The Evolving Role of Second- and Third-Generation Tyrosine Kinase Inhibitors in Gastrointestinal Malignancies: Advances in Targeted Therapy with Sunitinib, Regorafenib, and Avapritinib
Piotr Kawczak, Tomasz Bączek

TL;DR
This paper reviews how newer tyrosine kinase inhibitors like sunitinib, regorafenib, and avapritinib are improving treatment for gastrointestinal stromal tumors by targeting specific mutations and extending survival.
Contribution
The paper highlights the role of second- and third-generation tyrosine kinase inhibitors in mutation-specific therapy for gastrointestinal stromal tumors.
Findings
Sunitinib, regorafenib, and avapritinib offer improved disease control for imatinib-resistant gastrointestinal stromal tumors.
Avapritinib specifically targets PDGFRA D842V and KIT exon 17 mutations, addressing a previously untreatable subgroup.
Therapeutic sequencing and biomarker-driven decisions are transforming personalized treatment for advanced gastrointestinal stromal tumors.
Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. While imatinib revolutionized first-line therapy, resistance and specific mutation profiles necessitate subsequent generations of tyrosine kinase inhibitors (TKIs). Sunitinib, regorafenib, and avapritinib represent second-line, third-line, and mutation-specific therapies, respectively, offering improved precision and disease control. This review summarizes clinical trial evidence, real-world data, and translational studies evaluating the efficacy, safety, and mechanistic basis of second- and third-generation TKIs in GIST. Emphasis is placed on therapeutic sequencing, resistance mechanisms, and molecularly guided treatment selection. Sunitinib, a multitargeted TKI inhibiting KIT, PDGFR, and VEGFR, provides effective disease control in imatinib-resistant or intolerant patients.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
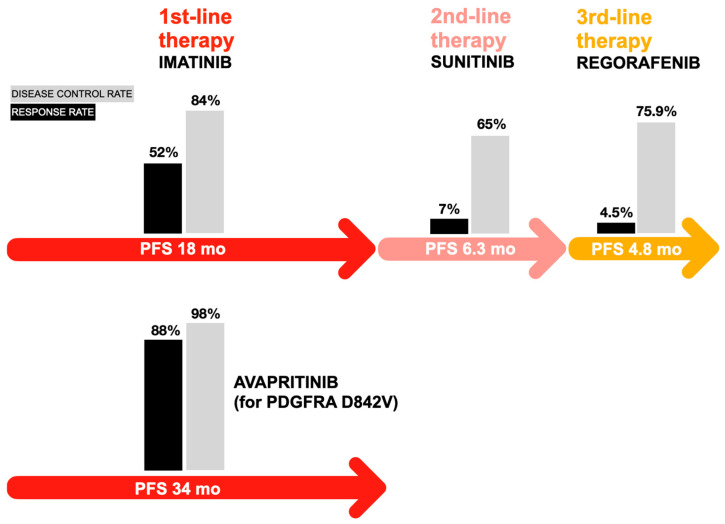 Figure 1
Figure 1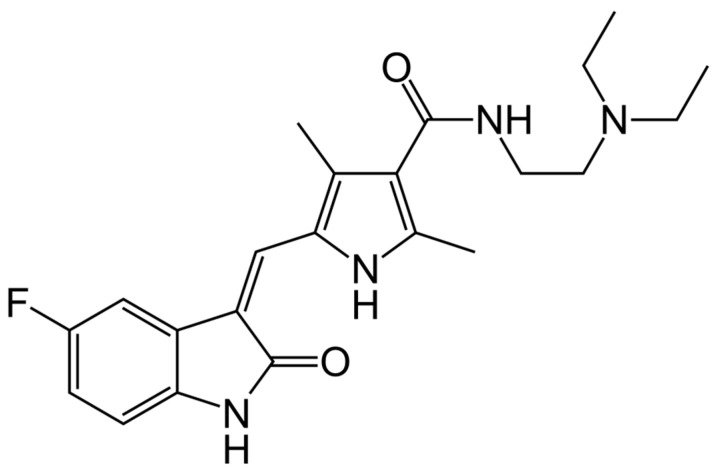 Figure 2
Figure 2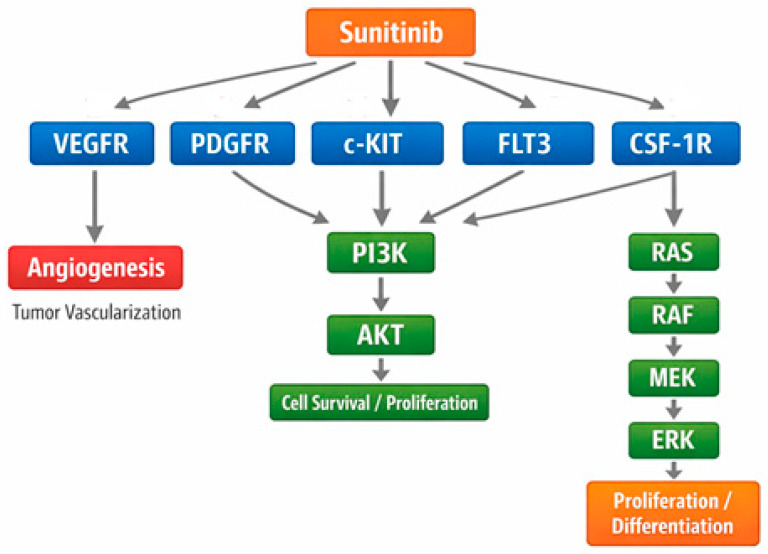 Figure 3
Figure 3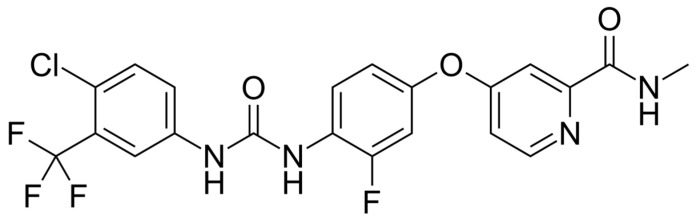 Figure 4
Figure 4 Figure 5
Figure 5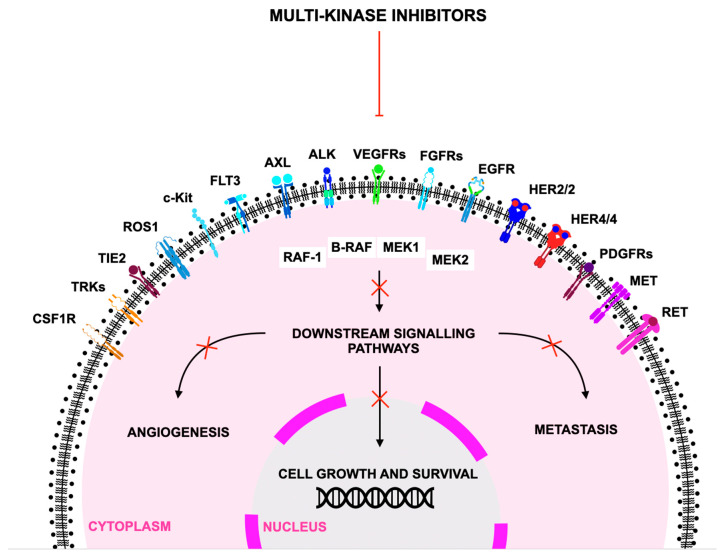 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGastrointestinal Tumor Research and Treatment · Sarcoma Diagnosis and Treatment · Gastrointestinal Bleeding Diagnosis and Treatment
1. Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract and originate from the interstitial cells of Cajal, with most driven by activating mutations in KIT or PDGFRA [1,2,3,4]. As the prototypical and most extensively characterized gastrointestinal mesenchymal tumor, GIST occupies a distinct position among gastrointestinal malignancies—most of which are epithelial cancers such as gastric and colorectal adenocarcinomas or neuroendocrine tumors, lymphomas, and other soft-tissue sarcomas [5,6,7].
