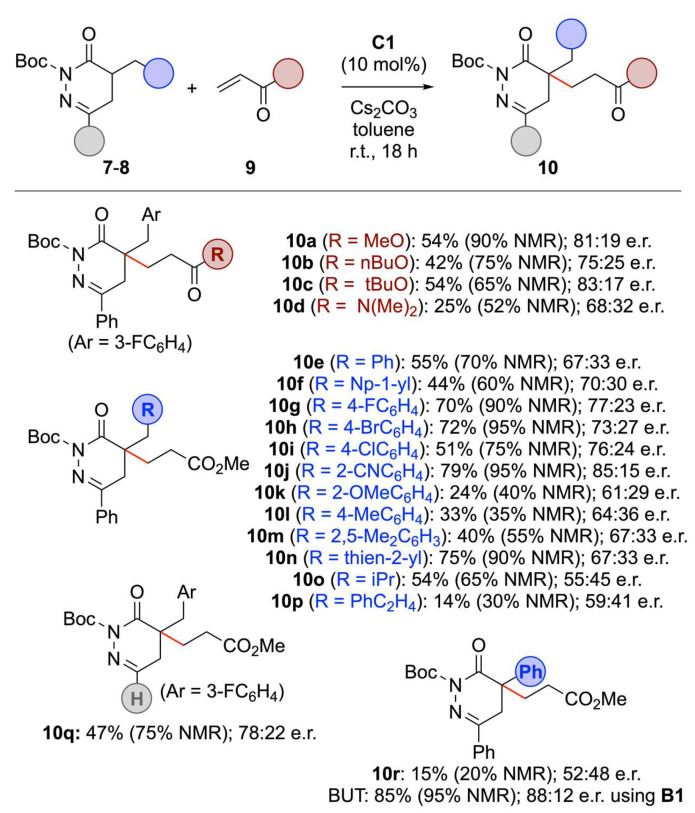 Figure 5
Figure 5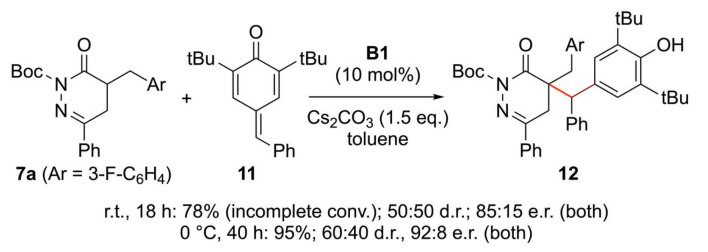 Figure 6
Figure 6- —Austrian Science Funds (FWF)
- —Université Rouen Normandie
- —INSA Rouen Normandie
- —Centre National de la Recherche Scientifique (CNRS)
- —European Regional Development Fund (ERDF)
- —Région Normandie
- —Labex SynOrg
- —Carnot Institute I2C
- —graduate school for research XL-Chem
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPhenothiazines and Benzothiazines Synthesis and Activities · Synthesis and Reactivity of Heterocycles · Synthesis and bioactivity of alkaloids
1. Introduction
The 4,5-dihydro-2H-pyridazin-3-one scaffold (DHPDO; Scheme 1A) represents an important structural motif that can be frequently found in a variety of synthetic and naturally occurring biologically active 6-membered *N-*containing heterocycles [1,2,3,4,5,6,7,8,9,10,11,12]. Not surprisingly, several research groups have developed catalytic asymmetric syntheses of DHPDO derivatives which are either mono-substituted at the C5- [13,14], or the C4-position (α-position of the carbonyl functionality) [15,16,17,18,19,20,21,22]. Furthermore, (chiral) substituted 4,5-dihydropyridazinone derivatives are useful building blocks and may be considered as masked γ-amino acid (γ-AA) derivatives [23,24], after reduction of the hydrazone moiety or cleavage of the N-N bond [16,25,26,27,28,29]. In that domain, several of these potentially medicinally interesting heterocycles contain one or two substituents at the 4-position [5,11]. However, to the best of our knowledge, the asymmetric catalytic elaboration of α,α-disubstituted DHPDOs, containing a quaternary stereocenter has not been covered yet. Thus, the development of reliable strategies to access different α-functionalized DHPDOs straightforwardly, even in an asymmetric manner, represents a worthwhile goal. Our groups have a longstanding interest in asymmetric synthesis and functionalization of chiral heterocycles, i.e., those that can be considered as masked α- and β-amino acids [30,31,32,33].
