Bulleidia extructa PP_925: Genome Reduction, Minimalist Metabolism, and Evolutionary Insights into Firmicutes Diversification
Peter V. Evseev, Irina V. Podoprigora, Andrei V. Chaplin, Zurab S. Khabadze, Artem A. Malkov, Lyudmila I. Kafarskaia, Dmitriy A. Shagin, Yulia N. Urban, Olga Yu. Borisova, Boris A. Efimov

TL;DR
This paper studies a bacteria with a very small genome, revealing insights into its minimalist metabolism and evolutionary adaptations.
Contribution
The paper presents a novel genome analysis of Bulleidia extructa PP_925, revealing its unique metabolic and evolutionary features.
Findings
PP_925 has a compact genome and lacks most biosynthetic pathways, relying on salvage and cross-feeding.
The genome retains systems for host interaction and defense, including CRISPR-Cas and competence systems.
Phylogenetic analysis shows independent peptidoglycan losses in related lineages and a small core genome with adaptive modules.
Abstract
Bulleidia extructa strain PP_925, isolated from the periodontal pocket of a patient with periodontitis, is a Gram-positive Bacillota with an unusually compact genome of 1.38 Mb. Phylogenomic analyses place PP_925 within Erysipelotrichales and show close relatedness of Bulleidia to Solobacterium and Lactimicrobium, as well as the existence of previously undescribed related clades. The metabolic repertoire of PP_925 is strongly reduced: it retains glycolysis, the phosphotransacetylase–acetate kinase pathway, and arginine catabolism but lacks the tricarboxylic acid cycle and most de novo biosynthetic pathways for amino acids, nucleotides, fatty acids, cofactors, and vitamins, implying reliance on salvage and cross-feeding. Phylogenetic inference indicates independent peptidoglycan losses in multiple mycoplasma Erysipelotrichia-related lineages, while PP_925 has retained an ancestral…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
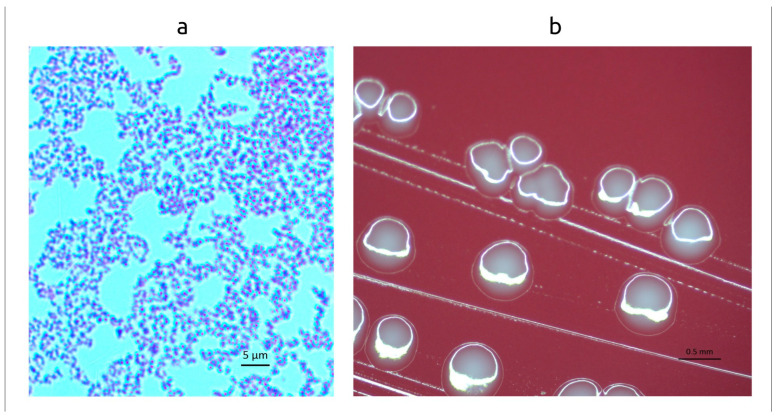 Figure 1
Figure 1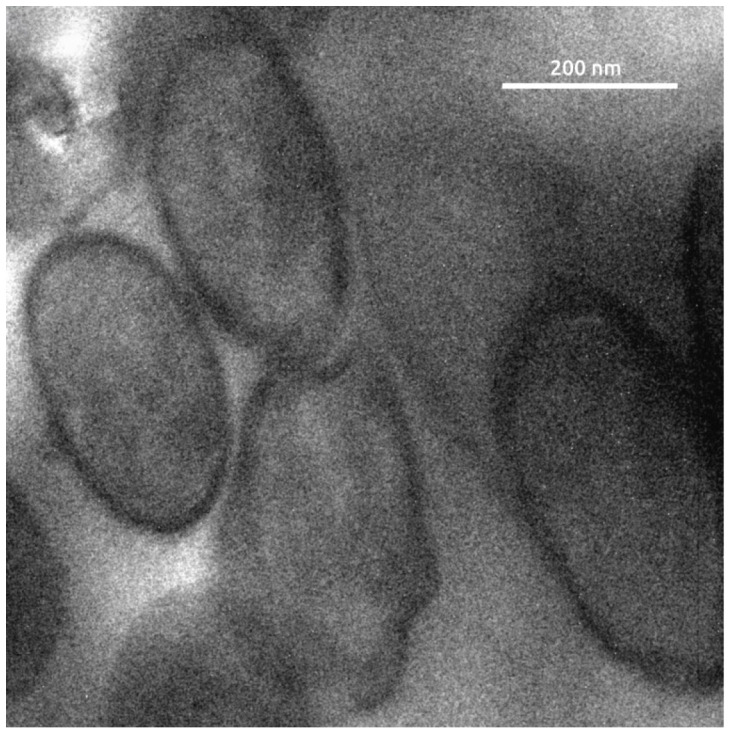 Figure 2
Figure 2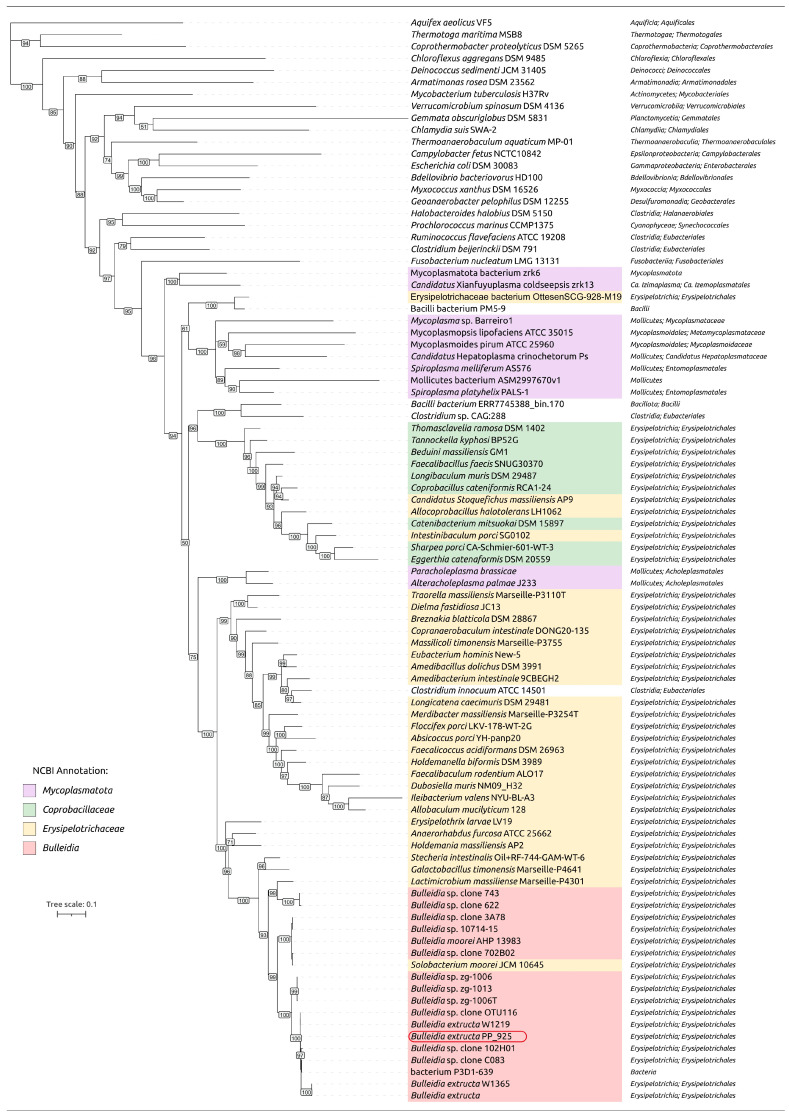 Figure 3
Figure 3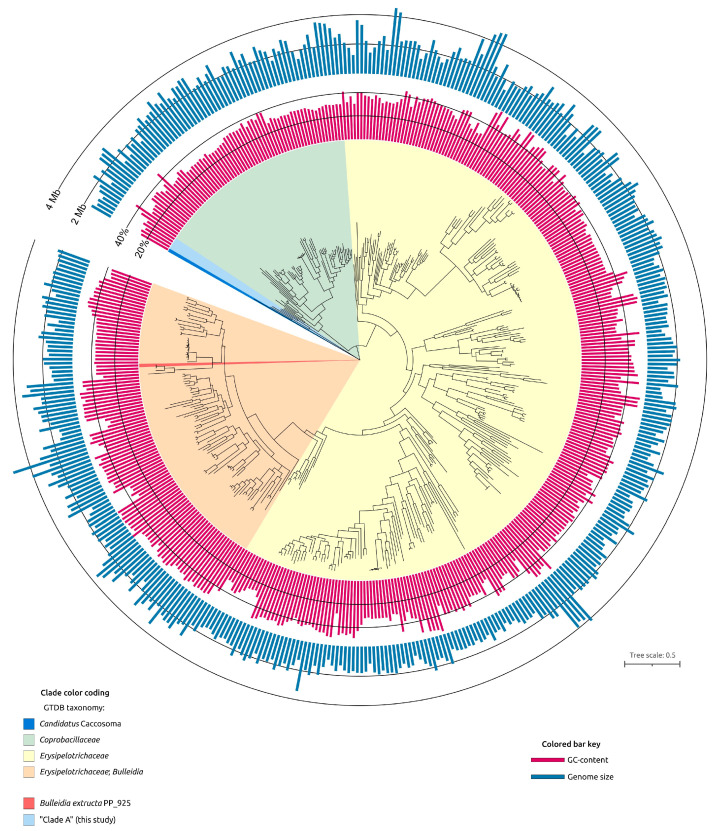 Figure 4
Figure 4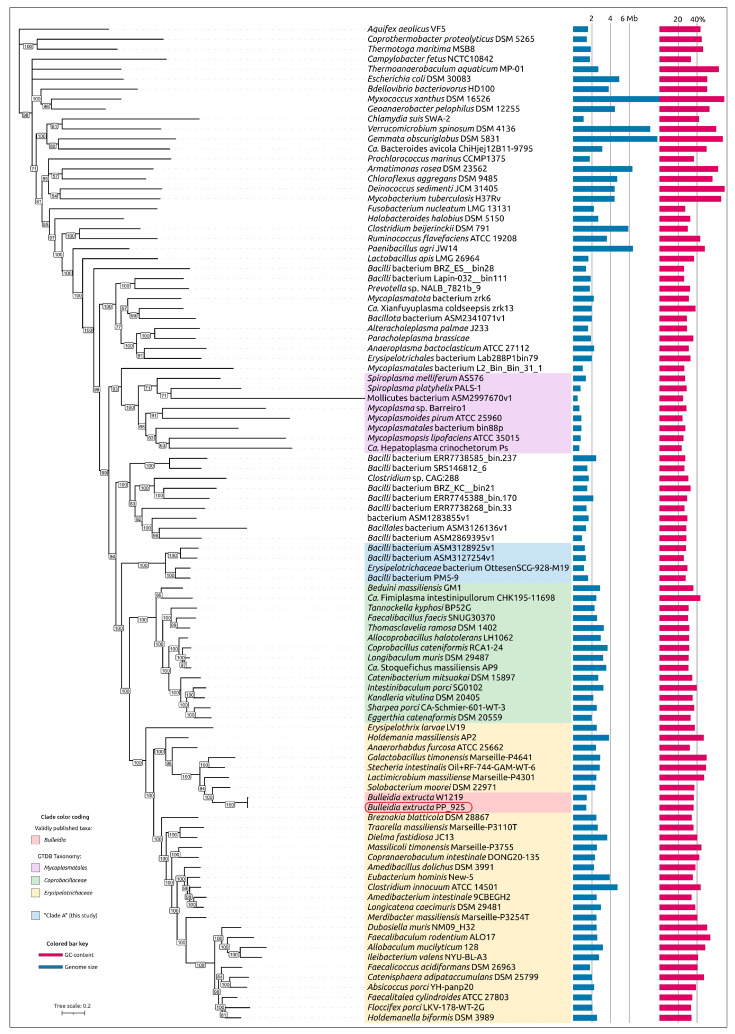 Figure 5
Figure 5 Figure 6
Figure 6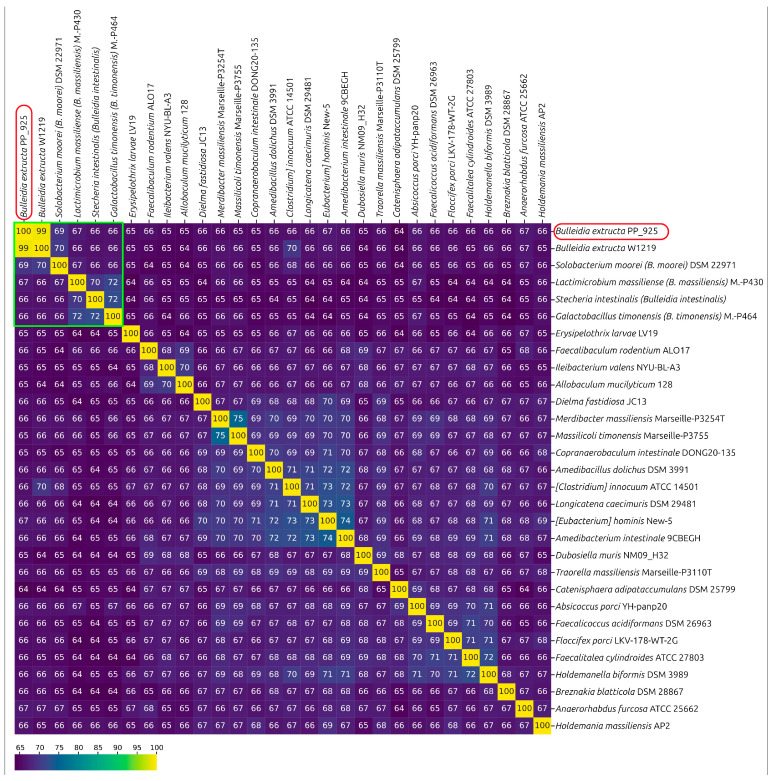 Figure 7
Figure 7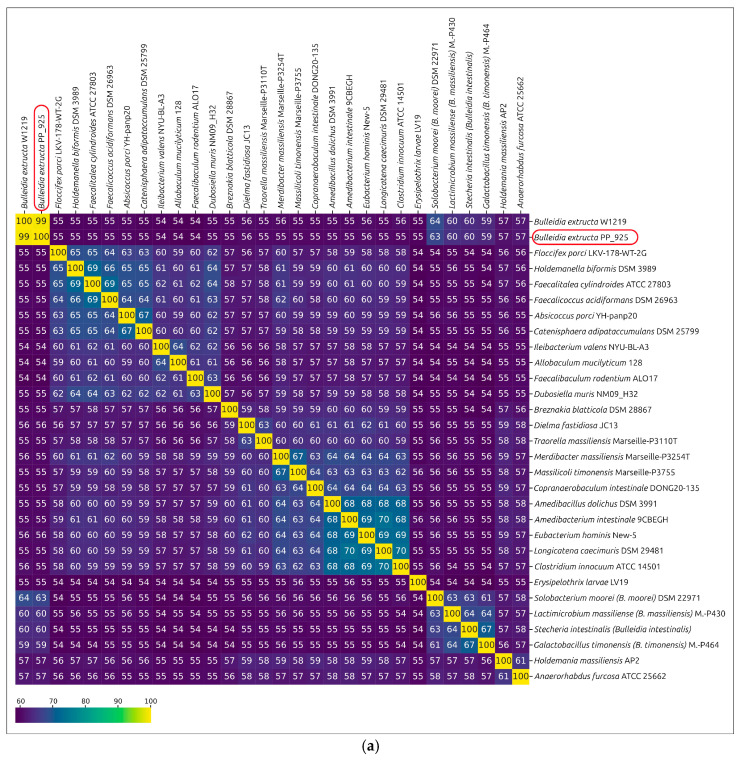 Figure 8
Figure 8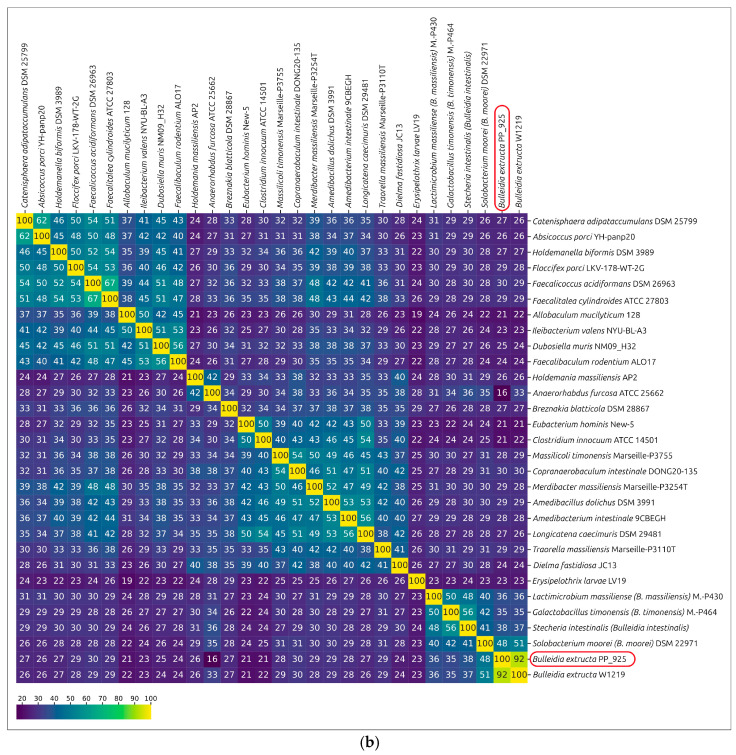 Figure 9
Figure 9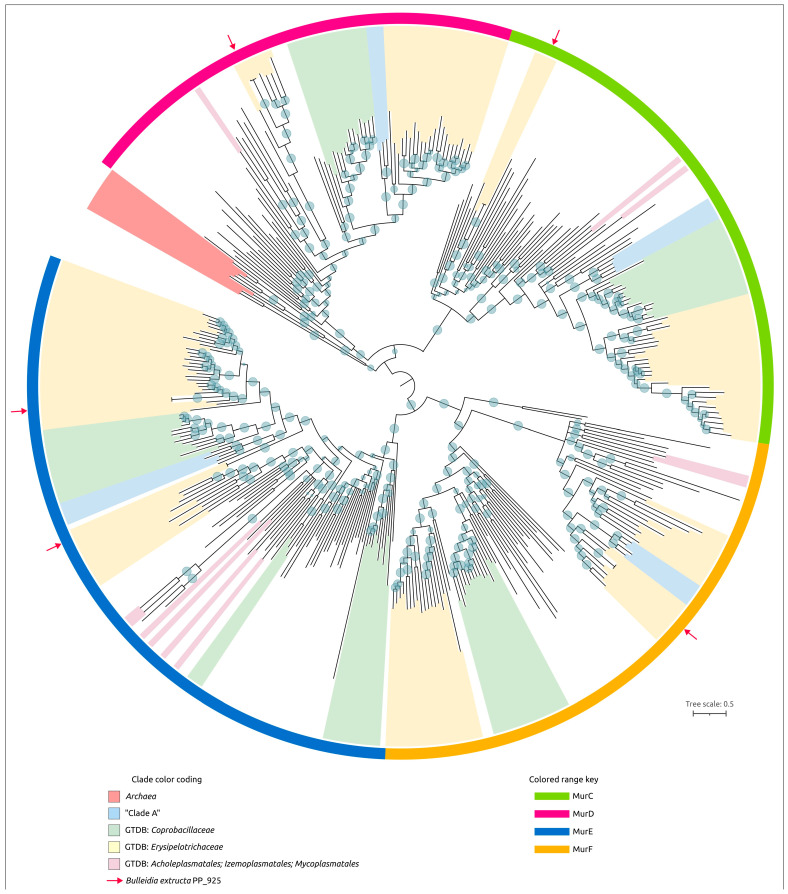 Figure 10
Figure 10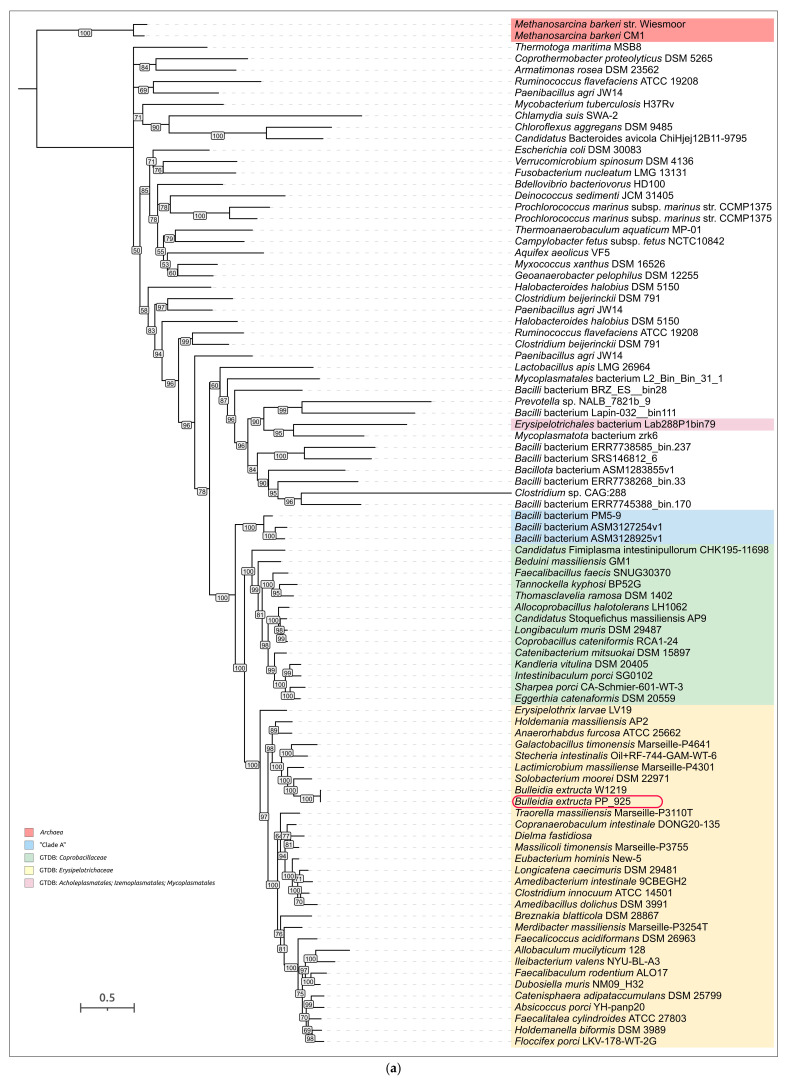 Figure 11
Figure 11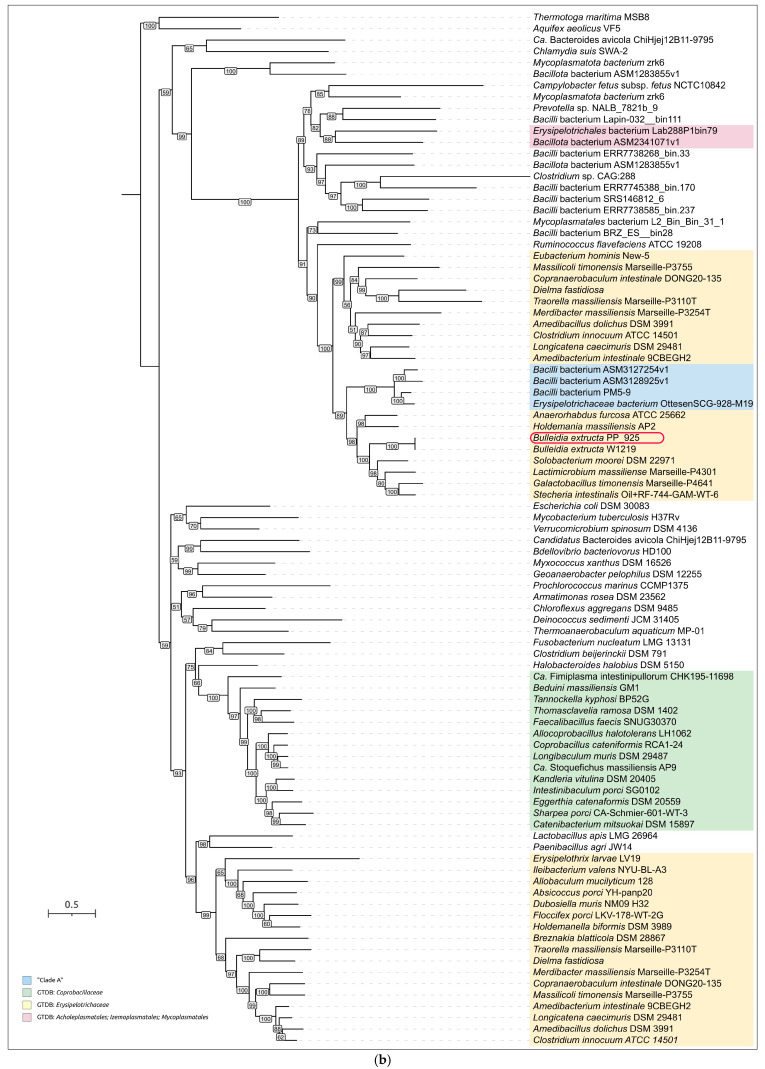 Figure 12
Figure 12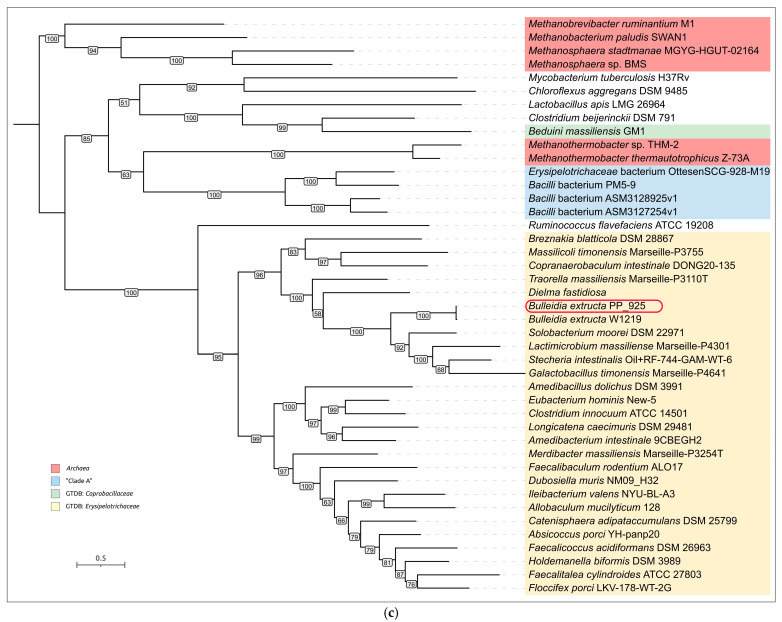 Figure 13
Figure 13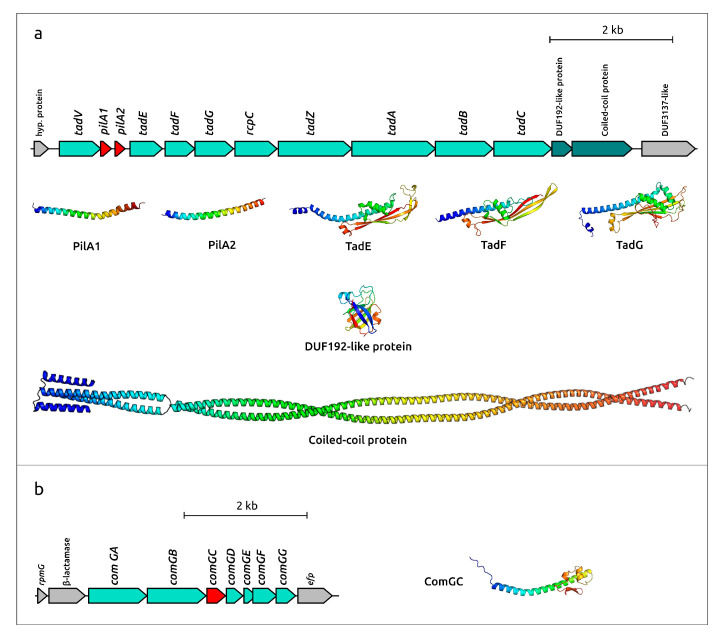 Figure 14
Figure 14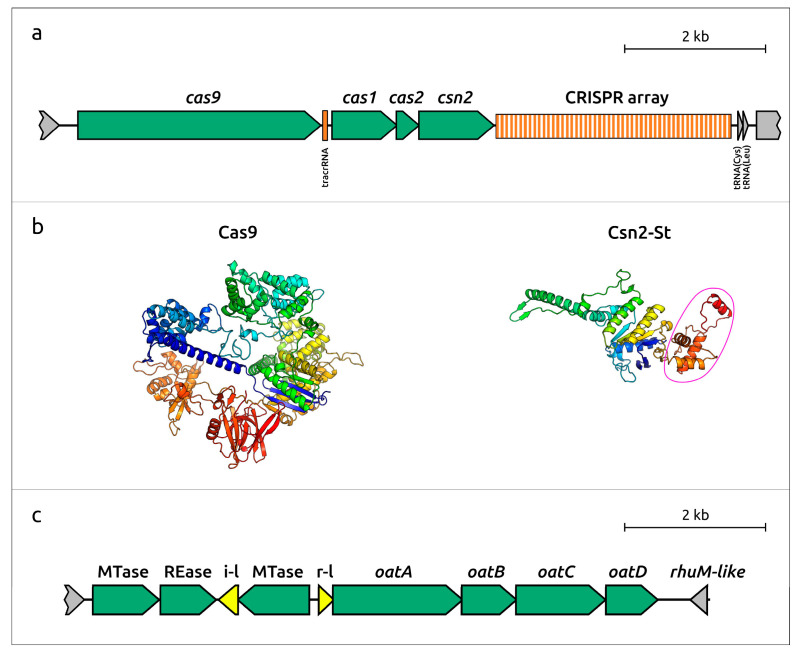 Figure 15
Figure 15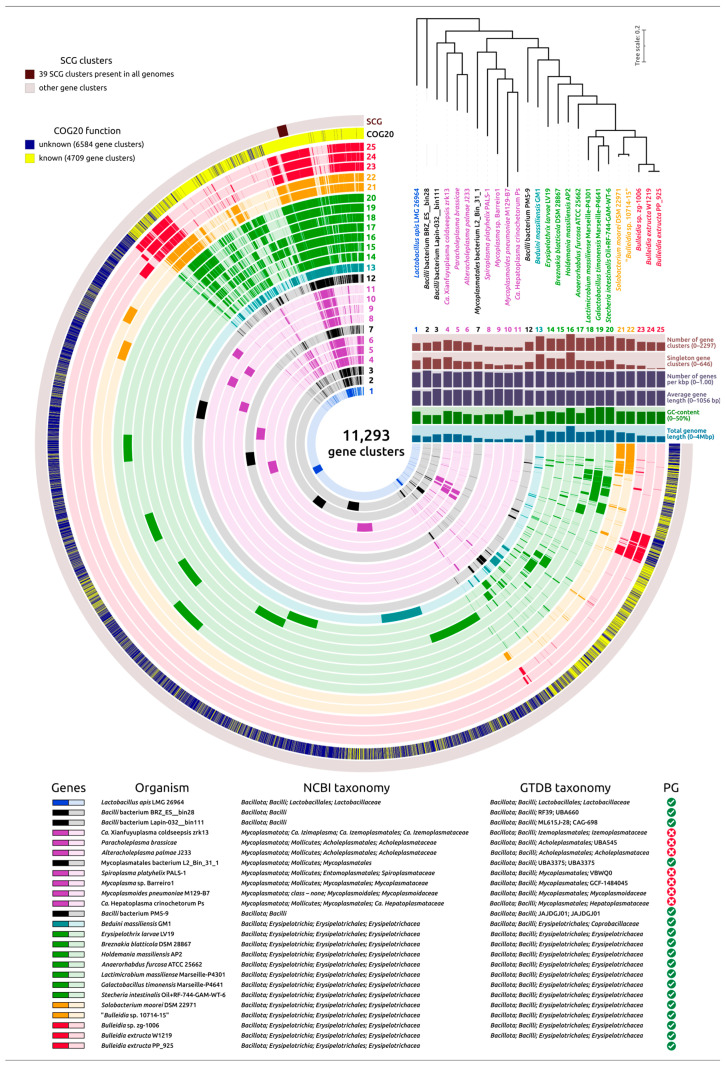 Figure 16
Figure 16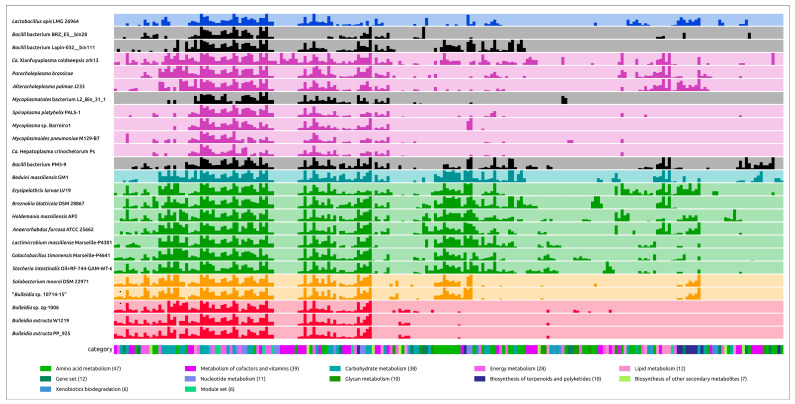 Figure 17
Figure 17Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsOral microbiology and periodontitis research · Genomics and Phylogenetic Studies · Bacterial Identification and Susceptibility Testing
1. Introduction