The majority of GISTs are driven by sporadic somatic KIT or PDGFRA mutations rather than inflammation-associated carcinogenesis; rare hereditary and syndromic forms of GIST (e.g., familial KIT/PDGFRA mutations, Carney–Stratakis syndrome, and neurofibromatosis type 1–associated GIST) do exist [8,9,10]. The identification of KIT and PDGFRA mutations fundamentally reshaped the biological classification and clinical management of GIST, distinguishing it from other sarcomas and enabling molecularly targeted therapeutic development [11,12,13]. This foundation positioned GIST as an early model of precision oncology within solid tumors and underscored the importance of genotype-directed therapy in gastrointestinal cancer care.
Tyrosine kinase inhibitors (TKIs) have transformed the treatment of GIST and other gastrointestinal malignancies, including metastatic colorectal cancer (mCRC), through selective targeting of oncogenic kinases. These agents are commonly categorized into first-, second-, and third-generation inhibitors based on progressive refinements in potency, selectivity, and activity against resistance mutations [14,15,16]. Multitargeted TKIs such as sunitinib, regorafenib, and sorafenib—originally developed and validated in renal cell carcinoma, hepatocellular carcinoma, and mCRC—exert antitumor effects primarily via inhibition of VEGFR, PDGFR, RAF, and KIT-related signaling pathways. Because these pathways are central to GIST pathogenesis and the emergence of imatinib resistance, insights from angiogenesis-driven malignancies have informed both the mechanistic rationale and clinical sequencing of TKIs in GIST [7,15,17]
First-generation TKIs such as imatinib, which inhibit KIT, PDGFR, and BCR-ABL, revolutionized first-line therapy for GIST and chronic myeloid leukemia (CML) [18,19,20]. Imatinib achieved unprecedented disease control in advanced GIST, which had historically been refractory to cytotoxic chemotherapy and radiotherapy [21,22,23]. However, primary resistance—particularly in tumors harboring the PDGFRA D842V mutation—and the near-universal emergence of secondary resistance mutations during treatment ultimately limited long-term efficacy [24,25,26]. These resistance dynamics parallel clonal evolution observed across advanced gastrointestinal malignancies, where selective pressure drives molecular adaptation and treatment failure.
Second-generation TKIs were developed to counter resistance and broaden inhibitory profiles. Multitargeted agents such as sunitinib [27], regorafenib [28], sorafenib [29], pazopanib [30], cabozantinib [31], and axitinib [32] inhibit a range of kinases, including VEGFR, PDGFR, KIT, and RET, producing both antitumor and antiangiogenic effects. These agents have demonstrated substantial benefit in imatinib-resistant GIST and in malignancies such as refractory mCRC, renal cell carcinoma (RCC), and hepatocellular carcinoma (HCC) [27,28,29,30,31,32]. Sunitinib became standard second-line therapy after improving outcomes in patients progressing on imatinib [33,34,35], with differential efficacy across molecular subsets—including enhanced activity in tumors with KIT exon 9 mutations and specific secondary mutations [36,37]—highlighting the heterogeneity of GIST evolution and reinforcing the need for molecular profiling.
