Novel SIM1 Variants Expanding the Spectrum of SIM1-Related Obesity
Idris Mohammed, Wesam S. Ahmed, Tara Al-Barazenji, Hajar Dauleh, Donald R. Love, Khalid Hussain

TL;DR
This study identifies new SIM1 gene variants linked to severe childhood obesity, expanding the genetic understanding of monogenic obesity and highlighting the gene's role in obesity-related disorders.
Contribution
The study reports novel SIM1 variants, including frameshift and missense mutations, and a recurrent synonymous variant with potential splicing effects, expanding the known genetic spectrum of SIM1-related obesity.
Findings
Five rare SIM1 variants were identified in eleven patients with severe early-onset obesity.
Structural modeling suggests that missense variants likely disrupt critical protein–protein interactions.
A recurrent synonymous variant was found in five unrelated patients and may affect splicing.
Abstract
Monogenic forms of severe early-onset obesity often involve genetic disruptions in the hypothalamic leptin-melanocortin pathway. Pathogenic variants in the SIM1 gene, a key transcription factor required for the development of the paraventricular nucleus, are a known cause of Prader–Willi-like syndrome, characterized by hyperphagia, severe obesity, and developmental delay. We performed targeted next-generation sequencing of 52 obesity-associated genes on a cohort of pediatric patients with severe early-onset obesity. Identified variants were analyzed for population frequency and predicted pathogenicity using in silico tools. The structural impact of the novel missense variants was assessed using protein domain modeling with AlphaFold3. We identified five rare SIM1 variants in eleven patients. Four were heterozygous nonsynonymous variants: one frameshift in the bHLH domain (p.Ser18Ter),…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
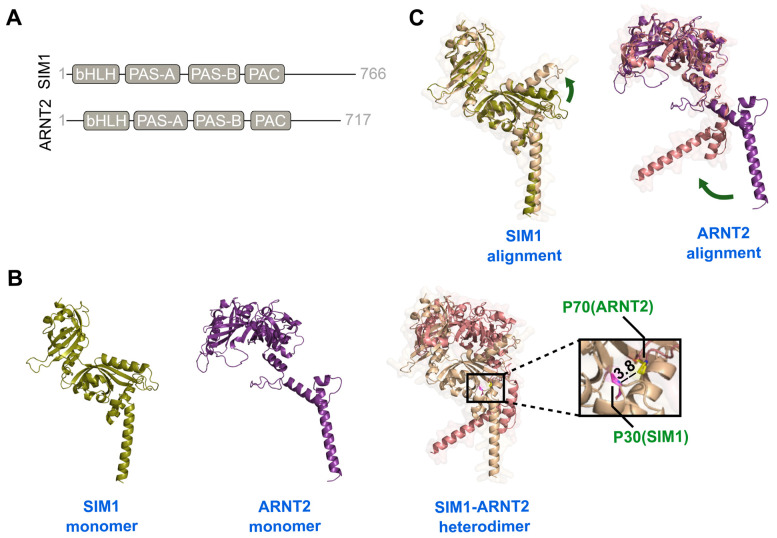 Figure 1
Figure 1- —Qatar National Research Fund
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Syndromes and Imprinting · Regulation of Appetite and Obesity · Wnt/β-catenin signaling in development and cancer
1. Introduction
Obesity is a worldwide epidemic with rates nearly tripled over the last three decades among adults and increased almost five times among children and adolescents [1]. Although the primary cause of obesity is complex, an interplay between environmental and polygenic factors, there is considerable evidence of a monogenic contribution [2]. The vast majority of monogenic forms of obesity are due to genetic defects in the hypothalamic leptin-melanocortin signalling pathway, a pathway crucial in regulating appetite, energy regulation, and body weight [3]. Pathogenic variants in monogenic obesity disrupt a single gene and lead to severe early-onset obesity, accompanied by hyperphagia and some endocrine disorders [4].