Progress in culturing methods over recent decades has significantly advanced the isolation of fastidious, strictly anaerobic bacteria from the human microbiota [1,2]. This has led to a substantial increase in the number of cultured and phenotypically characterized bacterial taxa that were previously considered ‘unculturable’. In the human oral and gut microbiota, particularly among adults, the highest biodiversity is observed within the phylum Bacillota, primarily represented by the families Lachnospiraceae, Oscillospiraceae, and Erysipelotrichaceae of the class Clostridia. Numerous studies are currently aimed at elucidating the potential beneficial roles of species from these families in host health [3,4]. These roles include their contribution to controlling the host’s energy balance and immune regulation. Furthermore, various genera and species within these families have been associated with several human diseases, including metabolic syndrome, obesity, diabetes, liver diseases, inflammatory bowel syndrome, and oral inflammatory diseases such as periodontitis and peri-implantitis [5,6]. Research into the potential of microbiome-targeted therapy is increasingly incorporating multi-omics approaches to study these slow-growing, low-GC, Gram-positive organisms. These studies are revealing their significant ecological versatility [7,8].
Among the various bacterial lineages within Bacillota, Erysipelotrichia remains one of the most understudied. Phylogenomic studies place Erysipelotrichia in a clade that also includes Mycoplasma and other wall-less organisms that branch from within the lineage [9,10]. Yet all characterized Erysipelotrichia retain a peptidoglycan cell wall [9,10,11,12,13,14,15,16,17], and even their smallest chromosomes, e.g., ≈1.4 Mbp in B. extructa, are substantially larger than the ≈0.5 Mbp genomes of the tiniest Mycoplasmatales.
Genome reduction is a widespread evolutionary strategy in bacteria, emerging under different ecological pressures. In free-living marine bacteria, particularly those in nutrient-poor ecosystems, selection favors compact genomes with reduced non-essential DNA. This process, known as genome streamlining, enhances replication efficiency in large populations and is well documented in models like Prochlorococcus and Pelagibacter ubique [18]. On the other hand, host-associated bacteria (e.g., symbionts, commensals) often lose key anabolic functions and rely on salvage mechanisms, driven by small population sizes and relaxed selection, as reviewed in studies of insect symbionts and other intracellular taxa [19,20]. Within the Bacillota, the class Erysipelotrichia exhibits a remarkable spectrum of genome sizes and metabolic capabilities. While some members maintain versatile genomes, others show significant reductions; yet, unlike Mycoplasmatota, they typically retain a Gram-positive cell wall. Phylogenomic evidence indicates Mycoplasmatota are polyphyletic, derived independently from Erysipelotrichia-like ancestors, suggesting multiple parallel instances of genome reduction in this lineage [9,21].
Bulleidia extructa is currently the sole validated species of the genus Bulleidia, the type strain of B. extructa is represented by strain W1219^T^ [22]. A Gram-positive, non-spore-forming obligate anaerobe, it was first isolated from advanced periodontitis pockets and assigned to the family Erysipelotrichaceae as a lineage distinct from Erysipelothrix and Holdemania [22]. 16S-rRNA surveys reveal B. extructa as a rare yet persistent member of the human oral microbiome, enriched in deep periodontal pockets and smoker-associated plaque, while shotgun metatranscriptomics associates its transcripts to metabolically active biofilms in periodontitis lesions [23]. Outside the oral cavity, the species appears, typically at low abundance, in faecal, duodenal, and vaginal microbiomes, suggesting broader anaerobic versatility [11]. Several studies report elevated relative abundance in cohorts with inflammatory bowel disease or schizophrenia, hinting at a possible gut–brain axis connection that warrants systematic investigation [24].