Regorafenib, a broad-spectrum inhibitor of angiogenic and oncogenic kinases including TIE2, RAF, RET, and FGFR, further extended therapeutic options in the third-line therapy setting. The phase III GRID trial established its efficacy in delaying disease progression following imatinib and sunitinib failure [38,39,40], and subsequent real-world studies confirmed its clinical utility and manageable toxicity across heterogeneous patient populations [41,42]. In this context, the term “diverse patient groups” encompasses heterogeneity in age, KIT/PDGFRA mutational profiles, extent of prior TKI exposure, and geographic background, as reflected in both pivotal clinical trials and post-approval cohorts [37,38,43]. Within the biological landscape of late-line GIST—characterized by the accumulation of multiple co-existing secondary resistance mutations—regorafenib’s activity across angiogenic and oncogenic signaling pathways has demonstrated consistent benefit despite increasing molecular complexity.
The advent of third-generation TKIs marked a shift toward mutation-specific precision therapy. Molecular analyses of imatinib-resistant GIST have demonstrated that secondary KIT mutations arise predominantly within the ATP-binding pocket (exons 13 and 14) or the activation loop (exons 17 and 18), with these distinct alterations differentially affecting sensitivity to subsequent TKIs [44,45].
Against this backdrop, avapritinib emerged as a highly potent type I inhibitor engineered to selectively target activation-loop mutations, including PDGFRA D842V and KIT exon 17 variants, providing a therapeutic breakthrough for molecular subsets previously resistant to all available TKIs [46,47,48,49]. Results from the NAVIGATOR and VOYAGER trials demonstrated exceptional efficacy in PDGFRA D842V-mutant GIST [50,51], leading to global regulatory approval.
As GISTs progress through successive lines of therapy, increasing molecular heterogeneity driven by the accumulation of secondary KIT mutations becomes a defining feature of late-line disease [52,53]. While earlier multitargeted TKIs such as sunitinib and regorafenib provide partial coverage of resistance mutations, their efficacy is often limited by differential sensitivity across ATP-binding pocket (exons 13/14) and activation loop (exons 17/18) alterations [52,53]. This therapeutic challenge prompted the development of next-generation inhibitors designed to more comprehensively suppress a broad spectrum of KIT and PDGFRA mutants. Ripretinib, a switch-control kinase inhibitor that stabilizes KIT and PDGFRA in an inactive conformation, represents a distinct mechanistic advance and has expanded treatment options for heavily pretreated patients [54,55,56]. The pivotal phase III INVICTUS trial demonstrated that ripretinib significantly improved progression-free survival compared with placebo, establishing its role as a standard late-line therapy in advanced GIST [55]. Collectively, these advances underscore the increasing importance of comprehensive molecular diagnostics in guiding mutation-tailored therapy, informing optimal treatment sequencing, and adapting therapeutic strategies as tumor biology evolves under selective pressure [57,58,59]. Table 1 summarizes first-, second-, and third-generation TKIs utilized in gastrointestinal malignancies and associated cancers, while Figure 1 depicts treatment strategies for advanced gastrointestinal stromal tumors employing FDA-approved tyrosine kinase inhibitors, including imatinib, sunitinib, regorafenib, and avapritinib.
Ongoing research into resistance mechanisms, kinase structural dynamics, and microenvironmental influences continues to inform next-generation inhibitor development and combination strategies [61,62].
The development of second- and third-generation TKIs—including sunitinib, regorafenib, and avapritinib—has markedly advanced the management of advanced GIST, allowing more precise, mutation-driven therapy and extending patient survival compared with earlier treatment eras. As precision oncology continues to evolve, the integration of molecularly targeted agents with emerging therapeutic modalities promises to further optimize care for patients with GIST.
First-generation TKI therapy (imatinib) has been extensively reviewed in the literature and is well established as first-line treatment. In contrast, therapeutic resistance to imatinib and the molecular heterogeneity of GIST have driven the development of second- and third-generation TKIs, which remain an area of rapid progress and clinical interest. Therefore, our review intentionally focuses on these later-generation agents to highlight advances in overcoming resistance mechanisms and improving precision treatment.
This review synthesizes current clinical, translational, and molecular evidence supporting precision TKI use in GIST, integrating data from pivotal trials, real-world studies, and mechanistic analyses. In addition, it highlights emerging challenges, including the management of polyclonal resistance, heterogeneity across KIT and PDGFRA mutations, and the need for dynamic monitoring of tumor evolution. By examining both established and next-generation therapies, this review emphasizes opportunities to optimize genotype-driven treatment strategies and to incorporate molecular insights into routine clinical practice, ultimately aiming to improve patient outcomes in advanced GIST.
This synthesis was informed by a narrative literature review of PubMed and Scopus using the search terms “sunitinib,” “regorafenib,” and “avapritinib” combined with “ targeted therapy” and “gastrointestinal stromal tumors.” Peer-reviewed publications from 2005 to 2025 were selected for relevance, methodological rigor, and contributions to understanding therapeutic efficacy, mechanisms of action, and resistance patterns. Both preclinical and clinical studies were included when mechanistic insights informed therapeutic strategy, enabling a comprehensive evaluation of the roles, benefits, and limitations of sunitinib, regorafenib, and avapritinib in contemporary gastrointestinal oncology.
2. Sunitinib
Sunitinib is an orally bioavailable, small-molecule, multi-targeted tyrosine kinase inhibitor (TKI) designed to block key signaling pathways that regulate tumor cell proliferation, angiogenesis, and metastatic progression [14,63]. It is classified as a second-generation multitarget TKI with activity against a broad panel of receptor tyrosine kinases, including vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2, VEGFR-3), platelet-derived growth factor receptors (PDGFR-α/β), and proto-oncogenic kinases such as KIT, RET, CSF-1R, and FLT3 [64,65,66]. This inhibitory spectrum enables sunitinib to simultaneously modulate angiogenic, stromal, and oncogenic signaling within the tumor microenvironment, disrupting critical processes required for tumor maintenance and dissemination [67,68,69]. Figure 2 shows structural formula of sunitinib.