One of the monogenic obesity genes is the single-minded homolog one gene (SIM1), which encodes a transcription factor located on the long arm of chromosome 6 (6q16.3) and required for neurogenesis, particularly in the development and function of the paraventricular nucleus (PVN) [5]. The paraventricular nucleus is a crucial component of the central nervous system (CNS), playing a vital role in maintaining energy balance [6]. Loss-of-function and haploinsufficiency of the SIM1 gene lead to a Prader–Willi-like phenotype (PWL). Patients with PWL syndrome carry deletions in the long arm of chromosome 6 (6q16), which contains approximately ten genes, including the SIM1 gene [7,8]. Recently, several studies have linked the PWL syndrome specifically to SIM1 haploinsufficiency. Some of the common phenotypes associated with PWL syndrome include severe early-onset obesity, hyperphagia, and developmental delay [5,9]. A study by Yang et al. demonstrated that mice with haploinsufficiency of SIM1 exhibited abnormal hypothalamic development, whereas overexpression of Sim1 led to reduced food intake [10]. Mice homozygous for Sim1 deficiency die prenatally, whereas heterozygosity and haploinsufficiency lead to the development of early-onset obesity, hyperphagia, hyperinsulinemia, and increased linear growth [7]. Haploinsufficiency and loss-of-function mutations in SIM1 in humans have also been shown to lead to severe early-onset obesity, hyperphagia, and developmental delay [8,11,12].
SIM1 heterodimerizes with aryl hydrocarbon receptor nuclear translocator 2 (ARNT2), which is essential for SIM1 to function in the development of the paraventricular nucleus [13]. The SIM1-ARNT2 heterodimer is crucial for proper hypothalamic development, particularly in regulating neuroendocrine and metabolic functions [14]. When either SIM1or ARNT2 is absent, the neuronal precursors develop normally, but they cannot assemble into the proper neuroendocrine structures, specifically the paraventricular and supraoptic nucleus (SON) in the anterior hypothalamus. This leads to failed neuronal organisation despite normal precursor development [15]. Hence, loss of function or haploinsufficiency of the SIM1 protein results in a reduction in the MC4R neuronal populations in the paraventricular nuclei, contributing to hyperphagia and obesity, similar to the MC4R deficiency [7,16].
To date, approximately 30 patients have been reported to have SIM1 variants associated with severe obesity (Table 1). In this study, we describe eleven patients with early-onset obesity who underwent genetic testing and were found to carry both nonsynonymous and synonymous variants in the SIM1 gene.
2. Methodology
This study included 246 probands with severe early-onset obesity (BMI percentile > 95th). One hundred thirty-nine probands were males (57%), and 107 (43%) were females. The inclusion criteria were an age at obesity onset before 10 years old. Two-thirds of the probands (n = 163, 68%) were under five years old, and the mean age of the probands was 4.3 years. The institutional research board (IRB) approved the study (IRB reference number 1702007592, approval date: 2 June 2023), and signed assent, consent forms, and parental permission were obtained. Peripheral blood was collected, and genomic DNA was extracted following the manufacturer’s protocol (#51104, Qiagen, Hilden, Germany). Genomic DNA concentrations and purities were assessed using a Nanodrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). For next-generation sequencing, exonic regions of all genes of interest were captured using an optimised set of DNA hybridisation probes. The captured DNA was sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA). We targeted regions, including all exons and flanking regions, of 52 genes associated with obesity—genes regulating energy balance in the leptin-melanocortin pathway, genes involved in the differentiation and proliferation of hypothalamic nuclei, genes linked to monogenic ciliopathy diseases, and genes essential for adipocyte differentiation. The sequencing of these cases was part of a cohort that underwent panel sequencing, specifically a comprehensive monogenic obesity panel by a company (Prevention Genetics, Marshfield, WI, USA). According to the company’s report, the average NGS coverage was 538× and the base coverage fraction was 100.0%. The minimum NGS coverage was ≥20× for all exons and ±10 bp in the flanking regions. A summary of all 52 genes included in the targeted gene panel, along with the analysis method, is provided in our previously published research [24].
To assess the structural impact of pathogenic SIM1 missense variants, the protein sequences of SIM1 (UniProt entry: P81133) and ARNT2 (UniProt entry: Q9HBZ2) were obtained [25], and the structures of their bHLH, PAS, and PAC domains were predicted using AlphaFold3 [26]. For SIM1, residues 1–350 were modeled; for ARNT2, residues 60–455 were modeled. Predictions were performed in both monomeric and heterodimeric states using the same seed value for all runs. The resulting structures were visualized and analyzed in PyMOL (Molecular Graphics System, Version 2.0.0, Schrödinger, LLC, New York, NY, USA; https://www.pymol.org).