Although isolation in pure culture is uncommon, B. extructa has been recovered from dento-alveolar abscesses, brain abscess material, and even a prosthetic hip infection. In the latter case, the strain proved uniformly susceptible to penicillin, clindamycin, and metronidazole, and clinical resolution followed hardware removal plus targeted therapy [25]. These scattered but telling observations, together with their frequent co-occurrence alongside Porphyromonas or Fusobacterium, point to an opportunistic rather than obligate pathogenesis: the bacterium seldom acts alone, yet when it joins a polymicrobial consortium, it can amplify tissue damage.
Robust conclusions about its pathogenic potential are hampered by the absence of controlled virulence assays, population-scale prevalence studies, and experimental validation of metabolic predictions. Closing these gaps will clarify to what extent B. extructa behaves as a low-abundance commensal hitchhiker within subgingival biofilms and/or as a slow-growing opportunistic contributor to chronic anaerobic infections. Comprehensive analysis, including in silico, in vitro, and in vivo approaches, is therefore required to elucidate its lifestyle, pathogenicity, and ecological roles.
An important aspect of understanding the lifestyle and potential pathogenicity of bacteria with reduced genomes is the presence of virulence-associated loci that mediate host interaction. Among these, type IV filament systems, such as the tad, are widely distributed in bacterial and archaeal genomes and frequently acquired via horizontal gene transfer [26]. Tad pili play critical roles in adherence, colonization, and biofilm formation in diverse organisms [26,27,28,29,30]. Even in highly reduced host-dependent lineages, type IV pili are often retained and serve essential functions in attachment and colonization [31,32,33,34]. In Gram-positive bacteria, type IV pili and competence-associated filament systems facilitate adhesion, motility, and DNA uptake, contributing to both colonization and horizontal gene transfer [32,33,34].
In this study, we describe the isolation, cultivation, and genomic characterization of Bulleidia extructa strain PP_925, a rare anaerobic, cell wall-containing bacterium with one of the smallest genomes in its class (approximately 1.4 Mb). Through phylogenetic, pangenome, and metabolic analyses, we uncover evidence of multiple genome reduction events within Erysipelotrichales and Mycoplasmatota, with different gene-loss strategies. We demonstrate that B. extructa retains a tad locus (likely horizontally acquired) and a competence-associated locus, along with multiple adhesion-related virulence factors and anti-phage defense systems, despite lacking intact prophages. CRISPR analysis reveals exposure to groups of phages infecting Bacillota. Metabolically, the organism retains only glycolysis, acetate fermentation (Pta-AckA), and arginine degradation, but has lost most biosynthetic pathways, instead relying on salvage. Together, these findings position B. extructa as a compelling model for minimalist adaptation in Gram-positive bacteria, highlighting parallel reductive paths to Mycoplasmatota and offering insights into host-associated microbial evolution.