Mechanistically, sunitinib functions as a competitive ATP-binding site inhibitor, preventing phosphorylation of downstream signaling molecules that govern cell survival (e.g., PI3K–AKT), proliferation (e.g., MAPK), endothelial cell migration, and vascular permeability [70]. By inhibiting VEGFR and PDGFR activity, sunitinib suppresses endothelial cell proliferation, reduces pericyte coverage of nascent vessels, and leads to regression and normalization of tumor vasculature [71]. Inhibition of KIT and FLT3 further impacts hematopoietic and stromal signaling, explaining both the antitumor activity in GIST and the hematologic toxicities observed clinically. This multitarget action distinguishes sunitinib from earlier angiogenesis inhibitors and contributes to a robust antitumor profile across genetically diverse malignancies. Figure 3 illustrates the molecular targets of sunitinib and the downstream signaling pathways it modulates.
Sunitinib’s initial clinical development in the early 2000s focused on metastatic renal cell carcinoma (mRCC) and gastrointestinal stromal tumors (GIST), two tumor types highly dependent on VEGF-, PDGF-, and KIT-mediated pathways [72]. In phase II and III trials in mRCC, sunitinib yielded substantial objective response rates and significantly improved progression-free survival compared with interferon-α, the prior standard of care [27]. These studies were the first to demonstrate that multitargeted angiogenesis inhibition could provide superior outcomes to immunotherapy alone in renal cancer. In GIST populations refractory to imatinib, sunitinib offered clinically meaningful disease stabilization and prolonged time to progression, highlighting its ability to overcome resistance mediated by secondary KIT mutations [33]. These pivotal data supported the drug’s initial regulatory approval in 2006 [73].
Subsequent research expanded sunitinib’s clinical utility to pancreatic neuroendocrine tumors (pNETs), where VEGF-driven angiogenesis is prominent. In a randomized trial, sunitinib significantly prolonged progression-free survival and improved disease control rates, providing a new targeted option for this previously underserved population [74]. In other malignancies—including differentiated thyroid cancer, soft-tissue sarcoma, and pediatric solid tumors—sunitinib demonstrated varying degrees of antitumor activity [75]. Nonetheless, differences in tumor biology, resistance patterns, and toxicity profiles have limited its widespread adoption beyond its primary indications [76]. Combination regimens of sunitinib with immune checkpoint inhibitors or other TKIs have shown biological synergy in preclinical studies but have often been restricted clinically due to overlapping hepatotoxicity, fatigue, and immune-mediated adverse events [77,78]. Comparative trials with next-generation agents suggest that while sunitinib retains strong efficacy, some newer TKIs may offer enhanced target specificity or reduced toxicity burdens [79].
The drug’s adverse-effect profile reflects its broad inhibitory activity. Class-related toxicities include fatigue, hypertension, mucosal inflammation, diarrhea, and dermatologic reactions, many of which derive from VEGFR/PDGFR blockade in normal tissues [80]. Chronic inhibition of thyroidal VEGF signaling is thought to contribute to the high incidence of hypothyroidism in sunitinib-treated patients, which may require thyroid hormone replacement [81]. More severe toxicities, although less frequent, include cardiotoxic events such as decreases in left ventricular ejection fraction—likely related to mitochondrial dysfunction and off-target kinase inhibition—along with hepatotoxicity and thromboembolic complications [82]. VEGF inhibition also impairs wound healing, vascular integrity, and tissue regeneration, creating risks of bleeding or delayed recovery following surgery [83]. These risks have prompted the incorporation of rigorous monitoring guidelines for blood pressure, thyroid function, cardiac status, and hepatic enzymes throughout treatment [84]. Table 2 presents treatment-emergent adverse events (TEAE) and management strategies for sunitinib.
Clinically meaningful associations have been observed between sunitinib toxicity markers and therapeutic outcomes. Treatment-emergent hypertension has been correlated with improved progression-free and overall survival, supporting its potential role as a mechanism-based biomarker of VEGF pathway inhibition [91]. Similarly, the development of hypothyroidism has been proposed as a surrogate marker for adequate systemic kinase inhibition, although prospective evidence remains limited [92]. Efforts to identify pharmacogenomic predictors of response or toxicity—such as polymorphisms in CYP3A5, ABCB1, and VEGF pathway genes—have yielded inconsistent results, suggesting multifactorial determinants of drug metabolism and resistance [93].
Beyond its approved uses, sunitinib occupies an important role in the evolution of multitargeted oncology therapeutics. It is widely employed in preclinical studies examining mechanisms of angiogenesis, tumor–stromal interactions, and acquired resistance pathways [94]. As a well-characterized benchmark TKI, sunitinib remains a reference compound in kinase-selectivity profiling, structural biology studies, and drug-development pipelines exploring next-generation VEGFR/PDGFR inhibitors [95]. Real-world longitudinal studies continue to refine their clinical positioning, providing insights into optimal dosing schedules, strategies for managing chronic toxicity, and biomarkers for patient selection [96]. Thus, sunitinib has not only shaped therapeutic paradigms within renal and gastrointestinal oncology but also contributed substantially to the broader scientific understanding of targeted multi-kinase inhibition. Table 3 illustrates major pivotal clinical trials of sunitinib.