3. Results
In this study, we identified five rare variants in the SIM1 gene (NM_005068.3) among severely obese children, most of whom were under the age of five years. Four of these variants were nonsynonymous: c. 52_53delAG, p.(Ser18Ter) (Chr6:100,911,293-100,911,294delTC), c.88C>G, p.(Pro30Ala) (Chr6:100911257G/C), c.1988C>T, p.(Ser663Leu) (Chr6:100838550G/A), and c.428_434del, p.(His143Ter) (Chr6.100897498-1008974504delTAGTTGG), each found in unrelated patients. At the same time, we identified one synonymous variant, c.1173G>A, p.(Ser391Ser) (Chr6:100841760-G/A), that was detected in five patients. All the above-mentioned variants are based on GRCh37/hg19 genomic coordinates.
CASES
Case1: c. 52_53delAG, p.(Ser18Ter)
A 13-year-old boy weighing 112.8 kg (BMI 35.7 kg/m^2^, z-score of 3.31), placing him in the 99.5th BMI percentile for his age and sex. The patient presented with severe obesity characterised by both peripheral and central adiposity and had prediabetes. His liver ultrasound revealed nonalcoholic steatohepatitis, persistent hepatomegaly, and diffuse fatty infiltration of the liver. He exhibited elevated liver enzymes (ALT, AST, and GGT). The patient also showed acanthosis nigricans on the neck and beneath the arms. Genetic testing identified a novel frameshift deletion in the SIM1 gene, c. 52_53del, p.(Ser18Ter). This variant is located within a highly conserved domain, the basic helix-loop-helix (bHLH) domain of SIM1, which plays a crucial role in hypothalamic development, particularly in the paraventricular nucleus, which regulates appetite and metabolism.
Case 2: c.88c>g, p.(Pro30Ala)
A case involving a 15-year-old boy with a birth weight of 3.3 kg who started gaining weight at age 5. His current weight is 159 kg (BMI 47.3 kg/m^2^, Z-score 4.11), placing him at the 100th percentile for his age and sex. Liver ultrasound revealed increased echogenicity, indicating diffuse fatty infiltration of the liver. The patient has acanthosis nigricans on his neck. Laboratory tests showed elevated ALT levels. His genetic analysis identified a novel heterozygous nonsynonymous variant, c.88c>g, p.(Pro30Ala), in the SIM1 gene, located within the conserved basic helix-loop-helix (bHLH) domain (Table 2A). This variant is predicted to be deleterious by several in silico tools, SIFT (https://sift.bii.a-star.edu.sg/ (accessed on 28 September 2025)), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/ (accessed on 28 September 2025)), and MutationTaster (https://www.mutationtaster.org/), and was not found in the gnomAD (https://gnomad.broadinstitute.org/) or TOPMed population databases (https://bravo.sph.umich.edu/).
Case 3. c.428_434del, p.(His143Ter18)
A 13-year-old child who started gaining weight at age two currently weighs 108.6 kg with a BMI of 48 kg/m^2^. The patient presented with severe obesity, hepatomegaly, hepatic steatosis, pre-diabetes, and dysmorphic features such as hydrocephalus, an elongated face, hypertelorism, and brachydactyly. Signs of insulin resistance and obstructive sleep apnea (OSA) were observed, along with severe acanthosis nigricans on the back of the neck. Genetic analysis revealed a pathogenic novel frameshift deletion, c.428_434del, p.(His143Ter18). The deletion of seven nucleotides occurs within a highly conserved domain among species, the PER-ARNT-SIM (PAS) domain, resulting in a truncated protein. This results in the loss of the PAS and transactivation motifs of the SIM1 protein.
Cases 4, 5 and 6 c.1988C>T, p.(Ser663Leu)
We detected a heterozygous missense variant, c.1988C>T, p.(Ser663Leu), in the SIM1 gene. The variant has a minor allele frequency (MAF) of 0.000003977 in gnomAD. In silico prediction tools predicted conflicting pathogenicity interpretations, with SIFT and MutationTaster predicting it is deleterious, whereas PolyPhen2 predicts it is likely benign. The variant lies within a conserved domain of the SIM1 transactivation domain (Table 2B).