2. Results
2.1. Phenotypic Features
Cells of strain PP_925 were obligately anaerobic, non-spore-forming, non-motile, Gram-positive, coccobacillus rods. Colonies on Anaerobe Basal Agar (Oxoid) plates after six days of incubation at 37 °C under anaerobic conditions were 0.6–0.8 mm in diameter, round, white, shiny, and flattened with an entire margin. In areas of dense growth, there was weak beta-hemolysis on the media with defibrinated sheep blood. In colonies from 96 h plates, Gram-stained cells are typically 0.4–0.45 × 0.5–0.6 μm in size and occur in pairs or clusters (Figure 1).
Transmission electron micrographs (Figure 2) of ultrathin sections of the cells revealed a typical Gram-positive monoderm cell envelope consisting of a cytoplasmic membrane and a peptidoglycan layer, and also demonstrated the absence of endospore formation.
From the API 20A test results, Bulleidia extructa PP_925 did not produce acid from any of the tested carbohydrates, including D-lactose, D-sucrose, D-maltose, D-xylose, L-arabinose, D-cellobiose, D-melezitose, D-raffinose, L-rhamnose, D-trehalose, D-glucose, D-mannitol, salicin, glycerol, D-mannose, and D-sorbitol. Additionally, tests for urease and indole production were negaative.
In the Rapid ID 32A identification panel, based on the use of chromogenic enzyme substrates, strain PP_925 demonstrated negative reactions for glycoside hydrolases, including α-galactosidase, β-galactosidase, β-galactosidase 6-phosphate, α-glucosidase, β-glucosidase, α-arabinosidase, β-glucuronidase, N-acetyl-β-glucosaminidase, and α-fucosidase. Carbohydrate fermentation reactions for mannose and raffinose, included in this panel, yielded negative results. Additionally, the strain demonstrated positive reactions for a number of arylamidases (e.g., arginine arylamidase, leucine arylamidase, histidine arylamidase, and tyrosine arylamidase (weak)). Alkaline phosphatase tested negative. Pyroglutamic acid arylamidase, nitrate reductase, arginine dihydrolase, and glutamate decarboxylase reactions were negative.
In disc-diffusion experiments, strain PP_925 was resistant to gentamicin and amikacin but sensitive to penicillin G, amoxicillin/clavulanic acid, ampicillin/sulbactam, ceftazidime, clindamycin, doxycycline hydrochloride, azithromycin, levofloxacin, metronidazole, and vancomycin (Supplementary Table S1).
2.2. Phylogeny and Taxonomy
2.2.1. Overview of Sequencing, Assembly, and Annotation for B. extructa PP_925
Whole-genome sequencing of Bulleidia extructa PP_925 resulted in an assembly of 20 contigs with a total sequence length of 1,377,588 bp (Contig N50, 217,216 bp) and 378.0 x coverage (NCBI GenBank accession number JBMPNY000000000.1). The GC content of the assembly was 36.1%. Annotation predicted 1335 protein-coding genes (coding sequences, CDSs); 41 tRNA genes; 1 tmRNA gene; and 3 genes corresponding to full-length 16S, 23S, and 5S rRNAs. In addition, one 3333 bp repeat region and two sequences corresponding to ncRNAs (RNase P RNA component class B and the small-type signal recognition particle sRNA) were identified in the genome. Functional assignment suggested putative functions or protein family affiliations for 1111 encoded proteins, while 224 genes were annotated as hypothetical protein CDSs. The coding density was 90.7%. Assembly completeness was estimated at 98.67%, and the predicted contamination level was 0.67%.
2.2.2. 16S rRNA Gene Sequence Analysis and Phylogeny
At present, only a single validly published species, Bulleidia extructa, is recognized within the genus, represented by the type strain W1219^T^ (https://bacdive.dsmz.de/strain/5365, accessed 15 September 2025) [22]. However, public sequence repositories contain several additional 16S rRNA gene sequences and genomic assemblies annotated as “Bulleidia”, many of them originating from culture-independent microbiome surveys, and have not yet been formally validated at the species level. To place strain PP_925 in this broader context, we extracted its 16S rRNA gene from the genome and used it as a query in BLAST+ 2.12.0 searches against the NCBI GenBank core nucleotide database, and then reconstructed phylogenies that also incorporated related sequences. These searches retrieved multiple homologous Bulleidia sequences (Supplementary Table S2), which were annotated as Solobacterium spp. and Lactimicrobium massiliense.
Phylogenetic analysis was carried out with these sequences and representatives of different bacterial taxa (Supplementary Table S3), including members of the class Erysipelotrichia (Figure 3). The analysis placed B. extructa PP_925 together with seven sequences showing 98.8–99.9% identity to it in a branch that was sister to another cluster containing three sequences annotated as Bulleidia spp., with identities of 97.1–97.3%. This combined clade was adjacent to a branch that included Solobacterium moorei JCM 10645^T^, the type strain of the officially recognized species S. moorei [35]. Sequences within this clade showed 93.3–93.8% identity to B. extructa PP_925.
Two further sequences, annotated as “Uncultured Bulleidia” spp. but sharing only 90.4% pairwise identity with B. extructa PP_925, clustered instead with Lactimicrobium massiliense. In this phylogenetic reconstruction, Solobacterium moorei and its closest relatives appear more closely related to Bulleidia than to other genera of Erysipelotrichia. Applying the frequently used thresholds of 98.5% for species-level similarity [36] and 94.5% for genus delineation [37], the available 16S rRNA gene sequences suggest the presence of three putative species-level lineages within the genus Bulleidia together with Solobacterium moorei. However, several sequences are partial, and the pairwise identities of the inferred new Bulleidia lineages to Bulleidia extructa W1219^T^ are close to these thresholds; therefore, any species-level assignments should be regarded as preliminary and will require more detailed analyses, including genome-wide similarity comparisons, to be substantiated (Supplementary Table S2). Notably, the analysis also highlights the polyphyly of Mycoplasmatota, a point of considerable interest for evolutionary discussions [38,39].
2.2.3. GTDB-Tk Analysis
A representative dataset for phylogenetic analyses based on signature proteins was compiled using the Genome Taxonomy Database (GTDB) and the GTDB-Tk taxonomic classification pipeline. Preliminary phylogenetic analysis of Bacillota bacteria was carried out with concatenated alignments generated by GTDB-Tk, followed by tree reconstruction using FastTree. The dataset included 4431 sequences, of which 371 were annotated by GTDB as members of the family Erysipelotrichaceae and 66 as members of the family Coprobacillaceae (both within the order Erysipelotrichales), giving a total of 437 sequences. These two families formed sister monophyletic branches that together constituted a clade adjacent to another clade comprising four unclassified bacterial genomes: Pq454_bin41 (genome size 1.2 Mbp), Md513_bin19 (1.3 Mbp), OttesenSCG-928-M19 (1.1 Mbp), and PM5–9 (1.6 Mbp).
These four unclassified bacteria, hereafter referred to as “Clade A,” together with their closest relative in the GTDB-Tk tree, Bacilli bacterium isolate HGM10900 assigned by GTDB-Tk to the genus Candidatus Caccosoma (recently proposed in [40]), formed the final dataset of 442 strains (Supplementary Table S4). This relative was designated as the outgroup in subsequent analyses.
GTDB-Tk phylogenetic analysis indicated the placement of B. extructa PP_925 within the family Erysipelotrichaceae and its close relatedness to the type strain Bulleidia extructa W1219^T^ (Figure 4). However, validly published classifications and NCBI taxonomy annotations differ from the GTDB-based taxonomy. For example, GTDB assigns several groups of bacteria that are usually considered distinct genera to the genus Bulleidia. These include Solobacterium moorei [41], annotated as “Bulleidia moorei”; Lactimicrobium massiliense [42], annotated as “Bulleidia massiliense”; Galactobacillus timonensis [42], annotated as “Bulleidia timonensis”; and Stecheria intestinalis [43], annotated as “Bulleidia intestinalis”.
General characteristics of the 437 genomes assigned by GTDB to the order Erysipelotrichales are highly variable. Genome sizes range from 916 kbp to 5.208 Mbp, with a median of 2.003 Mbp, and GC content ranges from 27.0% to 56.5%, with a median of 38.6%. Overall, the genome of Bulleidia extructa PP_925 (1.377 Mbp) is relatively small compared to most Erysipelotrichales genomes, while its GC content is typical for the order.
To show the phylogenetic position of Bulleidia extructa PP_925 and to resolve relationships at both higher and lower taxonomic ranks, a set of 100 genomes (Supplementary Table S3) was analyzed using the GTDB alignments of the BAC120 conserved protein marker set. This expanded dataset included representatives of higher bacterial taxa, such as Mycoplasmatota, which are evolutionarily related to the class Erysipelotrichia, and several genera within the family Erysipelotrichaceae. The analysis (Figure 5) indicated the closest affinity of Bulleidia to Solobacterium and to a lesser extent to Lactimicrobium, Galactobacillus, and Stecheria, in agreement with the 16S rRNA phylogeny (Figure 3). Both the GTDB phylogenies and the 16S rRNA phylogeny highlight the polyphyletic character of Mycoplasmatota and their close evolutionary relationship to Erysipelotrichia. Notably, the average genome size of bacteria within this clade, assigned by GTDB to Mycoplasmatota, is more than two times smaller than the average genome size of GTDB-classified Mycoplasmatota from the other clades, which also contain non-Mycoplasmatota bacteria.
2.2.4. Ribosomal Proteins Phylogeny. GTDB and NCBI Taxonomic Assignments
Phylogenetic analysis based on concatenated alignments of 42 ribosomal proteins from the same genomes used in the previously discussed GTDB tree revealed a topology similar to the GTDB-based reconstruction (Figure 6). The tree indicated the relatedness of Bulleidia to representatives of Solobacterium, Lactimicrobium, Galactobacillus, and Stecheria. It also showed that Mycoplasmatota are non-monophyletic and closely related to Erysipelotrichia. Importantly, taxonomic assignments obtained from NCBI taxonomy and GTDB are conflicting in several cases. The phylogenetic analysis, however, confirmed the consistency of tree topology in terms of the monophyly of corresponding branches according to the GTDB classification, while highlighting discrepancies between this topology and several NCBI taxonomy annotations.
2.2.5. Average Nucleotide Identity (ANI)
Calculations of ANI confirmed the close relatedness of Bulleidia extructa PP_925 to Bulleidia extructa W1219^T^, with an ANI value of 99.1% (Supplementary Table S5), which is significantly higher than the accepted species delineation threshold of 95–96% based on genomic data [36,44]. No other analyzed genomes reached a level of similarity sufficient to classify them within the B. extructa species. The next highest ANI values were observed for the genome labeled as “Bulleidia sp. zg-1006” (GCF_016812035, ANI 80.2%) and for a metagenomic assembly annotated as “uncultured Bulleidia sp. genome assembly SRR11749279_bin.12_metaWRAP_v1.3_MAG” (GCA_947087645, ANI 72.9%). All other analyzed sequences had ANI values below 70% (Figure 7).
2.2.6. Average Amino Acid Identity (AAI) and Percentage of Conserved Proteins (POCP)
As proposed earlier [45,46], AAI and POCP calculations were conducted to evaluate genus-level delineation. These metrics are generally correlated [47]. For AAI, genus delineation thresholds ranging from 58% to 65% have been suggested in different studies [47,48,49], while for POCP, a 50% threshold has been proposed [46]. However, the appropriateness of the 50% POCP threshold, as well as the applicability of this metric as the primary criterion, has been questioned [47].
Bulleidia extructa W1219^T^ and Solobacterium moorei DSM 2297^T^ show an AAI of nearly 64.5%, and approximately 60% when compared to Lactimicrobium massiliense, Galactobacillus timonensis, and Stecheria intestinalis (Figure 8a). Four genomes, including B. extructa PP_925, Bulleidia sp. zg-1006, B. extructa W1219^T^, and the metagenome-assembled genome annotated as “Bulleidia sp.” (GCA_947087645), formed a distinct cluster with AAI values above 70% (Supplementary Table S6). In a similar way, these four genomes also grouped together in a separate cluster with POCP values above 63%.
These four bacteria, together with Solobacterium moorei, Lactimicrobium massiliense, Galactobacillus timonensis, and Stecheria intestinalis, are members of a larger cluster, with POCP values between them apparently higher than with other analyzed genomes (Figure 8b, Supplementary Table S6).
The results of ANI, AAI, and POCP analyses are consistent with the phylogenetic reconstructions and support the delineation of the four analyzed strains (Bulleidia extructa PP_925, Bulleidia sp. zg-1006, B. extructa W1219^T^, and the metagenome-assembled genome annotated as “Bulleidia sp.” GCA_947087645) within the genus Bulleidia, based on the AAI and POCP values that are applied for the delineation of the closely related genera Solobacterium, Lactimicrobium, Galactobacillus, and Stecheria. Furthermore, based on the genomic data and ANI calculations, three Bulleidia species can be delineated. The first corresponds to the already classified B. extructa PP_925 and B. extructa W1219^T^, the second is represented by the isolated strain Bulleidia sp. zg-1006, and the third is represented by the metagenome-assembled genome “uncultured Bulleidia sp. genome assembly SRR11749279_bin.12_metaWRAP_v1.3_MAG” (GCA_947087645).
2.3. Common and Characteristic Gene Clusters
The genome of Bulleidia extructa PP_925 was analyzed for the presence of gene clusters associated with cellular processes that are important for the general characterization of the bacterium and related groups, as well as for distinguishing B. extructa PP_925 and closely related bacteria from other taxa. The genome was also examined for genes associated with antibiotic and phage resistance, which are of therapeutic relevance. Special attention was given to the relationships between the genes and proteins of B. extructa PP_925 and those of other organisms.