In line with our general research interests and the aforementioned value of chiral 4,5-dihydro-2H-pyridazin-3-ones, we now set out to establish versatile synthesis routes to access various α-functionalized DHPDOs III based on hydrazine and Meldrum’s acid chemistry, i.e., derivatives I [34,35,36,37] (Scheme 1). The easily accessible compounds I were believed to allow the elaboration of appropriately functionalized precursors II, which can be seen as 1,4-ketoester equivalents enabling the synthesis of DHPDOs III upon treatment with hydrazine (Scheme 1B). We herein furthermore report a strategy which demonstrates the organocatalytic asymmetric α-functionalization of DHPDO compounds III with different Michael-type acceptors under chiral ammonium salt phase-transfer catalytic conditions en route to the construction of new heterocyclic derivatives IV with an all-carbon stereocenter [38,39,40,41,42].
2. Results and Discussion
2.1. Syntheses of Pyridazinones
At the onset, we sought a straightforward and versatile route to α-functionalized DHPDOs in order to allow the investigation of the catalytic asymmetric α-alkylation reactions (III to IV, Scheme 1). Inspired by the pioneering contribution of Tóth and collaborators [43], we exploited the readily available substituted Meldrum’s acid derivatives 1 as convenient precursors of 1,4-ketoester equivalents (Scheme 2) [11,44,45]. After an alkylation step of 1 with α-bromoacetophenone under basic conditions, producing access to products 2, the facile condensation of hydrazine led to a series of free NH-containing DHPDOs 3 in good yields. On the other hand, in the purpose of expanding the scope of DHPDO substrates, we tackled the introduction of the CH_2_CH_2_CHO motif. However, this turned out to be not a trivial task on Meldrum’s acid derivatives 1, according to the weak nucleophilic properties of its corresponding anion [34,35,36,37]. By revisiting Parsons’ conditions, in the presence of manganese(III) acetate as a single-electron oxidant, we found that tert-butyl vinyl ether smoothly underwent a radical addition of the model benzylated Meldrum’s acid derivative 1a, yielding product 4 [46,47]. This tert-butyl derived acetal 4 was easily deprotected into the corresponding aldehyde 5 in 15 min using Sc(OTf)3 as a Lewis acid [48]. (We initially tried the commonly employed n-butyl vinyl ether as radical reaction partner [46], but the transformation proved to be less efficient than with tert-butyl vinyl ether, and the corresponding addition product was reluctant to be deprotected into aldehyde without leading to decomposition events under forced conditions). This rather unstable aldehyde has to be rapidly engaged in the subsequent condensation with hydrazine at 60 °C, affording the DHPDO 6 in excellent 70% yield over three steps. Eventually, in order to explore the reactivity of various molecular platforms, the N-Boc-protected DHPDOs 7 and 8 were also synthesized.
2.2. α-Functionalizations of Pyridazinones
With a robust route for the syntheses of differently functionalized pyridazinones 3 and 6, as well as the N-protected derivatives 7 and 8 at hand, we next focused on the asymmetric α-functionalization of these heterocycles. We started our investigations by testing the quaternary ammonium salt-catalysed Michael addition of the parent compound 7a to acrylate 9a (Table 1 gives a condensed overview of the most significant results obtained by using the catalysts depicted in Figure 1).
First experiments using the well-established Cinchona alkaloid-based Quatsalts A showed that the 1,4-addition of 7a to 9a is possible in principle, but the enantioselectivities obtained for product 10a were disappointing (entries 1–4) [38,39,40,41,42]. Also, varying the base and solvent did not allow us to achieve any notable levels of enantiodifferentiation (entries 5–7 give illustrative examples of a broader screening). Thus, we focused on alternative catalyst scaffolds. Unfortunately, the commercially available Maruoka catalysts B allowed for low conversions and low enantioselectivities only (entries 8 and 9; other conditions were tested as well, but did not lead to any improvement) [49].
Interestingly, the recently introduced spirobiindane-based salt C1 was the only catalyst scaffold that gave reasonable conversion combined with somewhat more promising levels of enantioselectivities under the initially chosen conditions (entry 10) [50,51,52]. Variation of base and solvent (entries 10–13) revealed that Cs_2_CO_3_ in toluene allows for notable enantioselectivities (up to 90:10 e.r.), but conversion was slow (entry 13). While increased reaction times resulted in more practical rates of conversion, this was accompanied by a slight decrease in enantioselectivity (entries 13–15). Such a decrease in enantioselectivity with increased reaction progress can most likely be rationalized by a change of the catalyst’s integrity, i.e., its counter anion or also some catalyst degradation over the course of several catalyst turnovers. Using a slightly larger excess of base (3 eq.) at room temperature finally gave product 10a with a high NMR yield of around 90% and a moderate enantioselectivity of 81:19 e.r. (entry 16). Unfortunately, the reaction became again much slower when carried out at 0 °C (entry 17). Noteworthy, the isolated yield of 10a was lower than expected from the NMR yield (54% vs 90%; entry 16), which can be explained by a partial retro-Michael reaction of 10a under the applied “classical” normal-phase silica gel column chromatography conditions (as observed by the formation of notable amounts of starting material 7a again; this obstacle can to some extent be reduced, but not totally suppressed by using basified high-quality silica gel).