Case 4:
This 17-year-old adolescent was born at term with a birth weight of 3.5 kg and started gaining weight around the age of 10 years. Currently, the adolescent weighs 99.6 kg (BMI of 35.4 kg/m^2^). The patient presented with mild acanthosis nigricans around the neck and elevated ALT levels. The patient has a strong family history of obesity, with the father, older brother, and sister being obese.
Case 5:
A 5-year-old boy born at term with a birth weight of 3.5 kg. He started gaining weight in infancy. His current weight is 40.75 kg (BMI of 28.7 kg/m^2^, corresponding to a z-score SDS of 3.48 at the 99th percentile). The patient had marked hyperphagia and vomited after overeating. He presented with prediabetes (HbA1c of 5.8%) and elevated insulin and C-peptide levels, 398 pmol/L and 1475 pmol/L, respectively. He has a strong family history of obesity; his 12-year-old sister weighs 90 kg.
Case 6:
A 15-year-old adolescent began a significant weight gain trajectory around age 7, with a marked acceleration over the past year, resulting in a 30 kg weight gain and a current weight of 102 kg (BMI 32.35 kg/m^2^, Z-score + 2.67). The patient was diagnosed with type 2 diabetes at age 13 (HbA1c 7.3%), which has since progressed to poor glycemic control, with a current HbA1c of 9.6%. Genetic testing identified a heterozygous SIM1 variant, c.988C>T (p.Ser663Leu), which is likely contributory to his severe, early-onset obesity and metabolic complications.
Cases 7, 8, 9, 10, 11. (c.1173G>A, p.Ser391Ser)
We detected a synonymous variant, c.1173G>A, p.(Ser391Ser) (6-100841760-C-T (GRCh37)) in five patients with early-onset obesity. The variant was identified in four unrelated families. Alamut Visual v2.11 predicts that the variant activates a cryptic acceptor site six base pairs downstream of the native acceptor site. The variant is rare, with MAF: 1/232,296 (0.000004305) in gnomAD.
Case 7:
An 11-year-old girl presented with childhood obesity, having begun gaining weight at age one. Her current weight is 71.7 kg (BMI: 32 kg/m^2^). The patient has obesity, hypothyroidism, and vitamin D deficiency. She has been taking Levothyroxine 25 mcg daily since age three for hypothyroidism and cholecalciferol 600 IU for vitamin D deficiency. Her mother is obese and diabetic, having developed gestational diabetes during her pregnancy.
Case 8:
A 10-year-old girl presented with childhood obesity and hyperphagia. She began gaining weight in infancy, and her current weight is 65 kg (BMI: 31.5 kg/m^2^).
Case 9:
A 15-year-old girl presented with severe obesity (136 kg, BMI: 48.8 kg/m^2^, and SDS: 3.2) and type 2 diabetes (HbA1c: 8.3). She had normal growth and development, with rapid weight gain beginning at age 10. Following this rapid weight gain, she developed breathing abnormalities and severe OSA. The patient experienced alveolar hypoventilation during sleep and showed evidence of hypothalamic dysfunction, including corticotropin deficiency. She had a strong family history of obesity, with a sister who developed early-onset obesity and a mother who underwent sleeve surgery for obesity.
Case 10:
A 17-year-old girl with severe obesity and insulin resistance. This patient is the sister of Case 8, mentioned above. Her weight is 103 kg, with a BMI of 37 kg/m^2^ and a BMI SDS of 2.85. She began gaining weight around age five. Her abdominal ultrasound showed a mild, diffuse increase in liver echogenicity, consistent with mild fatty infiltration. The pancreas appeared echogenic but otherwise normal, likely due to pancreatic lipomatosis (fatty infiltration). Her liver function tests revealed elevated ALT and AST levels at 33 IU/L and 34 IU/L, respectively.