2.3.1. Cell Wall Biosynthesis
To the best of current knowledge, the family Erysipelotrichaceae includes Gram-positive bacteria with a cell envelope consisting of a cytoplasmic membrane covered by a distinct layer of peptidoglycan (PG) cell wall. This structure is clearly visible in the earlier published TEM images of Bulleidia extructa and Solobacterium moorei [22,50] as well as in the present study. Although cell-wall-less mycoplasmas are phylogenetically close to Erysipelotrichia, no cell-wall-less representatives of Erysipelotrichia are known at present. The absence of peptidoglycan closer to the root of phylogenetic trees raises questions about the evolutionary origin of these genes in Erysipelotrichia.
To investigate the evolutionary history of peptidoglycan biosynthesis in B. extructa PP_925, twelve enzymes known to participate in the stepwise synthesis of the UDP-MurNAc-pentapeptide precursor or peptidoglycan assembly were analyzed [51,52,53] (Table 1).
Homologs of these proteins encoded in the genome of B. extructa PP_925 were searched using BLAST (E-value cut-off 10^−5^) across 100 representative genomes of Erysipelotrichia, Mycoplasmatota, and other bacterial taxa used in the previous analyses (Figure 3, Figure 4, Figure 5 and Figure 6). The searches for MurC, MurD, MurE, and MurF yielded partially overlapping results, reflecting their functional similarity and possible evolutionary relatedness. Some annotations conflicted with those of homologous sequences, and therefore a preliminary clustering was performed by constructing a phylogenetic tree that included all four proteins (Figure 9, with organism names removed; the complete tree is shown in Supplementary Figure S4). In addition, several homologs from complete archaeal genomes (non-metagenomic), identified in the NCBI nr/nt database, were included to facilitate rooting of the trees. The resulting trees are presented in Figure 10a,b, as well as in Supplementary Figures S5–S14.
Interestingly, the MurCDEF tree groups homologous archaeal sequences apart from bacterial sequences, apparently reflecting differences in the structure of murein and pseudo-murein stem peptides, and places them close to each other, although they are not monophyletic in this reconstruction. The branch containing MurC and MurD is closer to archaeal sequences than the MurE and MurF branch (Figure 9, Supplementary Figure S4).
Homologs of MurA, MurB, MurT, and MraY were also found in genomic sequences of cultured haloarchaeal species, and several were included in phylogenetic analyses for rooting the trees (Figure 10a,b). Homologs of FemA and MurG were not identified in the genomes of cultured archaea using TBLASTN searches against the NCBI nt database.
The MurT phylogeny shows a complex evolutionary history of this protein, which may involve genetic exchanges between archaea and bacteria (Figure 10c). Comparisons of the GTDB and ribosomal protein phylogenies, on the one hand, and the phylogenies of peptidoglycan biosynthesis-associated genes, on the other hand, indicate multiple events of horizontal transfer and gene duplication.
Genomes of several presumably wall-less mycoplasma-like organisms, assigned by GTDB to the order Mycoplasmatales, do not contain complete sets of peptidoglycan-associated genes. Notably, the assembly labeled Erysipelotrichales bacterium Lab288P1bin79 (GCA_009784525), assigned by GTDB to the order Acholeplasmatales, encodes nearly the complete set of peptidoglycan biosynthesis genes, including a non-homologous alanine racemase compared to that of B. extructa PP_925. The genome labeled Bacillota bacterium ASM2341071v1 (GCA_023410715) also contains most of the examined peptidoglycan-associated genes, despite its GTDB classification to the order Izemoplasmatales. It has been proposed that Candidatus Izemoplasma represents an intermediate stage in the reductive evolution from Bacillota to Mycoplasmatota [54].
In contrast, several genomes assigned by GTDB to taxa outside the recognized wall-less orders Acholeplasmatales, Haloplasmatales [55], Candidatus Izemoplasmatales and Mycoplasmatales, but phylogenetically close to them, were not found to encode the homologs of twelve analyzed peptidoglycan biosynthesis proteins. These include Bacillales bacterium ASM3126136v1 (GCA_031261365), Bacilli bacterium ASM2869395v1 (GCA_028693955) and “uncultured Bacilli bacterium BRZ_KC__bin21” (GCA_944332075). This group diverged after the wall-less taxa and before Clade A; Erysipelotrichia diverged later, after Clade A (Figure 5 and Figure 6). Interestingly, neighboring genomes appear to retain the peptidoglycan-biosynthesis genes that these bacteria lack. These data may indicate the need to investigate whether a cell wall is present in some currently unclassified, uncultured bacteria that occupy a phylogenetically intermediate position between Mycoplasmatota and Erysipelotrichia.
It is possible that some of these observations result from erroneous assemblies. CheckM2 completeness analysis of the genomic assemblies indicated possible loss of information about certain genes or contamination in some cases. Nevertheless, the reliability of completeness metrics is not absolute, as illustrated by the genome of Spiroplasma platyhelix PALS-1 (GCF_021496725, circular chromosome), which was labeled as complete but yielded a CheckM2 completeness estimate of only 92.94%.
Importantly, the group of wall-less taxa (Acholeplasmatales, Haloplasmatales, Candidatus Izemoplasmatales, and Mycoplasmatales) is not monophyletic in either the GTDB tree or the ribosomal protein tree. In both cases, the clades containing wall-less branches are preceded by earlier-diverging lineages that include at least several peptidoglycan-associated genes, which are positioned closer to the root of the entire clade with meaningful statistical support. These include assemblies that are misclassified in the NCBI annotations as “Prevotella sp. NALB_7821b_9” (GCA_023665755), “Mycoplasmatota bacterium zrk6” (GCA_018394315), and “uncultured Mycoplasmatales bacterium L2_Bin_Bin_31_1” (GCA_937936465) (Figure 10).
2.3.2. Search for Genes Associated with the Biosynthesis of Teichoic Acids
A search for genes encoding proteins typically involved in the biosynthesis of teichoic and lipoteichoic acids (TAs and LTAs) was conducted using BLAST searches against a custom database, in a manner similar to the search for peptidoglycan-associated genes. Amino acid sequences of proteins encoded by the genes tagA, tagB, tagC, tagD, tagE, tagF, tagG, and tagO [56,57] were used to identify homologs in the genome of Bulleidia extructa PP_925 and other bacteria. Only a glycerol-3-phosphate cytidylyltransferase domain homologous to the TagD protein was identified in the encoded proteins of B. extructa PP_925 as well as in B. extructa W1219^T^. The corresponding protein is located within the locus 00310–00350, involved in the biosynthesis of surface polysaccharides. The presence of the phosphotransferase gene 00330 within this locus suggests that negative charges may be incorporated into the biosynthetic product. This modification could allow the product to functionally complement tag-synthesized teichoic acids. In contrast, a substantial proportion of representatives of other genera within the families Coprobacillaceae and Erysipelotrichaceae (21 out of 43 analyzed genomes) encoded nearly the complete set of tag proteins.
2.3.3. Tight Adherence and Competence Systems
Annotation of the genome of B. extructa PP_925, performed with different annotation pipelines and complemented by HHblits and BLAST searches, identified two loci encoding pilin-like proteins. Detailed analysis of the genetic context of these loci suggested that one of them belongs to the tight adherence system (tad operon), while the other is part of the competence system (comG operon) (Figure 11).
Tad operons show noticeable diversity among Gram-positive bacteria [58,59]. The tad operon of Bulleidia extructa PP_925 displays several important features in its organization and composition, including a unique arrangement of the constituent genes. The operon in B. extructa PP_925 apparently comprises 13 genes. It begins with the prepilin peptidase tadV gene, followed by two genes, pilA1 and pilA2, encoding the major pilin subunits (Figure 11a). As in other Tad pilins [60], structural models of PilA1 and PilA2 predict the absence of the C-terminal globular β-sheet domain (for comparison, see AF3 models of PilA1 and PilA2 and the model of the competence system major pilin ComGC in Figure 11a,b). Predicted minor pilins TadE and TadF, as modeled with AF3, share the same basic structural architecture but possess additional C-terminal domains compared to the major pilins. TadG is characterized by an even larger C-terminal domain, although it retains a structural organization similar to that of TadE and TadF.
A BLAST search of the amino acid sequences of tad operon proteins against NCBI and custom databases identified homologous proteins encoded within a similar locus (pairwise identity > 90%) in the genome of B. extructa W1219^T^. No homologs were found in closely related genomes of Solobacterium, Lactimicrobium, Galactobacillus, and Stecheria, except for PilA1. Numerous homologs of the conserved TadA ATPase were identified across different Erysipelotrichia bacteria. However, many of them were less similar to the sequences from Bulleidia than to homologs from other, more distantly related groups, including Helcococcus ovis and members of Clostridia. This suggests that the tad locus in B. extructa was acquired horizontally.
Functions of eleven gene products of the tad operon can be predicted by HMM-based searches or AF3 modeling and comparison with published structures [61,62,63]. Two genes, however, located at the 3′ end of the operon, show no clear sequence or structural similarity to functionally characterized Tad-system proteins. Nevertheless, these genes are consistently found adjacent to Tad-related genes in B. extructa W1219^T^, in two metagenomic sequences classified by GTDB as Erysipelotrichales, and in three complete genomes of Helcococcus ovis. The penultimate gene encodes a protein of 112 amino acids similar to DUF192 domain-containing proteins (Pfam entry PF02643). Its predicted structure (Figure 11a) closely resembles that of a “hypothetical signal peptide protein” or “putative transcription regulator” from Sinorhizobium meliloti (PDB code 3pjy, DALI Z-score 15.8) and of a “protein of unknown function” from Novosphingobium aromaticivorans (PDB code 3m7a, DALI Z-score 14.5). It also shows weaker similarity to structures such as the “putative exported protein” from Burkholderia pseudomallei (PDB code 6ozd, DALI Z-score 8.8). The last gene of the operon encodes a protein predicted to have a coiled-coil structure (Figure 11a). HHpred analysis revealed a strong similarity (>99% probability) to the chromosome partitioning protein Smc from Bacillus subtilis (PDB code 5nnv [64]) and to α-helical regions of several eukaryotic proteins, including dynein. However, DALI searches using the predicted structure did not return close structural homologs. These two proteins may play important and unique roles in the functioning of the Tad system in B. extructa PP_925.
Another genomic locus associated with the formation of type IV pili corresponds to the competence operon comG. This operon is flanked upstream by a gene encoding a β-lactamase-like protein and downstream by the elongation factor efp. BLAST searches using the predicted sequences of ComGA-ComGG identified homologous loci in the genomes of Solobacterium moorei DSM 2297, Bulleidia sp. zg-1006, and other Erysipelotrichaceae and Erysipelotrichales, suggesting a vertical origin of the locus. Interestingly, the β-lactamase-like gene has its closest homologs not only in Bulleidia sp. zg-1006, S. moorei DSM 2297, and other Erysipelotrichales, but also in several strains of Fusobacterium and other anaerobic bacteria, suggesting horizontal gene exchange among evolutionarily distant organisms inhabiting the same ecological niche.
2.3.4. Anti-Phage Defense Systems
The search for anti-phage defense systems in the genome of Bulleidia extructa PP_925 identified several loci potentially encoding such systems. These include the following:
- Abi systems. Two single-gene defense systems of the Abi family were identified [65]. Notably, both of them are located near genes for cellular non-coding RNAs, specifically tRNA-Thr and tmRNA. This genomic arrangement suggests they may be organized as a type III toxin–antitoxin module, in which a regulatory RNA inhibits the toxin protein under normal conditions. The closest homologs were found in the complete genome of “Bulleidia sp. 10714-15” (identified using TBLASTN and the NCBI Core_nt database), which is likely misclassified and more plausibly represents Solobacterium moorei. This suggests that these genes might have been inherited vertically and subsequently lost in other related bacteria, including Bulleidia and Solobacterium. The next closest homologs of the Abi protein encoded by gene locus tag 01180 were found in genomes of two strains of Faecalibacillus intestinalis. Homologous sequences to the Abi protein encoded by gene locus tag 02335 were identified in Staphylococcus gallinarum strain X16P4, Lacticaseibacillus paracasei strain PC-H1, different strains of Lactobacillus helveticus and other bacteria.
- CRISPR-Cas system. A locus comprising genes 02725–02740 and a downstream repeat region was identified as a type II-A CRISPR-Cas system, characterized by a minimal set of core components. These include the signature effector protein Cas9 as a single multidomain nuclease, the CRISPR array, tracrRNA, Cas1 and Cas2 adaptation proteins, and the auxiliary protein Csn2 [66]. The Csn2 protein in B. extructa PP_925 belongs to the Csn2 subfamily St, previously described in Bacillota, largely in Streptococcus and Enterococcus. These proteins are longer than canonical Csn2 due to an additional C-terminal domain [67] (Figure 12a,b). The CRISPR array spacers were searched against the GenBank PHG database. No exact matches were found in phage genomes. The most similar sequences (identity up to 95%, query coverage ≤ 80%) belonged to phages infecting Gram-positive bacteria from the families Herelleviridae, Salasmaviridae, and Vilmaviridae; subfamily Trabyvirinae; and genus Coventryvirus, all within the class Caudoviricetes. BLAST searches identified the closest homologs of all four proteins of the B. extructa PP_925 CRISPR-Cas system in various species of the genus Streptococcus. However, no significant homologs were found in NCBI Core_nt genomes attributed to Bulleidia or closely related genera, except for limited similarity (33%) of Cas1 to a protein in “Bulleidia sp. 10714-15”.