Having identified operationally reliable conditions that represent a reasonable compromise between conversion (yield) and enantioselectivity, we next investigated the generality of this protocol (Scheme 3). Testing different acrylates 9 first revealed that the ester functionality does not massively influence the outcome of this transformation (products 10a–c). Interestingly, even the poorly electrophilic dimethylacrylamide was transformed into the corresponding Michael adduct 10d in 32:68 er while in moderate NMR yield of 52%. Variations of the (hetero)benzylic α-substituents of starting materials 7 were investigated next (products 10e–k and 10n). Unfortunately, enantioselectivities were only modest as well, while conversion rates and yields somewhat depended on the nature of the benzylic groups (i.e., Me-containing electron-rich aryl groups led to lower yields as exemplified for products 10m and 10n). Other alkyl substituents were less well tolerated in terms of yield and e.r. (10l, 10p).
The use of the 6-H containing DHPDO 8 was also reasonably well tolerated, producing product 10q in comparable yield and enantiopurity as for the 4-Ph-substituted analogue 10a. Furthermore, we also tested an α-Ph-containing DPHDO and hereby made an interesting observation (product 10r). While the standard conditions using Quatsalt C1 resulted in low yield and racemic product formation only, the use of Maruoka’s catalyst B1 resulted in high yield and good e.r. of 88:12. This result, which is in clear contrast to the observations made when optimizing the parent reaction using α-alkylated pyridazinone 7a (Table 1), clearly underscore the difficulties in predicting a suited catalyst for a given transformation and the importance of proper catalyst screening for differently substituted starting materials!
In addition to using classical 1,4-acceptors 9, we also tested if compounds 7 may undergo 1,6-additions to para-quinone methides (p-QMs) such as compound 11 (Scheme 4) [53,54,55,56]. We quickly realized that the addition of 7a to 11 is a rather rapid and high-yielding process under quaternary ammonium salt catalysis conditions, affording the corresponding adduct 12 in up to 95% yield. When using chiral Quatsalts, the diastereoselectivity was always low (<60:40 d.r.)—this limitation is not rare. Reactions of enolate species to p-QMs could also not be overcome by any alteration of the reaction conditions. In contrast, enantioselectivities for both diastereomers were up to 92:8 e.r. when using Maruoka’s catalyst B1 at 0 °C.
3. Experimental Details
Detailed experimental procedures and analytical details can be found in the online supporting information.
General procedure for the synthesis of N-Boc 4,5-dihydropyridazinone derivatives 7. Compounds 2: C5-monosubstituted Meldrum’s acid derivatives 1 (4.9 mmol), 2-bromo acetophenone (1.05 g, 5.3 mmol) and sodium acetate (435 mg, 5.3 mmol) were introduced into a round-bottom flask (N_2_ atmosphere). The mixture was dissolved in DMF (5 mL, 0.98 M), and acetic acid (0.29 mL, 4.9 mmol) was added dropwise at room temperature (vigorous stirring). After a night, the solvent was evaporated under reduced pressure. The crude product was dissolved in DCM and washed with a mixture of H_2_O/Na_2_CO_3_ (sat.) (9:1), and then with brine. The resulting organic phase was dried over anhydrous Na_2_SO_4_, filtrated, and finally the solvent was evaporated under reduced pressure. The crude reaction mixture was triturated with Et_2_O to give products 2a–o (exact experimental and analytical details can be found in the online supporting information).Compounds 3: C5-disubstituted Meldrum’s acid derivative 2 (3.3 mmol) was solubilized in DMF (11 mL, 0.3 M) first (N_2_ atmosphere). At 0 °C, under vigorous stirring, hydrazine monohydrate (0.65 mL, 13.3 mmol) was added dropwise. The reaction was stirred for 24 h at room temperature, then the solvent was evaporated under reduced pressure. The crude was dissolved in DCM and washed with an aqueous acidic solution (pH 3–4) and then with brine. The combined organic phases were dried over anhydrous Na_2_SO_4_ and