Case 11:
A 14-year-old girl presented with early-onset obesity, subclinical hypothyroidism, snoring, acanthosis nigricans, and prediabetes (HbA1c 5.9%). The patient began gaining weight before age five. Her current weight is 106 kg (BMI 35 kg/m^2^). An abdominal ultrasound showed a mildly enlarged liver with hepatic steatosis. She has a strong family history of obesity, with her mother and three paternal uncles undergoing bariatric surgery for severe obesity. The patient’s demographics, genetic variants, in silico predictions, and ACMG classifications for all cases identified in this cohort are summarized in Table 3.
Structural Predictions and Domain Organization of the Missense Variants
AlphaFold 3 modelling of SIM1 (residues 1–350) and ARNT2 (residues 60–455) reveals that both proteins possess the canonical bHLH–PAS architecture, comprising bHLH, PAS-A, PAS-B, and PAC domains. The N-terminal region of ARNT2 (residues 1–59) and the C-terminal segments of both proteins are predicted to be intrinsically disordered (Figure 1A). Structural predictions were generated for both monomeric and heterodimeric states of SIM1 and ARNT2. The resulting SIM1–ARNT2 heterodimer model exhibits extensive interdomain contacts across the bHLH and PAS regions. Notably, SIM1 residue P30 is located within a loop of the bHLH domain at the dimer interface, approximately 3.8 Å from the bHLH/PAS-A loop of ARNT2 (Figure 1B). This proximity suggests that P30 may contribute to the amino acid substitution and could alter the locus responsible for heterodimer formation. The substitution with alanine (P30A) could weaken this interface, impairing dimer formation. Conversely, residue Ser663 of SIM1 resides within the intrinsically disordered C-terminal region, precluding reliable structural inference. Nonetheless, since transcriptional regulation occurs through this disordered tail, the Ser663Leu variant might influence transcriptional activity rather than dimer formation.
Furthermore, comparison of monomeric and dimeric models showed conformational changes in both proteins that appear to facilitate dimerization. In SIM1, this involved movement of the first α-helix of PAS-A, while in ARNT2, a shift in the positioning of the bHLH domain was observed (Figure 1C). This indicates that direct dimer predictions, which we applied here, may more closely capture physiologically relevant conformations than docking approaches based solely on monomer structures.
4. Discussion
In this study, we identified five rare variants in the SIM1 gene among eleven pediatric patients with severe early-onset obesity. Four were nonsynonymous variants: p.(Ser18Ter), p.(Pro30Ala), p.(Ser663Leu), and p.(His143Ter). One was a synonymous variant, c.1173G>A, p.(Ser391Ser), detected in five patients. These findings reinforce the critical role of SIM1 in hypothalamic regulation of appetite and energy homeostasis, further supporting its involvement in monogenic obesity.
The SIM1 gene encodes a transcription factor essential for the development of the PVN of the hypothalamus, a key region in regulating satiety and energy expenditure [7,15]. Our study identified frameshift deletions, p.(Ser18Ter) and p.(His143Ter), that likely cause haploinsufficiency, consistent with previous reports linking SIM1 loss-of-function mutations to severe obesity and hyperphagia [8,9,11]. Notably, p.(His143Ter) truncates the PAS domain, which is crucial for protein–protein interactions and transcriptional activity [15]. Similarly, p.(Ser18Ter) is predicted to produce a null allele due to a premature termination codon, which is expected to cause a complete loss of the protein either through nonsense-mediated decay (NMD) or the generation of a non-functional, severely truncated protein, thereby abolishing its function. A non-functional SIM1 protein impairs DNA binding and dimerization with ARNT2, a partner necessary for proper hypothalamic development [13,15]. These findings are consistent with those from murine models, where Sim1 haploinsufficiency causes hyperphagia, obesity, and a reduction in PVN neuronal populations [7,16].
Additionally, we report a missense variant, p.(Ser663Leu), in the transactivation domain in two unrelated patients with severe obesity and strong family histories of the condition. Although in silico predictions for this variant are conflicting (deleterious according to SIFT/MutationTaster but benign according to PolyPhen-2), its absence in population databases (gnomAD, TOPMed) and co-segregation with obesity in affected siblings suggest a potential pathogenic role. Previous studies have demonstrated that SIM1 missense variants can impair transcriptional activity, contributing to the development of obesity.