3. Regorafenib
Regorafenib is an orally administered, small-molecule multikinase inhibitor belonging to the class of second-generation angiogenesis and oncogenic signaling inhibitors [15,98]. Structurally derived from sorafenib by the addition of a fluorine atom, regorafenib was rationally designed to provide broader target selectivity and stronger potency across multiple kinase families [99,100]. Its inhibitory spectrum encompasses vascular endothelial growth factor receptors (VEGFR-1, VEGFR-2, VEGFR-3), platelet-derived growth factor receptors (PDGFR-α/β), fibroblast growth factor receptor (FGFR), KIT, RET, TIE2, DDR2, and RAF kinases [101,102]. This multilevel blockade affects both the tumor microenvironment and the cancer cells themselves, including angiogenic, stromal, and oncogenic pathways essential for tumor survival and proliferation [103,104,105]. Figure 4 depicts structural formula of regorafenib.
Mechanistically, regorafenib functions as a competitive ATP-binding inhibitor that simultaneously disrupts angiogenesis, oncogenic signaling, and metastatic processes [106]. Inhibition of VEGFR and TIE2 reduces endothelial cell proliferation, migration, and vessel maturation, thereby suppressing tumor angiogenesis and inducing vascular normalization [107]. Through PDGFR and FGFR blockade, regorafenib interferes with pericyte recruitment and fibroblast activation, weakening tumor stroma and nutrient supply [108]. Additionally, RAF/MEK/ERK pathway inhibition contributes to antiproliferative effects in tumors with MAPK dependence, although this pathway is not the primary driver of its clinical efficacy [28]. Inhibition of RET and KIT supports its therapeutic role in thyroid cancers and gastrointestinal stromal tumors (GIST), particularly in the context of resistance to earlier-generation TKIs [109]. This systems-level inhibition distinguishes regorafenib from earlier anti-angiogenic agents by expanding its activity across multiple malignant phenotypes.
Although regorafenib was initially developed for metastatic colorectal cancer (mCRC), accumulating evidence has firmly established its role in GIST, particularly in the post–imatinib and sunitinib setting. Early clinical development focused on mCRC, where resistance to chemotherapy and anti-VEGF therapies represented a major unmet need [110,111,112]. The pivotal CORRECT phase III trial demonstrated a statistically significant overall survival benefit in heavily pretreated mCRC patients, establishing regorafenib as an effective option in refractory disease [113]. This study highlighted the capacity of broad multikinase inhibition to overcome resistance mechanisms that limit the efficacy of VEGF-specific monoclonal antibodies and conventional chemotherapies [114].
Building on earlier clinical development, regorafenib was approved for GIST following failure of imatinib and sunitinib on the basis of the pivotal phase III GRID trial. In this study, regorafenib significantly improved progression-free survival compared with placebo (median PFS 4.8 vs. 0.9 months; hazard ratio 0.27), leading to its establishment as standard third-line therapy for advanced GIST [38,115]. Even though objective response rates were low, a substantial proportion of patients achieved durable disease control, consistent with the predominantly cytostatic activity of later-line tyrosine kinase inhibitors in GIST. Subsequent analyses of the GRID dataset demonstrated clinical benefit across a broad spectrum of KIT and PDGFRA mutational subtypes, including secondary mutations associated with resistance to earlier-line therapies, reinforcing regorafenib’s role as a broadly active agent in the post–imatinib and post–sunitinib setting [34,116,117]. Collectively, these findings provided definitive evidence supporting regorafenib monotherapy and solidified its position as a key component of sequential KIT-targeted therapy, as reflected in international treatment guidelines from ESMO and NCCN [118,119].
With the introduction of newer agents such as ripretinib and avapritinib, treatment sequencing beyond second line has continued to evolve. Nevertheless, regorafenib remains an integral element of contemporary GIST management owing to its proven efficacy, well-characterized safety profile, and ability to achieve durable disease stabilization in a subset of patients. Ongoing clinical experience and retrospective analyses continue to refine optimal sequencing strategies, particularly in relation to mutational status and prior treatment exposure. Beyond GIST, regorafenib’s clinical utility was further expanded in hepatocellular carcinoma (HCC), where the phase III RESORCE trial confirmed its benefit as second-line therapy after sorafenib, representing the first agent to demonstrate a survival advantage in patients progressing on prior VEGFR-targeted treatment [120].
Outside of the pivotal trials, regorafenib has been investigated across a range of tumor types, generating an expanding body of evidence with particular relevance to GIST resistance mechanisms and therapeutic sequencing. In HCC, real-world studies have corroborated survival benefits observed in randomized trials and emphasized the importance of patient selection, particularly with respect to liver function and performance status [121]. In mCRC, combination strategies incorporating regorafenib with immune checkpoint inhibitors—most notably PD-1 inhibitors—have shown encouraging activity, especially in microsatellite-stable tumors that are typically refractory to immunotherapy alone [122]. In GIST, regorafenib has demonstrated sustained benefits for patients harboring secondary KIT mutations associated with imatinib or sunitinib resistance, with continued research focusing on optimizing sequencing strategies among KIT-targeted TKIs [123]. Exploratory trials in glioblastoma, ovarian cancer, and sarcomas have shown variable results, often limited by toxicity profiles or by intrinsic tumor biology [124]. Nevertheless, regorafenib remains a benchmark multikinase inhibitor in translational oncology research.