- Restriction-modification (RM) type II system. A locus encoding proteins reminiscent of the type II RM system of Haemophilus parahaemolyticus HhaI [68,69] was identified. It includes three genes: two encoding DNA (cytosine-5-)-methyltransferases (locus tags 04715 and 04730) and one encoding a restriction endonuclease (locus tag 04720) (Figure 12c). The protein encoded by gene 04725, located between the restrictase (locus tag 04720) and the second methyltransferase (locus tag 04730), showed no significant similarity (>90% probability) in HHpred searches. However, AF modeling and subsequent DALI analysis revealed structural similarity to tyrosine recombinases, including the site-specific integron recombinase IntI4 from Vibrio cholerae (Z-score 7.3) and phage integrases. The gene appears to be degraded, since the encoded protein is at least twice as small as functional integrases. This suggests that the second methyltransferase may have arisen through recombination and that gene 04725 encodes a degraded integrase-like protein. Notably, the orientation of genes 04725 and 04730 is opposite to that of the rest of the RM locus. TBLASTN searches identified close homologs (up to 99% pairwise identity) of all these proteins in genomes of Campylobacter concisus, Fusobacterium animalis, Streptobacillus moniliformis, Fusobacterium pseudoperiodonticum, Haemophilus haemolyticus, and Metamycoplasma arthritidis. No Erysipelotrichia representatives were among the first 100 hits.
- Gao_Qat system. A complete Gao_Qat system was identified, comprising the four characteristic proteins QatA, QatB, QatC, and QatD encoded by genes 04740, 04745, 04750, and 04755. This locus is located next to the RM type II system, separated by a small gene encoding a repressor similar to phage repressors and to the competence regulator ComR from Streptococcus suis (HHpred probability > 98%) [70]. Adjacent to this region, a gene was found encoding a protein with strong similarity to virulence protein RhuM (HHpred probability 99.05%). However, the encoded protein is three times smaller than the canonical RhuM protein encoded within the SPI-3 pathogenicity island of Salmonella typhimurium [71], suggesting that it may represent a degraded copy. This gene is unlikely to be part of a mobile genetic element, since BLAST searches revealed different patterns of homology compared to the RM and Gao_Qat proteins. Searches of genes surrounding the RM and Gao_Qat loci revealed other distinct homologs not shared with these systems.
In addition, the genome of B. extructa PP_925 encodes the Wadjet system, which provides resistance to plasmid transformation [72,73]. This locus includes all four characteristic genes, jetA, jetB, jetC, and jetD (locus tags 01950-01965).
2.3.5. Search for Prophage Regions and Phage-Derived Sequences
Search for prophage regions, conducted using PHASTEST and Phigaro, did not find prophages in the genome of B. extructa PP_925. Furthermore, analysis of the results of the HHblits search involving all predicted proteins encoded in the genome did not find the sequences that could represent the phage major capsid protein of Duplodnaviria (HK-97) or other viruses, portal protein, or terminase large subunit. The HHblits search (probability threshold 90%) found sequences similar to phage and other integrases and recombinases, which could belong to other mobile elements or represent non-functional remnants of phages or other mobile elements.
2.3.6. Search for Antibiotic Resistance Genes
In the genome of Bulleidia extructa PP_925, we identified a gene encoding an aminoglycoside N(3)-acetyltransferase (AAC(3)) (locus tag 02460). BLASTp analysis showed that its closest homologs (up to 89.6% PI) occur in multispecies proteins annotated as Bacillati and in closely related Erysipelotrichia, including Solobacterium spp., indicating possible lineage-level conservation rather than recent acquisition from distant taxa. AAC(3) enzymes acetylate the 3-amino group of 2-deoxystreptamine aminoglycosides and are commonly associated with reduced susceptibility to agents such as gentamicin and related compounds, in agreement with experimental data obtained in this study; however, the substrate spectrum is variant-dependent [74,75].
A search for antibiotic resistance genes (ARGs) using the Resistance Gene Identifier (RGI) server identified only four vanY-like vancomycin resistance genes, potentially associated with the vanB, vanF, and vanG gene clusters. However, analysis of the genomic context of these vanY-like genes did not reveal the presence of the typical additional genes that constitute the vanB [76], vanF [77], vanG [78], or vanA [79] clusters. Furthermore, AMRFinderPlus analysis did not detect ARGs.
A BLAST search of the 1335 predicted proteins against the latest version of the CARD database identified 110 proteins associated with antibiotic resistance that are homologous to B. extructa PP_925 proteins. The best hits were distributed as follows: 15 proteins each from Enterococcus spp. and Streptomyces spp.; 10 from Staphylococcus aureus; 9 from Neisseria gonorrhoeae; 8 each from Escherichia coli, Streptococcus spp., and Paenibacillus spp.; 7 from Riemerella anatipestifer; 4 each from Bacillus licheniformis, Clostridioides difficile, and Desulfitobacterium hafniense; 2 each from Acinetobacter baumannii, Bifidobacterium adolescentis, and Pseudomonas aeruginosa; and 12 from other organisms. No proteins from Erysipelotrichia were present among the top hits. This scarcity of matches may reflect a gap in current knowledge of the molecular mechanisms of drug resistance in Erysipelotrichiaceae. It also likely reflects the bias in existing datasets toward widespread pathogens. HHblits searches additionally identified 32 proteins with similarity to β-lactamases and 161 proteins with similarity to diverse bacterial proteins that could potentially be associated with antibiotic resistance, highlighting the need for experimental studies to clarify the functional significance of these candidate ARGs. However, this may also reflect the susceptibility of B. extructa PP_925 to antibiotics observed in our experiments.
2.4. Search for Virulence Factors
BLASTP searches against the Virulence Factor Database (VFDB) core dataset, which includes representative genes associated with experimentally verified virulence factors (https://www.mgc.ac.cn/VFs/ accessed 20 August 2025), identified similarities between several hundred proteins of Bulleidia extructa PP_925 and the VFDB core set. In total, 230 predicted proteins matched with an E-value cut-off of 10^−5^. However, such sequence-level matches cannot be directly interpreted as evidence of virulence-related functions without experimental validation or more detailed in silico analysis. This is particularly important since several proteins within the VFDB core set are multifunctional and not always directly associated with virulence. Examples include elongation factor Tu in Francisella tularensis, which interacts with host nucleolin [80]; the nucleoside ABC transporter substrate-binding protein BmpD in Borrelia burgdorferi; and the DNA repair protein RecN in Neisseria meningitidis.
Bacterial pathogenicity typically depends on several core strategies: adherence to host tissues, evasion or modulation of host immune responses, secretion of exoenzymes that promote tissue invasion and nutrient acquisition, and stress-survival mechanisms that enable persistence and regulation of virulence expression [81,82,83,84]. The search revealed multiple B. extructa PP_925 proteins homologous to experimentally verified factors from these functional categories. Representative homologs of virulence factors in B. extructa PP_925 include the following:
- Stress survival: Clp ATP-binding chain C (ClpC, VF0072; Listeria monocytogenes EGD-e).
- Exoenzymes: Hyaluronidase (HysA, VF0146; Streptococcus pneumoniae TIGR4).
- Adherence: Listeria adhesion protein Lap (Lap, VF0444; L. monocytogenes EGD-e); chaperonin GroEL (GroEL, VF0594; Clostridium difficile 630); Tad pilus protein (CpaF/TadA, VF0612; Vibrio vulnificus CMCP6) encoded in the aforementioned tad locus; fibronectin-binding protein (FbpA, VF0349; L. monocytogenes EGD-e); collagen adhesin precursor (CNA, VF0005; Staphylococcus aureus MW2); laminin-binding surface protein (Lmb, VF0275; Streptococcus agalactiae NEM316).
- Immune modulation: Cap8D (VF0003; S. aureus MW2); phosphomannomutase ManB/YhxB (VF0044; Haemophilus influenzae Rd KW20); undecaprenyl diphosphate synthase CpsA/UppS (VF0361; Enterococcus faecalis V583); LCP family protein GBS_RS06610 (VF0274; S. agalactiae NEM316); Bcs1′ (VF0043; H. influenzae 1007); CapA (VF0141; Bacillus anthracis); aminotransferase Cj1437c (VF0323; Campylobacter jejuni NCTC 11168); HasC (VF0244; Streptococcus pyogenes M1 GAS). Interestingly, the LCP family protein and a capsular polysaccharide synthesis-like protein are encoded in a genomic region apparently responsible for PG modifications or exopolysaccharide synthesis.
- Exotoxins: Hemolysin B (HlyB/HlyA, VF0225; Escherichia coli CFT073); Toxin A (TcdA, VF0376; C. difficile 630).
BLAST searches also indicated that the closest homologs of 8 out of these 18 B. extructa PP_925 proteins were found in bacteria other than the closest relatives of Bulleidia (Solobacterium moorei and Lactimicrobium massiliense).
2.5. Pangenome Analysis
An anvi’o pangenome analysis of 25 genomes spanning the Erysipelotrichales, Lactobacillales, Mycoplasmatales, and atypical Bacillales (Figure 13) revealed a total of 11,293 gene clusters, of which only 39 single-copy core gene clusters (SCGs) were present in all genomes. This small universal core reflects the deep evolutionary divergence across the dataset and highlights the high degree of lineage-specific adaptation. Within the Bulleidia group, 66 gene clusters were found to be unique to the genus (B. extructa PP_925, B. extructa W1219^T^, and Bulleidia sp. zg1006), while 93 clusters were shared exclusively between the two B. extructa strains, suggesting significant lineage-specific lateral gene flow. The overall functional composition of the pan-genome was skewed toward poorly annotated regions: approximately 58% of clusters could not be assigned to known COG20 functional categories, forming “genomic dark matter” often found in lineage-specific arcs in Figure 13. Nevertheless, Bulleidia extructa stood out for its relatively high fraction of annotated functions compared with both larger and smaller genomes. Despite its minimal size, the B. extructa genome has a surprisingly high fraction of functionally characterized genes relative to many other bacteria with reduced genomes. Only roughly 24–27% of its genes are of unknown function, meaning ~73–76% could be assigned to known COG functional categories in annotations. This proportion of “unknown” genes is markedly lower than that in larger relatives or other minimal bacteria. For example, the 3.8-Mbp genome of Holdemania massiliensis (another Erysipelotrichaceae member) has about 34% of genes with no known function in COG analysis, the genome of Mycoplasmoides pneumoniae M129-B7 encodes 35% or more such proteins (despite their importance and intensive study as an important pathogen), and Mycoplasma sp. Barreiro1 has 30% of such genes, as was revealed by the present analysis. This low content of functionally uncharacterized genes can be a consequence of the specifics of Bulleidia’s gene repertoire: it appears to have retained mostly well-characterized core functions and shed dispensable or novel gene families above the limit necessary for its lifestyle. Turning to singleton gene clusters, B. extructa PP_925 presented only 38, fewer than any other genome in the dataset (e.g., M. pneumoniae M129-B7 has 161, Erysipelothrix larvae LV19 has 309, H. massiliensis AP2 has 609). Given the close evolutionary relationship between PP_925 and W1219^T^, this low singleton count plausibly reflects their recent common ancestry and limited divergence. Finally, average gene lengths and coding densities varied by only ~10–20% across genomes, implying that genome reduction occurred predominantly through gene loss, rather than through compaction of gene or intergenic regions.
To further interpret these genomic patterns, a phylogenetic analysis was performed using the 39 single-copy core genes (SCGs) identified using the 30% identity threshold (other parameters are clarified in Section 4). A concatenated SCG tree was constructed, with Lactobacillus apis included as an outgroup to root the topology (Figure 13). The evolutionary history reflected in the topology of the SCGs tree follows that of the GTDB tree (Figure 5) and, to a lesser degree, the ribosomal proteins tree (Figure 6). In the SCG tree, all Erysipelotrichales (including Bulleidia) cluster together on a distinct branch, separate from the mollicutes (cell-wall-less mycoplasma-related clades) and representing a strikingly versatile group, by genomic size and content. As in other trees discussed earlier in the article, the cell-wall-less bacteria in the analysis do not form a single monophyletic group; instead, they appear in two different clades, each branching within or next to cell-wall-bearing relatives, suggesting at least two independent origins of the mollicute lifestyle. In one clade, a group of mycoplasmas is phylogenetically interwoven with peptidoglycan-containing relatives at its deepest branches; in another clade, a separate lineage of wall-less bacteria (possibly an anaeroplasma or asteroleplasma group) branches from within a big clade containing unclassified Bacillalli and Erysipelotrichales. For completeness, we note that this placement reiterates a well-established result: the class Mollicutes (the only member of the phylum Mycoplasmatota) is polyphyletic, having arisen from Firmicute (phylum Bacillota) ancestors multiple times [9]. Combining pangenome and phylogenetic results, we infer two contrasting trajectories within this clade: genome minimization (mycoplasma-like bacteria) and genome expansion/innovation. Both trajectories are present within Erysipelotrichales, with minimization in some lineages (Bulleidia) and expansion/innovation in others (e.g., H. massiliensis).