Interestingly, we identified a synonymous variant, c.1173G>A, p.(Ser391Ser), in five patients. Although synonymous variants are often presumed benign, in silico splicing prediction tools yield conflicting results regarding their potential effect on mRNA processing. For instance, Alamut Visual v2.11 predicted the creation of a cryptic acceptor site six base pairs downstream of the native splice site, suggesting possible aberrant splicing. However, other splice prediction algorithms, such as SpliceAI (low predicted probability of causing a major splice defect) and MaxEntScan (no strong signal for creating or destroying a splice site at this position), did not support a strong splice-altering effect. These discrepancies underscore the importance of further functional validation to determine whether this variant influences SIM1 splicing in vivo. Similar observations of synonymous variants influencing splice sites have been reported previously, and these variants may influence splicing, protein function, and disease susceptibility [27,28,29,30]. These studies reinforce the notion that synonymous variants should not be ignored as neutral. Instead, integrative approaches that combine bioinformatics predictions, RNA-seq validation, and functional assays are essential for assessing their potential pathogenicity.
Our findings support previous evidence that SIM1 disruptions lead to PWL phenotypes, including hyperphagia, early-onset obesity, and developmental delay [5,7,11]. The phenotypic overlap between SIM1 and MC4R deficiency, another key component of the leptin-melanocortin pathway, further underscores the importance of SIM1 in appetite regulation [31,32]. Notably, some of our patients exhibited endocrine abnormalities, including hypothyroidism and hypothalamic dysfunction, consistent with earlier reports of patients with SIM1 deficiency [31,33].
To assess the structural impact of pathogenic SIM1 missense variants, we analyzed both the monomeric and heterodimeric forms of SIM1. SIM1 forms heterodimers with ARNT or ARNT2 [34]. However, because ARNT2 is preferentially expressed in the hypothalamus and the variants under investigation alter hypothalamic function, we focused on its interaction with ARNT2. This dimerization is a prerequisite for DNA binding and transcriptional activity [13]. Since experimental structures of full-length SIM1 and ARNT2 are unavailable, we used AlphaFold3 to predict their domain structures. The AlphaFold3 models provide a structural perspective for interpreting the potential effects of SIM1 missense variants in the context of its heterodimerization with ARNT2. Both proteins belong to the bHLH–PAS (basic helix–loop–helix/Per-Arnt-Sim) family of transcription factors and function as heterodimeric partners in neurodevelopment and hypothalamic regulation. They share typical domain architecture, comprising bHLH, PAS-A, PAS-B, and PAC [8,35]. The N-terminal region (first 59 residues) of ARNT2, along with the C-terminal regions of both SIM1 and ARNT2, are predicted to be intrinsically disordered.
Heterodimerization and DNA binding are mediated by the bHLH and PAS domains, while the PAC motif, which stabilizes the PAS domain fold, links the PAS tandem to the intrinsically disordered C-terminal transcriptional regulatory region [8,36]. To focus on the structured elements, we modeled the bHLH, PAS, and PAC domains of both proteins, excluding intrinsically disordered segments that do not contribute to heterodimerization and remain challenging to predict accurately in AlphaFold. This corresponded to residues 1–350 in SIM1 and 60–455 in ARNT2.
The identification of P30 at the dimer interface highlights a residue likely to play a stabilizing role in the bHLH-mediated heterodimer. Substituting this proline with alanine (P30A) could disrupt optimal packing at the interface, thereby weakening dimerization and diminishing subsequent transcriptional regulation. In fact, several studies have identified obesity-associated missense variants in the SIM1 bHLH (Thr46Arg, Glu62Lys) [8,36], and PAS (Ser71Arg, Ile128Thr, Gln152Glu, Arg171His, Leu238Arg) domains, all of which are involved in forming the heterodimer with ARNT2 [9].