The adverse-effect profile of regorafenib reflects its broad kinase selectivity. The most frequently reported toxicities include hand–foot skin reaction (HFSR), hypertension, fatigue, diarrhea, anorexia, and hepatotoxicity [125]. HFSR is particularly characteristic of regorafenib and occurs more frequently than with many other TKIs, likely due to off-target inhibition of kinases involved in keratinocyte function and microvascular repair [126]. Hypertension reflects on-target VEGFR blockade, while diarrhea and mucosal toxicities result from gastrointestinal epithelial injury and stromal VEGF signaling inhibition [127]. Hepatotoxicity—including elevations in transaminases and bilirubin—remains clinically significant and necessitates close monitoring, especially during the first two treatment cycles [128]. Less common but serious adverse events, such as hemorrhage, gastrointestinal perforation, and myocardial ischemia, are clinically relevant, particularly in patients with underlying vascular disease [129]. Table 4 summarizes treatment-emergent adverse events and management strategies for regorafenib.
Clinically meaningful associations between regorafenib-related toxicities and therapeutic outcomes have been reported. Early onset of HFSR has been correlated with improved survival in both mCRC and HCC, suggesting a potential exposure–response relationship indicative of adequate systemic drug levels [136]. Hypertension may similarly function as a mechanism-based biomarker of effective angiogenesis inhibition, consistent with observations across other VEGFR-targeted TKIs [137]. Pharmacogenomic investigations examining UGT1A9, ABCB1, and CYP3A4 variants have explored interindividual variability in regorafenib metabolism and toxicity, though results remain inconsistent and have not yet translated into routine clinical practice [138]. Dose-optimization studies have demonstrated that alternative dosing strategies—such as reduced starting doses with subsequent escalation—can improve tolerability without compromising efficacy, informing evolving real-world treatment approaches [139].
Complementing its approved clinical indications, regorafenib is widely employed in preclinical models to study angiogenesis, tumor–stromal interactions, and resistance to targeted therapies [140]. Its function as a reference multikinase inhibitor has made it invaluable for kinase profiling, structural analyses, and comparative drug-development research [95]. Additionally, regorafenib is increasingly evaluated in combination with immune checkpoint inhibitors, anti-FGFR agents, MAPK inhibitors, and cytotoxic chemotherapies to explore synergistic activity and overcome resistance mechanisms across diverse malignancies [141]. By integrating angiogenesis inhibition with blockade of oncogenic signaling pathways, regorafenib continues to serve as a cornerstone agent in the evolving landscape of multikinase therapeutics. Table 5 summarizes the major pivotal clinical trials of regorafenib.
4. Avapritinib
Avapritinib is an orally administered, selective small-molecule inhibitor of receptor tyrosine kinases, specifically engineered to target oncogenic mutations in KIT and platelet-derived growth factor receptor alpha (PDGFRA) [146,147,148]. Unlike broader-spectrum tyrosine kinase inhibitors such as sunitinib or regorafenib, avapritinib was designed to exhibit high selectivity and potency against mutant forms of PDGFRA, particularly the D842V mutation, which is associated with primary resistance to conventional TKIs [47,149,150]. Its specificity minimizes off-target inhibition while maximizing activity against resistant gastrointestinal stromal tumor (GIST) clones, distinguishing it within the therapeutic landscape of TKI therapy [151,152]. Figure 5 illustartes structural formula of avapartinib.
Mechanistically, avapritinib functions as a type I ATP-competitive inhibitor that preferentially binds to the active conformation of KIT and PDGFRA mutants, blocking phosphorylation and downstream MAPK, PI3K–AKT, and STAT signaling pathways [153]. By directly inhibiting mutant-driven oncogenic signaling, avapritinib suppresses cell proliferation, promotes apoptosis, and reduces tumor vascular support indirectly, given that PDGFRA-mutant tumors often induce pro-angiogenic microenvironments [154]. The high affinity for activation-loop mutations enables the drug to overcome resistance that commonly develops with first- and second-generation TKIs in GIST [155].
Avapritinib’s clinical development initially targeted patients with unresectable or metastatic GIST harboring PDGFRA exon 18 mutations—particularly D842V—a population historically refractory to imatinib, sunitinib, and regorafenib [47,156,157]. The pivotal phase I NAVIGATOR trial assessed dose escalation, safety, and efficacy across multiple lines of prior therapy with a strong emphasis on molecularly defined subgroups, establishing 300 mg once daily as the recommended phase II dose [158]. In patients with PDGFRA exon 18 mutations, including D842V, avapritinib demonstrated remarkable clinical activity characterized by high objective response rates, deep and durable responses, and prolonged progression-free survival regardless of prior treatment exposure [43,158]. These outcomes led to accelerated regulatory approval of avapritinib for PDGFRA exon 18–mutant GIST and underscored the importance of molecular profiling for patient selection [159]. Phase II expansion cohorts further confirmed robust efficacy across PDGFRA-mutant populations with generally manageable toxicity profiles [160]. NAVIGATOR also showed activity in heavily pretreated KIT-mutant GIST, particularly in tumors with activation loop mutations, although dose-dependent neurocognitive adverse events were observed, informing subsequent dose-modification strategies and clinical management recommendations [158].
The phase III VOYAGER trial directly compared avapritinib with regorafenib in patients with advanced GIST who had received at least two prior tyrosine kinase inhibitors. Although avapritinib did not significantly improve progression-free survival compared with regorafenib in the overall study population, exploratory subgroup analyses suggested differential efficacy according to mutational status [161]. Importantly, VOYAGER confirmed regorafenib as the preferred third-line therapy for unselected GIST, while further refining the role of avapritinib as a mutation-driven therapy rather than a broadly applicable later-line agent.
In the aggregate, findings from the NAVIGATOR and VOYAGER trials highlight the critical importance of molecular stratification in GIST and clarify the optimal clinical use of avapritinib. Current international guidelines recommend avapritinib primarily for patients with PDGFRA exon 18–mutant GIST, while alternative tyrosine kinase inhibitors remain preferred in KIT-mutant disease in later-line settings [57,119].