The metabolic genomic analysis of B. extructa PP_925, visualized in the summary heatmap of KEGG pathway comparison (Figure 14) and detailed in Supplementary Figure S15, reveals a profound reduction in anabolic and energy-generating functions, with retention of only a narrow fermentative core. At the global scale, the Bulleidia strains clustered with other Erysipelotrichales, sharing a number of common features in metabolism and, like the closest relatives of Bulleidia extructa, showing a reduction in metabolism. However, the Bulleidia extructa rows were particularly sparse. Both strains of B. extructa exhibit a strongly reduced metabolic repertoire compared to most other Bacillota analyzed. The KEGG pathway comparison highlights that B. extructa retains only a limited subset of central metabolic functions, with conspicuous losses in amino acid biosynthesis, carbohydrate utilization, and energy metabolism. This pattern indicates a streamlined physiology consistent with adaptation to a nutrient-rich, host-associated environment. When contrasted against other Erysipelotrichales (e.g., Solobacterium, Holdemania), B. extructa shows a shared loss of oxidative phosphorylation, the TCA cycle, and broad amino acid biosynthesis, but stands out as one of the most reduced in nucleotide and lipid pathways. In the broader comparison including mycoplasmas, the pangenome analysis (Figure 13) illustrates how Bulleidia retains large blocks of clusters absent in mycoplasmas, while converging with them in genome reduction and losing many biosynthetic functions.
Carbohydrate metabolism. Despite broad gene loss, glycolysis was retained in full, providing a backbone for ATP generation. The analysis (Figure 14, Supplementary Figure S15) indicated the presence of the Embden–Meyerhof–Parnas pathway, as well as sugar transport modules, including phosphotransferase systems (PTS). The presence of the phosphotransacetylase–acetate kinase (Pta-AckA) pathway [85] enables conversion of acetyl-CoA to acetate with ATP generation via substrate-level phosphorylation. Experimental studies confirm fermentation of glucose and maltose, yielding acetate, lactate, and succinate as end-products [22]. The ability to generate ATP from acetate production places Bulleidia in an intermediate position: it shares fermentative minimalism with mycoplasmas, but retains additional flexibility through this ancestral pathway.Energy metabolism. No respiratory complexes or cytochromes were detected. TCA modules are nearly absent, leaving only isolated enzymatic fragments without cycle functionality. Thus, B. extructa is strictly fermentative, relying on substrate-level phosphorylation. Despite lacking respiratory complexes, the genome retains F- and V-type ATPases that can reverse-operate, hydrolyzing ATP to pump H^+^ and maintain a membrane potential.Amino acid metabolism. The genome lacked nearly all de novo amino acid biosynthetic pathways. Most KEGG pathways are absent or incomplete, suggesting that B. extructa relies heavily on exogenous sources or metabolic cross-feeding within microbial communities. By contrast, several related taxa (e.g., Holdemanella, Anaerorhabdus) still retain partial biosynthetic capacities. Notably, arginine biosynthesis genes were absent, yet B. extructa retains the arginine dihydrolase (ADI) pathway for catabolism, consistent with experimental evidence of arginine hydrolysis [22].Nucleotide metabolism. B. extructa lacks genes for both purine and pyrimidine biosynthesis, and a complete loss of de novo purine and pyrimidine biosynthesis for B. extructa W1219 was predicted earlier [11]. Instead, Bulleidia seemingly relies on salvage reactions: rather than synthesizing nucleotides from scratch, the bacterium recycles free bases and nucleosides imported from the environment via phosphoribosyltransferases and kinases. These salvage pathways apparently compensate for the absence of de novo routes, but enforce strict dependence on exogenous nucleotide sources.Lipid metabolism. Fatty acid (Fab) biosynthetic genes are absent, making Bulleidia auxotrophic for fatty acids. This explains its growth requirement for Tween 80 (a source of oleic acid) [22].Cofactor and vitamin metabolism. Most B-vitamin and cofactor pathways are absent (e.g., biotin, thiamine, riboflavin), forcing reliance on host-derived vitamins.
3. Discussion
The currently available data are consistent with the presence of the genus Bulleidia, which includes more than ten sequenced representatives. Based on 16S rRNA gene similarity, these sequences appear to fall into three tentative species-level clusters. The type species Bulleidia extructa is represented by strains PP_925, the type strain W1219(T), and several 16S rRNA sequences; a second putative lineage is represented by the draft genome Bulleidia sp. zg-1006 and a 16S rRNA sequence; and a third group currently corresponds to 16S rRNA sequences only and thus remains to be confirmed by genome-based analyses.
A complicating factor is the inconsistent classification in public databases. In NCBI GenBank several representatives of the related genus Solobacterium are annotated as Bulleidia. This reflects not only errors in deposited sequence labels but also the close relatedness of these two genera, as confirmed by phylogenetic analyses with different gene sets. Comparisons based on AAI and POCP show values of about 64% and 51% between type representatives of Bulleidia and Solobacterium. These values are close to the commonly accepted genus-level thresholds of about 60% for AAI and 50% for POCP [46,48]. Nevertheless, pangenome analysis and metabolic profiling reveal clear differences in gene content and functional potential, supporting the separation of Bulleidia and Solobacterium as distinct genera despite numerical proximity to these thresholds. Consistent with this assignment, ANI calculations confirm the close relatedness of PP_925 and the type strain W1219^T^.
In the Genome Taxonomy Database (GTDB), several closely related genera, including Bulleidia, Solobacterium, Lactimicrobium, Galactobacillus, and Stecheria, are placed together under the genus Bulleidia. This reflects the principles of GTDB taxonomy, which relies on genome-wide phylogenies constructed from a standardized set of 120 conserved marker genes (BAC120) and applies rank normalization to ensure comparable divergence levels across clades [86,87]. In our analysis, these taxa form a cluster with pairwise similarities of up to 67% AAI and 56% POCP, and phylogenomic reconstructions recover them as a monophyletic and distinct lineage within Erysipelotrichiaceae. The coherence of this block and its treatment in GTDB suggest that its formal delineation as a higher-order taxonomic unit might merit consideration in the future.
Phylogenetic reconstructions based on GTDB marker sets and ribosomal protein sequences, including metagenome-derived representatives, confirm the monophyly of the order Erysipelotrichales, but also reveal a more complex structure of the larger clade that includes cell-wall-less mycoplasma-like bacteria. This topology refines the current view of Erysipelotrichales evolution. In particular, it highlights the proximity of Erysipelotrichales to a group of small-genome (1.1–1.6 Mbp) unclassified bacteria, including one assembly erroneously annotated in NCBI as Erysipelotrichaceae bacterium OttesenSCG-928-M19, which in fact lies outside the family Erysipelotrichaceae. At greater phylogenetic distance, the analyses also indicate relatedness to the small-genome lineage Candidatus Caccosoma. Consistent with previous work [9], cell-wall-less mycoplasma-like bacteria are not recovered as a monophyletic group. Instead, they are distributed among separate clades, each of which contains basal branches represented by cell-wall-bearing organisms. This pattern supports the view that the loss of peptidoglycan occurred multiple times independently from peptidoglycan-containing ancestors, providing additional resolution to the evolutionary trajectory of the broader Erysipelotrichales–Mollicutes assemblage.
Within this context, the phylogenetic proximity of Bulleidia extructa to Solobacterium moorei aligns with a lifestyle typical of opportunistic pathogens. S. moorei is repeatedly linked to halitosis and oral pathology and is documented in wound and bloodstream infections in susceptible hosts, supporting an opportunistic profile [88,89,90]. B. extructa, though infrequently reported, was originally described from the oral cavity and has been documented in rare serious infections such as periprosthetic joint infection [25]. Genomic features reinforce this profile, including adhesion-associated loci such as the tad operon and a competence system, homologs of adherence factors, and a broad suite of anti-phage defense systems. These properties are typical of low-abundance commensals that may expand under dysbiotic conditions and contribute to opportunistic infections.
The analysis of Bulleidia extructa PP_925 demonstrates the recurrent theme of reductive evolution among host-associated Bacillota. Genome erosion in such bacteria is not accidental but reflects adaptive tuning to nutrient-rich environments. The ~1.4 Mbp genome of B. extructa ranks among the smallest in cell-wall-containing Bacillota, illustrating a balance between metabolic necessity and redundancy removed due to host provisioning [91]. Despite wide gene loss, core fermentation pathways persist, signaling selective pressure for energy extraction. Glycolysis remains intact, and the phosphotransacetylase–acetate kinase route enables anaerobic ATP generation. The arginine deiminase pathway (ADS) is notable both for its energy-yielding capacity and its buffering effect on environmental pH, particularly in oral habitats where acidification drives cariogenic processes. Elevated ADS activity has been observed in caries-free individuals compared with those with active lesions, pointing to a protective role of arginine metabolism in dental microbial communities [92]. Experimental models and clinical observations show that arginine metabolism increases ammonia production, raises pH, and suppresses acidogenic bacteria, which can limit caries development [93]. These observations support the interpretation that retention of arginine catabolism in Bulleidia extructa, despite its streamlined genome, reflects a conserved strategy of environmental adaptation relevant to its ecological niche. Notably, our systematic survey of antibiotic resistance determinants in PP_925 revealed only a single clearly identifiable aminoglycoside N(3)-acetyltransferase and a small number of candidate loci with distant similarity to curated ARGs, consistent with its predominantly susceptible phenotype. Given the paucity of experimentally characterized resistance determinants in Erysipelotrichia and the bias of existing databases toward well-studied pathogens, robust links between genotype and the antibiotic susceptibility profile of B. extructa will have to come from dedicated functional work, including targeted gene knockouts, expression analyses, and biochemical characterization in future studies.
Phylogenomic and pangenomic analyses place B. extructa among Erysipelotrichales, highlighting different trajectories of genome evolution within Bacillota. Mycoplasmatota independently evolved wall loss in association with a host-dependent lifestyle, whereas B. extructa indicates that retention of peptidoglycan remains adaptive in dynamic host-associated niches. Furthermore, in Mycoplasmatota such as Mycoplasmoides pneumoniae and related bacteria, the evolutionary trajectory is largely unidirectional, with progressive gene loss leading to extremely small genomes lacking most biosynthetic and envelope functions. In contrast, within Erysipelotrichales, the pattern is more variable. Some lineages, such as Bulleidia, show reduced genomes, while others retain comparatively large repertoires. Gene content can both contract and expand, reflecting multiple independent episodes of genome reduction [9]. Similar observations have been made for gut-associated Bacillota, where adaptation to host environments frequently coincides with loss of sporulation and biosynthetic capacities, but with heterogeneous outcomes across lineages [94].
The retention of a horizontally acquired tad locus underscores the importance of adherence systems even in highly reduced genomes since tad pili facilitate host colonization in a range of bacteria [33,95], while competence loci can enhance genetic adaptability in streamlined genomes. Presence of homologs of known adherence-associated virulence factors further supports active rather than passive host interaction [96].
Anti-phage defense mechanisms of B. extructa PP_925 are unusually comprehensive. Restriction-modification systems, Wadjet-like loci, and other defense systems are retained despite apparent pressures for gene loss. CRISPR spacer analysis provides possible evidence of exposure to phages, which can include SPO1-like Herelleviridae myoviruses and φ29-like podoviruses, yet no prophages are found, suggesting robust defense rather than frequent viral integration. However, the presence of unique gene clusters suggests ongoing horizontal gene exchange. These patterns reinforce the view that reductive evolution in B. extructa is highly selective and modular.
Ecologically, B. extructa functions as a community specialist, depending on metabolic cross-feeding while ensuring persistence through adhesion and defense. Despite not being a common pathogen, Bulleidia possesses a genomic profile indicative of opportunistic behavior in compromised hosts. Furthermore, ecological investigations underscore that underappreciated taxa like Bulleidia can significantly influence microbial community dynamics, particularly in dysbiotic environments [97].
In summary, B. extructa illustrates that Gram-positive bacteria can undergo substantial genome reduction while retaining structural and adaptive features. From an evolutionary perspective, B. extructa exemplifies how members of Erysipelotrichia can occupy highly specialized roles through metabolic contraction. Its streamlined genome reflects the trade-off between independence and specialization: by discarding biosynthetic versatility, it achieves tighter integration into the metabolic networks of host-associated microbial ecosystems. Future experimental validation of substrate utilization and metabolite exchange will be essential to confirm these genomic predictions and to clarify the ecological role of B. extructa within host-associated consortia.
4. Materials and Methods
4.1. Strain Isolation and Cultivation
Strain Bulleidia extructa PP_925 was isolated in March 2024 from the periodontal pocket of a 64-year-old woman (in Moscow, Russia) with periodontitis, where it was present at a concentration of ~2.0 × 10^8^ CFU/mL in the sample. A sample was collected from periodontal pockets using sterile paper points (#30; three per site) [98]. To prevent salivary contamination, the sampling area was isolated with cotton rolls. The points were inserted to the full depth of the pocket and retained for 10 s to allow for absorption. They were then immediately transferred into sterile tubes containing 1 mL of thioglycollate transport medium (HiMedia Laboratories, Mumbai, India). In the laboratory, microbiological examination was performed according to a previously described method [99]. The isolation of strictly anaerobic bacteria was carried out on Schaedler Anaerobe Agar (Oxoid, Basingstoke, UK) supplemented with 5% (v/v) defibrinated sheep blood, Anaerobe Basal Agar (Oxoid, Basingstoke, UK) with sheep blood, and Wilkins-Chalgren Anaerobe Agar (Oxoid, Basingstoke, UK) with sheep blood. Inoculation was performed using 10^−6^, 10^−7^, and 10^−8^ dilutions of the sample. Following inoculation, the plates were placed into anaerobic jars (Schütt Labortechnik GmbH, Göttingen, Germany) with an atmosphere of 85% N_2_, 10% H_2_, and 5% CO_2_, using platinum catalysts, and incubated at 37 °C for 72 to 240 h (3–10 days). After incubation, the plates were examined macroscopically. Colony morphology was assessed, and colonies of each type were enumerated. Selected colonies were then examined microscopically following Gram staining. Distinct colonies were subcultured onto fresh plates of the same media and incubated anaerobically to obtain sufficient biomass for identification and preservation. Selected strains were preserved by lyophilization. Microbial suspensions were prepared in a cryoprotectant solution (10% sucrose and 1% gelatin (w/v)), frozen, and lyophilized using an SB1 freeze dryer (Chemlab, Barnsley, UK). The lyophilized strains were stored at –80 °C. Protocol No. 13 (15 December 2022) for the study was approved by the Ethics Committee of the Medical Institute at RUDN University (Moscow, Russia).