In contrast, Ser663 lies within the intrinsically disordered C-terminal region of SIM1, which functions as a transcriptional regulatory region. Although AlphaFold predictions provide limited insight into disordered regions, previous functional studies have shown that C-terminal SIM1 variants frequently disrupt transcriptional regulation rather than DNA binding or dimerization. For example, Arg550His, Pro692Leu, Asp707His, Gly715Val, and Asp740His have all been associated with obesity and varying effects on transcriptional activity [8,9,36,37,38]. These mutations cluster within the transactivation domain, underscoring the functional sensitivity of this region. It is therefore plausible that Ser663Leu, although structurally unresolvable, could similarly alter the recruitment of transcriptional cofactors or interfere with post-translational modifications required for SIM1-mediated gene regulation.
Overall, the modeling results indicate distinct mechanistic effects for the two variants analyzed. While the P30A mutation may compromise heterodimer formation, the Ser663Leu mutation likely affects transcriptional regulation by disrupting the C-terminal regulatory functions. These interpretations align with known pathogenic mechanisms of SIM1 variants implicated in hypothalamic dysfunction and monogenic obesity. Further validation through co-immunoprecipitation, BRET/FRET interaction assays, and luciferase reporter experiments would be valuable for confirming the structural predictions and elucidating the specific molecular consequences of these variants.
Limitations
Our study has limitations; functional assays are needed to confirm the pathogenicity of variants. Trio analysis was not available to investigate co-segregation of the variants. Such analyses would strengthen the evidence for causality, especially for variants with conflicting in silico predictions. The influence of polygenic background, epigenetic factors, or environmental influences on the obesity phenotype among carriers was not examined. These factors might contribute to the variable expressivity and penetrance of SIM1-related obesity.
5. Conclusions
This study broadens the range of SIM1 variants linked to monogenic obesity, underscoring their role in hypothalamic development and energy regulation. Functional studies are needed to better understand how these variants, particularly the synonymous change, affect SIM1 function. Early genetic testing for SIM1 mutations in children with severe obesity and hyperphagia could facilitate personalized interventions, leading to improved long-term metabolic outcomes.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1World Health Organization 2021 Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight(accessed on 2 November 2025)
- 2Hinney A. Vogel C.I.G. Hebebrand J. From monogenic to polygenic obesity: Recent advances Eur. Child Adolesc. Psychiatry 20101929731010.1007/s 00787-010-0096-620127379 PMC 2839509 · doi ↗ · pubmed ↗
- 3Oswal A. Yeo G.S.H. The leptin melanocortin pathway and the control of body weight: Lessons from human and murine genetics Obes. Rev.2007829330610.1111/j.1467-789X.2007.00378.x 17578380 · doi ↗ · pubmed ↗
- 4Farooqi I.S. O’Rahilly S. Monogenic Obesity in Humans Annu. Rev. Med.20055644345810.1146/annurev.med.56.062904.14492415660521 · doi ↗ · pubmed ↗
- 5Faivre L. Cormier-Daire V. Lapierre J.M. Colleaux L. Jacquemont S. Geneviéve D. Saunier P. Munnich A. Turleau C. Romana S. Deletion of the SIM 1 gene (6q 16.2) in a patient with a Prader-Willi-like phenotype J. Med. Genet.20023959459610.1136/jmg.39.8.59412161602 PMC 1735217 · doi ↗ · pubmed ↗
- 6Kim M.S. Rossi M. Abusnana S. Sunter D. Morgan D.G. Small C.J. Edwards C.M. Heath M.M. Stanley S.A. Seal L.J. Hypothalamic localization of the feeding effect of agouti-related peptide and alpha-melanocyte-stimulating hormone Diabetes 20004917718210.2337/diabetes.49.2.17710868932 · doi ↗ · pubmed ↗
- 7Michaud J.L. Boucher F. Melnyk A. Gauthier F. Goshu E. Lévy E. Mitchell G.A. Himms-Hagen J. Fan C.M. Sim 1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus Hum. Mol. Genet.2001101465147310.1093/hmg/10.14.146511448938 · doi ↗ · pubmed ↗
- 8Bonnefond A. Raimondo A. Stutzmann F. Ghoussaini M. Ramachandrappa S. Bersten D.C. Durand E. Vatin V. Balkau B. Lantieri O. Loss-of-function mutations in SIM 1 contribute to obesity and Prader-Willi–like features J. Clin. Investig.20131233037304110.1172/JCI 6803523778136 PMC 3696559 · doi ↗ · pubmed ↗