finally, the solvent was evaporated under reduced pressure, producing products 3 (exact experimental and analytical details can be found in the online supporting information).Compounds 7: Boc_2_O (938.5 mg, 4.3 mmol) and DMAP (87.5 mg, 716 µmol) were introduced in a round-bottom flask (N_2_ atmosphere). The N-unsubstituted 4,5-dihydropyridazinone 3 (3.58 mmol) was dissolved in anhydrous DCM (24 mL, 0.15 M) and introduced slowly into the round-bottom flask at room temperature. The solution was vigorously stirred overnight. Then, the solvent was evaporated under reduced pressure and the crude mixture was purified by silica gel column chromatography producing 7 (exact experimental and analytical details can be found in the online supporting information). General procedure for the quaternary ammonium salt-catalyzed Michael addition of pyridazinones to Michael acceptors. Spirobiindane-based ammonium salt C1 (9.2 mg, 0.01 mmol) and Cs_2_CO_3_ (98 mg, 0.3 mmol) were introduced into a 2 mL vial at room temperature. The N-Boc dihydropyridazinone derivative 7 or 8 (0.1 mmol) was dissolved in toluene (1 mL, 0.1 M) and added to the vial followed by the Michael acceptor 9 (0.3 mmol). The mixture was stirred at room temperature for 18 h (1100 rpm). The crude mixture was filtered through a pad of silica gel using EtOAc as an eluent. Then, the crude mixture was purified by silica gel column chromatography producing the corresponding α,α-difunctionalized pyridazinone derivatives 10 (exact experimental and analytical details can be found in the online supporting information).
4. Conclusions
We herein introduced a straightforward route for the synthesis of various 4-monofunctionalized 4,5-dihydro-2H-pyridazin-3-ones. These heterocycles can then be employed as starting materials to access various 4,4-disubstituted dihydropyridazin-3-ones in an asymmetric fashion upon using chiral ammonium salt phase-transfer catalysts. More specifically, several conjugate addition reactions of these scaffolds to acrylate-based Michael acceptors (1,4-additions) as well as quinone methides (1,6-additions) were carried out with moderate to good enantioselectivities and in reasonable yields, allowing the construction and control of pyridazinone derivatives with an all-carbon stereocenter. Parts of this research have been presented at the 29th International Electronic Conference on Synthetic Organic Chemistry [57].
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ma X. Sun P. Wang J. Huang X. Wu J. Pyridazine and pyridazinone compounds in crops protection: A review Mol. Divers 2024295085510010.1007/s 11030-024-11083-539724455 · doi ↗ · pubmed ↗
- 2Bansal R. Thota S. Pyridazin-3(2H)-ones: The versatile pharmacophore of medicinal significance Med. Chem. Res.2013222539255210.1007/s 00044-012-0261-1 · doi ↗
- 3Dubey S. Bhosle P.S. Pyridazinone: An important element of pharmacophore possessing broad spectrum of activity Med. Chem. Res.2015243579359810.1007/s 00044-015-1398-5 · doi ↗
- 4Shaik B.B. Chandrasekaran B. Obakachi V.A. Mokoena S. Mohite S.B. Kushwaha B. Kushwaha N.S. Karpoormath R. Pyridazinone: A privileged scaffold for synthetic and biomedical applications J. Mol. Struct.20251326140948
- 5Singh J. Sharma D. Bansal R. Pyridazinone: An Attractive Lead for Anti-Inflammatory and Analgesic Drug Discovery Future Med. Chem.201799512710.4155/fmc-2016-019427957866 · doi ↗ · pubmed ↗
- 6Yu S. Yu J.-T. Pan C. Advances in the synthesis of functionalized tetrahydropyridazines from hydrazones Org. Biomol. Chem.2024227753776610.1039/D 4OB 01147 C 39206967 · doi ↗ · pubmed ↗
- 7Nieminen M.S. Fruhwald S. Heunks L.M.A. Suominen P.K. Gordon A.C. Kivikko M. Pollesello P. Levosimendan: Current data, clinical use and future development Heart Lung Vessels 2013522724524364017 PMC 3868185 · pubmed ↗
- 8Ünlüa S. Banoglua E. Küpelib E. Yeşilada E. Şahina M.F. Ökçelika B. Investigations of New Pyridazinone Derivatives for the Synthesis of Potent Analgesic and Anti-Inflammatory Compounds with Cyclooxygenase Inhibitory Activity Arch. Pharm. Pharm. Med. Chem.200333640641210.1002/ardp.20030077814528488 · doi ↗ · pubmed ↗