Biochemical reactions were determined in triplicate by using the API 20A anaerobe test kit (bioMérieux, Marcy-l’Étoile, France) and Rapid ID 32A anaerobe identification kit (bioMérieux) using incubation for 48 h and 4 h, respectively. Disc-diffusion tests were carried out using Antimicrobial Susceptibility Test Discs (Bioanalyse, Ankara, Türkiye) [100,101].
4.2. Electron Microscopy
For transmission electron microscopy (TEM), the Bulleidia extructa PP_925 was grown for 72 h in a liquid medium of the following composition (g l^−1^): tryptone (Difco, Detroit, MI, USA), 10.0; papaic digest of soybean meal (BBL Phytone Peptone, BD Biosciences, Sparks, MD, USA), 5.0; yeast extract (Difco, Detroit, MI, USA), 10.0; glucose, 0.2; NaCl, 5.0; and arginine hydrochloride, 1.0; pH 6.5. To obtain a bacterial pellet, cultures were centrifuged at 8000× g for 3 min in 0.1 M sodium cacodylate buffer (pH 7.2; Honeywell Fluka, Buchs, Switzerland) containing 4% sucrose (PanReac AppliChem, Darmstadt, Germany), followed by three washes in the same buffer. Primary fixation was performed for 1 h at room temperature in 0.1 M sodium cacodylate buffer supplemented with 4% sucrose and 0.05% ruthenium red (Merck KGaA, Darmstadt, Germany), containing 2% paraformaldehyde (PanReac AppliChem, Darmstadt, Germany) and 2.5% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA). After three washes in 0.1 M sodium cacodylate buffer, pellets were post-fixed in 1% osmium tetroxide prepared in 0.1 M sodium cacodylate buffer for 60 min at 4 °C on ice. Dehydration was carried out in ethanol (70% and 90% for 5 min each, then 100% twice for 10 min), followed by propylene oxide (Acros Organics, Geel, Belgium) for 2 × 10 min. Pellets were infiltrated with epoxy resin (Epon 812; Electron Microscopy Sciences, Hatfield, PA, USA), first with a 1:1 (v/v) mixture of resin and propylene oxide for 60 min, then with pure resin overnight, and polymerized for 24 h at 60 °C. Ultrathin sections (~90 nm) were cut on a Reichert-Jung Ultracut E ultramicrotome (C. Reichert AG, Vienna, Austria) using a 45° glass knife, mounted on 200-mesh copper grids, air-dried, and stained with 2% uranyl acetate for 5 min in the dark. Samples were imaged on a LEO 912 AB Omega transmission electron microscope (Carl Zeiss, Oberkochen, Germany) operated at 80 kV under standard conditions.
4.3. Genome Sequencing
Genomic DNA was extracted using the ExtractDNA Blood & Cells kit (Evrogen, Moscow, Russia) according to the manufacturer’s instructions. DNA integrity was assessed by electrophoresis in a 1.5% agarose gel, and DNA concentration was measured with the Spectra Q HS Plus kit (Raissol Bio, Moscow Region, Russia) on a Qubit 2 fluorometer (Invitrogen, Carlsbad, CA, USA). Sequencing libraries were prepared with the MGIEasy Fast PCR-FREE FS DNA Library Prep Set v2.0 (MGI, Shenzhen, China) following the manufacturer’s protocol. Libraries were pooled equimolarly to ensure uniform coverage across samples, and the pooled library was circularized using the DNBSEQ MGIEasy Dual Barcode Circularization Kit (MGI, Shenzhen, China). Whole-genome sequencing was performed on a DNBSEQ-G50 platform (MGI, Shenzhen, China), generating 150 bp paired-end reads on an FCL PE150 flow cell (MGI, Shenzhen, China).
4.4. Functional Annotation, Protein Structure Prediction and Analysis, Search for Antibiotic Resistance Genes and Virulence Factors
Genomes were downloaded from the NCBI GenBank databases (https://www.ncbi.nlm.nih.gov/genbank/, accessed 20 August 2025). Annotation procedures were carried out using Bakta version 1.10 [102]. Additional analyses included BLAST searches in the NCBI databases (https://blast.ncbi.nlm.nih.gov, accessed 20 August 2025) [103], searches with InterPro [104], HHpred [105] searches against the databases PDBmmCIF70, NCBI_conserved_domains, Pfam-A, and UniProt_Swiss_Prot_viral70 [105], as well as HH-suite version 3.3.0 searches [106]. Structural predictions, homology searches, and comparisons are described below.
CRISPR loci were identified with MinCED version 0.4.2 (https://github.com/ctSkennerton/minced, accessed 20 August 2025) using the “-gffFull” setting. Spacer sequences were extracted with MinCED using the “-spacers” setting. Prophage-derived regions were identified using PHASTEST version 1.0.1 [107] and Phigaro version 2.4.0 [108], both implemented in the Proksee server [109]. HH-suite version 3.3.0 searches were additionally used to screen for phage major capsid protein and large terminase subunit sequences against the databases PDBmmCIF70, NCBI_conserved_domains, Pfam-A, and UniProt_Swiss_Prot_viral70. Genetic maps were visualized using clinker [110].
All protein structures were modeled with AlphaFold 3 [111] and visualized using PyMOL version 2.5.4 (Schrödinger Inc., New York, NY, USA). Structural similarity searches were performed with the DALI server using default settings, and similarity was evaluated with the DALI Z-score [112,113].
The search for antibiotic resistance genes was performed using AMRFinderPlus version 4.2.5 [114] and Resistance Gene Identifier (RGI) version 6.0.3 in conjunction with the Comprehensive Antibiotic Resistance Database (CARD) [115] using default settings. Virulence factors were identified by BLAST+ version 2.12.0 searches against a custom database containing the VFDB core dataset [116].
4.5. Phylogenetic Analysis Using 16S rDNA, Ribosomal Proteins, and 120 Bacterial Marker Genes
Nucleotide sequences of 16S rRNA genes were identified using BLAST+ version 2.12.0 or extracted from annotated bacterial genomes and aligned with Geneious Prime version 2025.0.3 (Biomatters, Inc., Auckland, New Zealand) and MAFFT version 7.48 [117] applying the L-INS-i algorithm with default settings. Ribosomal protein amino acid sequences were identified using the UBCG2 tool [118], aligned with MAFFT, and concatenated in Geneious Prime. Concatenated alignments of 120 bacterial marker genes were obtained using GTDB-Tk version 2.4.0 [119] and GTDB Release 220, applying the classify workflow with default settings.
All alignments were used for phylogenetic reconstruction with IQ-TREE version 2.4.0 [120], using automatic best-fit substitution model selection and bootstrap analysis with 1000 replicates (command line parameters: -m TEST -ninit 1000 -bb 1000). Preliminary analyses were performed with FastTree version 2.1.11 [121] under default settings. Genome completeness and contamination were estimated with CheckM2 version 1.1.0 [122] under default settings.
4.6. ANI, AAI, and POCP Calculations
Average nucleotide identity (ANI) was calculated with orthoANIu version 1.40 [123] using default settings and fasta files downloaded from NCBI. Clustering was performed using the UPGMA hierarchical clustering method. Average amino acid identity (AAI) calculations and clustering were performed with the EzAAI calculator version 1.2.3 [124] using default settings and genomic fasta sequences. The percentage of conserved proteins (POCP) was calculated with POCP-nf version 2.3.6 [125] under default settings using protein sequences predicted by Bakta.
4.7. Anvi’o Pangenome and Metabolism Analysis
Pangenome and metabolic analyses were conducted with anvi’o version 8 [126]. The pangenome analysis was performed with the program anvi-pan-genome following the workflow described in [127] and in online tutorials (https://merenlab.org/2016/11/08/pangenomics-v2, accessed 20 August 2025). Phylogenetic analysis based on concatenated alignments of single-copy core genes (SCGs) was used to order layers in the anvi’o visualization schemes. Programs and settings included anvi-get-sequences-for-gene-clusters, trimal -gt 0.50, and iqtree -s -m WAG -bb 1000.
KEGG functions and metabolic pathways [128] were annotated with anvi-run-kegg-kofams. The completeness of each KEGG pathway in every genome was assessed with anvi-estimate-metabolism, which uses previously assigned KEGG ortholog (KO) annotations. Pathway completeness matrices were visualized as heatmaps using anvi-matrix-to-newick, anvi-interactive, and other anvi’o tools following the tutorials (https://merenlab.org/tutorials/infant-gut/, accessed 20 August 2025) [129].
5. Conclusions
The genome of Bulleidia extructa PP_925 is compatible with pronounced reduction under retention of a peptidoglycan-containing cell wall. In contrast to Mycoplasmatota that have lost peptidoglycan, this lineage shows that streamlining can proceed without envelope loss. Loss of most amino acid, nucleotide, fatty acid, and cofactor biosynthesis pathways indicates a strongly restricted metabolic repertoire and suggests dependence on salvage pathways and metabolic support from surrounding microbiota.
A complete tad locus and a competence comG system indicate that functions related to adhesion, colonization, and genetic plasticity are preferentially retained. The defense repertoire combines CRISPR-Cas II-A with Csn2-St, a type II restriction–modification system adjacent to Gao_Qat-like elements, and the Wadjet module in a genome lacking detectable prophages. CRISPR spacer content indicates recurrent exposure to Bacillota phages, particularly Herelleviridae and φ29-like groups. Taken together, these features are consistent with a predominant role of active immunity rather than prophage-mediated exclusion, although this remains a working hypothesis requiring experimental validation.
Pangenome analysis reveals a very small core and few strain-specific genes, consistent with recent shared ancestry and functional focusing, whereas lineage-specific modules related to adhesion and defense underscore adaptive flexibility. Phylogenomic placement within Erysipelotrichales and the presence of multiple Bulleidia species accord with current evidence for mollicute polyphyly. Overall, B. extructa PP_925 represents a reductive strategy that maintains envelope integrity, host-interaction systems, and multiple defense modules while compressing biosynthetic capacity. It is further hypothesized that this genome organization may contribute to opportunistic behavior, a possibility that should be examined in future ecological and clinical studies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Khelaifia S. Virginie P. Belkacemi S. Tassery H. Terrer E. Aboudharam G. Culturing the Human Oral Microbiota, Updating Methodologies and Cultivation Techniques Microorganisms 20231183610.3390/microorganisms 1104083637110259 PMC 10143722 · doi ↗ · pubmed ↗
- 2Liu C. Du M.-X. Abuduaini R. Yu H.-Y. Li D.-H. Wang Y.-J. Zhou N. Jiang M.-Z. Niu P.-X. Han S.-S. Enlightening the Taxonomy Darkness of Human Gut Microbiomes with a Cultured Biobank Microbiome 20219119 Correction in Microbiome 2022, 10, 163. https://doi.org/10.1186/s 40168-022-01370-410.1186/s 40168-021-01064-334020714 PMC 8140505 · doi ↗ · pubmed ↗
- 3Dias B.d.C. Lamarca A.P. Machado D.T. Kloh V.P. de Carvalho F.M. Vasconcelos A.T.R. Metabolic Pathways Associated with Firmicutes Prevalence in the Gut of Multiple Livestock Animals and Humans Anim. Microbiome 202572010.1186/s 42523-025-00379-y 40033444 PMC 11874851 · doi ↗ · pubmed ↗
- 4Salvadori M. Rosso G. Update on the Gut Microbiome in Health and Diseases World J. Methodol.2024148919610.5662/wjm.v 14.i 1.8919638577200 PMC 10989414 · doi ↗ · pubmed ↗
- 5Nath S. Pulikkotil S.J. Weyrich L. Zilm P. Kapellas K. Jamieson L. Effect of Periodontal Interventions on Characteristics of the Periodontal Microbial Profile: A Systematic Review and Meta-Analysis Microorganisms 202210158210.3390/microorganisms 1008158236014000 PMC 9416518 · doi ↗ · pubmed ↗
- 6Cui Z. Wang P. Gao W. Microbial Dysbiosis in Periodontitis and Peri-Implantitis: Pathogenesis, Immune Responses, and Therapeutic Front. Cell. Infect. Microbiol.202515151715410.3389/fcimb.2025.151715440007610 PMC 11850578 · doi ↗ · pubmed ↗
- 7Hu W. Chen S. Zou X. Chen Y. Luo J. Zhong P. Ma D. Oral Microbiome, Periodontal Disease and Systemic Bone-Related Diseases in the Era of Homeostatic Medicine J. Adv. Res.20257344345810.1016/j.jare.2024.08.01939159722 PMC 12225918 · doi ↗ · pubmed ↗
- 8Aggarwal N. Kitano S. Puah G.R.Y. Kittelmann S. Hwang I.Y. Chang M.W. Microbiome and Human Health: Current Understanding, Engineering, and Enabling Technologies Chem. Rev.2022123317210.1021/acs.chemrev.2c 0043136317983 PMC 9837825 · doi ↗ · pubmed ↗