Subsequent studies explored avapritinib in other clinical contexts. For example, patients with GIST harboring secondary KIT mutations resistant to earlier TKIs exhibited variable responses, indicating that avapritinib’s activity is strongest against specific PDGFRA activation-loop mutations [162]. Additionally, avapritinib is being investigated in combination with other targeted therapies or in early lines of therapy to determine whether synergistic antitumor effects can broaden its clinical utility [163]. Preclinical models suggest potential activity in rare KIT-driven malignancies beyond GIST, including systemic mastocytosis and certain melanomas, although clinical translation remains limited [164].
The adverse-effect profile of avapritinib is generally favorable compared with multikinase TKIs, reflecting its high selectivity, but it is not without significant toxicities [165]. Common adverse events include nausea, fatigue, anemia, edema, and cognitive effects such as memory impairment and confusion, collectively referred to as “cognitive adverse events” [166]. Intracranial hemorrhage and severe cutaneous reactions have been reported in rare cases, requiring careful monitoring [167]. Dose modifications or temporary interruptions are frequently employed to manage these toxicities without compromising efficacy [168]. Table 6 presents TEAE and management strategies for avapartinib.
Several clinically relevant associations have been observed. Rapid onset of response in PDGFRA D842V-mutant GIST is highly predictive of durable disease control, highlighting the mutation as both a therapeutic target and a predictive biomarker [172]. Cognitive adverse events may correlate with drug exposure and can be partially mitigated with dose adjustment [173]. Pharmacokinetic studies indicate that food intake increases systemic exposure modestly, which can inform administration recommendations [174].
Avapritinib has additional mentions in preclinical and translational research. Its high selectivity makes it a useful tool for dissecting the biology of PDGFRA-driven tumors and understanding mechanisms of resistance [175]. The drug is also under investigation in combination with KIT inhibitors, MEK inhibitors, or immune checkpoint blockade in early-phase trials to expand its utility beyond PDGFRA D842V-mutant GIST [176]. As a precision TKI, avapritinib represents a paradigm shift in targeted oncology, emphasizing the importance of mutation-specific drug design and personalized therapy approaches. Table 7 shows major pivotal clinical trials of avapartinib.
5. Emerging Trends and Opportunities
The development of second- and third-generation tyrosine kinase inhibitors (TKIs) such as sunitinib, regorafenib, and avapritinib has markedly transformed the management of gastrointestinal malignancies, particularly in gastrointestinal stromal tumors (GIST) and colorectal cancer (CRC) [178]. Despite substantial advances, emerging trends suggest that the full potential of these agents is still being realized, offering opportunities for improved clinical outcomes and precision therapy [47,179]. Figure 6 illustrates how multi-kinase inhibitors function by targeting a spectrum of related kinases.
One prominent trend is the rational design of mutation-specific TKIs, exemplified by avapritinib. By selectively targeting PDGFRA D842V and other activation-loop mutations, avapritinib overcomes intrinsic resistance to earlier-generation inhibitors, underscoring the importance of genomic profiling in therapy selection [181,182]. This paradigm highlights a shift from broad-spectrum multikinase inhibition, as seen with sunitinib and regorafenib, toward precision therapeutics tailored to specific molecular drivers, thereby enhancing efficacy and minimizing off-target toxicity [183,184].
Combination therapy represents another major opportunity. Preclinical and early-phase clinical studies suggest that pairing TKIs with immune checkpoint inhibitors, cytotoxic chemotherapies, or other targeted agents may achieve synergistic antitumor effects [185,186,187]. In mCRC, regorafenib combined with PD-1 inhibitors has shown promise in microsatellite-stable tumors that are otherwise resistant to immunotherapy [188,189]. Similarly, rational combinations of sunitinib or regorafenib with other antiangiogenic or MAPK-pathway inhibitors may overcome adaptive resistance and improve long-term disease control [190,191].
Biomarker-driven therapy and real-time pharmacodynamic monitoring are also emerging as critical tools to optimize treatment. Treatment-emergent adverse events, such as hypertension with sunitinib or hand-foot skin reaction with regorafenib, have been correlated with improved outcomes, suggesting potential use as mechanism-based biomarkers for patient stratification [192,193]. In avapritinib-treated GIST, rapid objective responses in PDGFRA D842V-mutant tumors serve as predictive biomarkers for durable disease control, reinforcing the role of early genomic and phenotypic assessment in guiding therapy [194].
Advances in drug formulation and dosing strategies provide additional opportunities to enhance tolerability and patient adherence. Intermittent dosing schedules, dose-escalation strategies, and pharmacokinetic-guided administration of TKIs are increasingly being explored to mitigate toxicity without compromising efficacy [195]. Such strategies are particularly relevant for chronic therapy settings, as these agents are often administered long-term for disease stabilization.
The integration of TKIs into earlier lines of therapy and adjuvant settings is another growing area of investigation. Clinical trials are evaluating sunitinib and regorafenib in neoadjuvant or adjuvant settings for high-risk GIST and advanced CRC, aiming to prevent recurrence and improve long-term survival [196,197]. Avapritinib may similarly expand into earlier treatment lines for patients with PDGFRA-mutant GIST, potentially reshaping the standard-of-care landscape [198].
Finally, advances in understanding resistance mechanisms are driving the development of next-generation inhibitors and rational sequencing strategies. Secondary mutations in KIT and PDGFRA, compensatory activation of parallel signaling pathways, and tumor microenvironment-mediated resistance continue to limit long-term efficacy of current TKIs. Research focused on overcoming these mechanisms through structural-guided drug design, combination therapy, and biomarker-informed therapy selection represents a fertile area for future innovation [199,200].
The evolving landscape of second- and third-generation TKIs in gastrointestinal malignancies is characterized by precision targeting, rational combination therapy, biomarker-guided strategies, and adaptive dosing. These trends highlight an ongoing shift toward highly individualized treatment paradigms, offering opportunities to improve outcomes, reduce toxicity, and expand the therapeutic potential of sunitinib, regorafenib, and avapritinib. Continued integration of molecular insights, translational research, and clinical innovation promises to further refine the role of TKIs in the management of gastrointestinal malignancies [201,202]. Table 8 presents contemporary management of gastrointestinal malignancies—gastrointestinal stromal tumours: current methods of treatment.
6. Conclusions
Second- and third-generation TKIs such as sunitinib, regorafenib, and avapritinib have substantially reshaped the therapeutic landscape of gastrointestinal malignancies, particularly in GIST and mCRC. These agents exemplify the evolution from broad-spectrum multikinase inhibition toward precision-targeted therapy, where molecular profiling and mutation-specific design guide optimal treatment selection.
Sunitinib remains a cornerstone for patients with imatinib-resistant GIST, with a well-characterized safety profile and documented efficacy in targeting VEGFR, PDGFR, and KIT-driven pathways. Regorafenib, as a multikinase inhibitor, extends therapeutic options for refractory mCRC and GIST, demonstrating the value of multitargeted inhibition of angiogenic, stromal, and oncogenic pathways. Avapritinib, a third-generation selective TKI, represents a paradigm shift by specifically addressing PDGFRA D842V mutations, a population historically refractory to first- and second-generation TKIs.
Emerging trends highlight several opportunities to further enhance patient outcomes. These include combination strategies with immune checkpoint inhibitors or pathway-specific agents, biomarker-guided therapy for individualized dose optimization and toxicity management, and rational sequencing to overcome acquired resistance. Precision dosing, real-time pharmacodynamic monitoring, and predictive adverse event biomarkers further underscore the importance of personalized therapy in maximizing efficacy while minimizing toxicity.
Despite these advances, challenges remain. Resistance mechanisms, including secondary mutations, compensatory signaling, and tumor microenvironment adaptation, continue to limit long-term efficacy. Ongoing translational research and clinical trials investigating novel combinations, early-line interventions, and mutation-driven strategies are critical to addressing these gaps and expanding the therapeutic potential of TKIs.
In conclusion, the integration of molecularly targeted TKIs into contemporary treatment paradigms exemplifies the shift toward personalized oncology in gastrointestinal malignancies. Continued innovation in drug design, patient stratification, and combination therapy is likely to refine their role, improve long-term outcomes, and broaden the applicability of TKIs in both GIST and other gastrointestinal cancers. The evolving landscape underscores a promising future where mutation-specific, mechanism-informed therapy becomes standard practice, optimizing efficacy while reducing adverse effects.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hirota S. Isozaki K. Moriyama Y. Hashimoto K. Nishida T. Ishiguro S. Kawano K. Hanada M. Kurata A. Takeda M. Gain-of-Function Mutations of c-Kit in Human Gastrointestinal Stromal Tumors Science 199827957758010.1126/science.279.5350.5779438854 · doi ↗ · pubmed ↗
- 2Corless C.L. Fletcher J.A. Heinrich M.C. Biology of Gastrointestinal Stromal Tumors J. Clin. Oncol.2004223813382510.1200/JCO.2004.05.14015365079 · doi ↗ · pubmed ↗
- 3Nishida T. Blay J.Y. Hirota S. Kitagawa Y. Kang Y.K. The Standard Diagnosis, Treatment, and Follow-up of Gastrointestinal Stromal Tumors Based on Guidelines Gastric Cancer 20161931410.1007/s 10120-015-0526-826276366 PMC 4688306 · doi ↗ · pubmed ↗
- 4Foo T. Goldstein D. Segelov E. Shapiro J. Pavlakis N. Desai J. Yip D. Zalcberg J. Price T.J. Nagrial A. The management of unresectable, advanced gastrointestinal stromal tumours Target Oncol.2022179511010.1007/s 11523-022-00869-y 35290591 PMC 8995292 · doi ↗ · pubmed ↗
- 5Florou V. Trent J.C. Wilky B.A. Precision medicine in gastrointestinal stromal tumors Discov. Med.20192826727632053767 · pubmed ↗
- 6Bauer S. George S. von Mehren M. Heinrich M.C. Early and next-generation KIT/PDGFRA kinase inhibitors and the future of treatment for advanced gastrointestinal stromal tumor Front. Oncol.20211167250010.3389/fonc.2021.67250034322383 PMC 8313277 · doi ↗ · pubmed ↗
- 7Nghiem E. Friedman B. Srivastava N. Takchi A. Mohammadi M. Dedushi D. Edelmann W. Kuang C. Bteich F. Emerging strategies for targeting angiogenesis and the tumor microenvironment in gastrointestinal malignancies: A comprehensive review Pharmaceuticals 202518116010.3390/ph 1808116040872551 PMC 12389621 · doi ↗ · pubmed ↗
- 8Al-Share B. Alloghbi A. Al Hallak M.N. Uddin H. Azmi A. Mohammad R.M. Kim S.H. Shields A.F. Philip P.A. Gastrointestinal stromal tumor: A review of current and emerging therapies Cancer Metastasis Rev.20214062564110.1007/s 10555-021-09961-733876372 · doi ↗ · pubmed ↗