Distinct Regulation of the ARF and TAp73 Tumor Suppressor Genes by the Transcription Factor E2F1 Enables Discrimination of Cancer Cells from Normal Growing Cells
Yaxuan Zhou, Rinka Nakajima, Mashiro Shirasawa, Mariana Fikriyanti, Ako Watanabe, Caiwei Yang, Ritsuko Iwanaga, Andrew P. Bradford, Kenta Kurayoshi, Keigo Araki, Kiyoshi Ohtani

TL;DR
This study shows that cancer cells can be distinguished from normal cells based on unique activity of the E2F1 protein, which activates tumor suppressor genes only in cancer cells.
Contribution
The study identifies a unique E2F1 activity in cancer cells that activates tumor suppressor genes, enabling discrimination from normal cells.
Findings
Distinct E2F1 activity activates tumor suppressor genes ARF and TAp73 in cancer cells but not in normal cells.
Reporter assays confirmed that all 33 cancer cell lines tested have this unique E2F1 activity.
Normal cell lines lack this distinct E2F1 activity, suggesting it is a unique feature of cancer cells.
Abstract
Discrimination of cancer cells from normal growing cells is crucial to specifically target cancer cells. The transcription factor E2F1 is the principal target of the tumor suppressor pRB. E2F1 activated by growth stimulation activates cell cycle-related genes and facilitates cell proliferation. E2F1 activated by loss of pRB control, such as forced inactivation of pRB, activates tumor suppressor genes such as ARF and TAp73 and induces apoptosis. We show here that these genes are specifically activated by exogenously expressed E2F1 or forced inactivation of pRB but not by growth stimulation in epithelial cells. This observation indicates that E2F1 activity induced by forced inactivation of pRB contains distinct E2F1 activity that activates these tumor suppressor genes. Cancer cells survive with concomitant dysfunction of apoptosis-inducing pathways, suggesting the presence of distinct…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
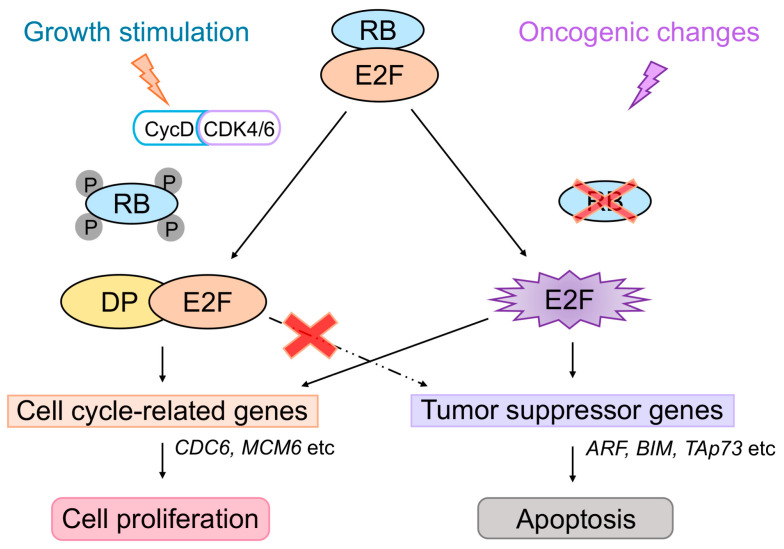 Figure 1
Figure 1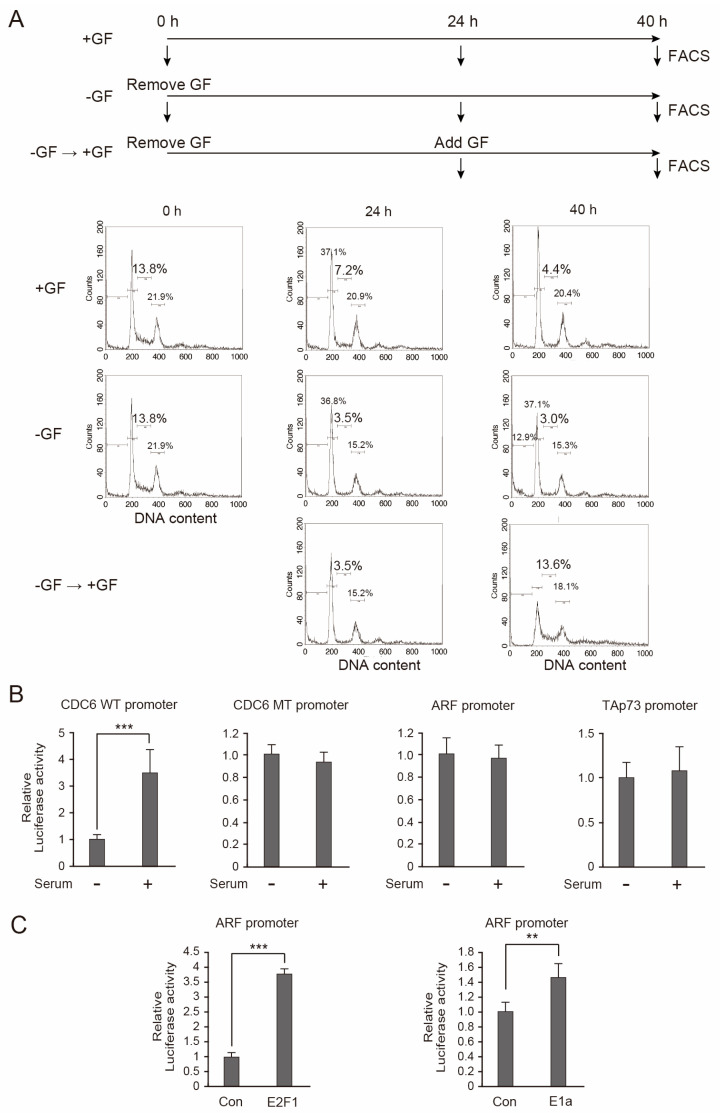 Figure 2
Figure 2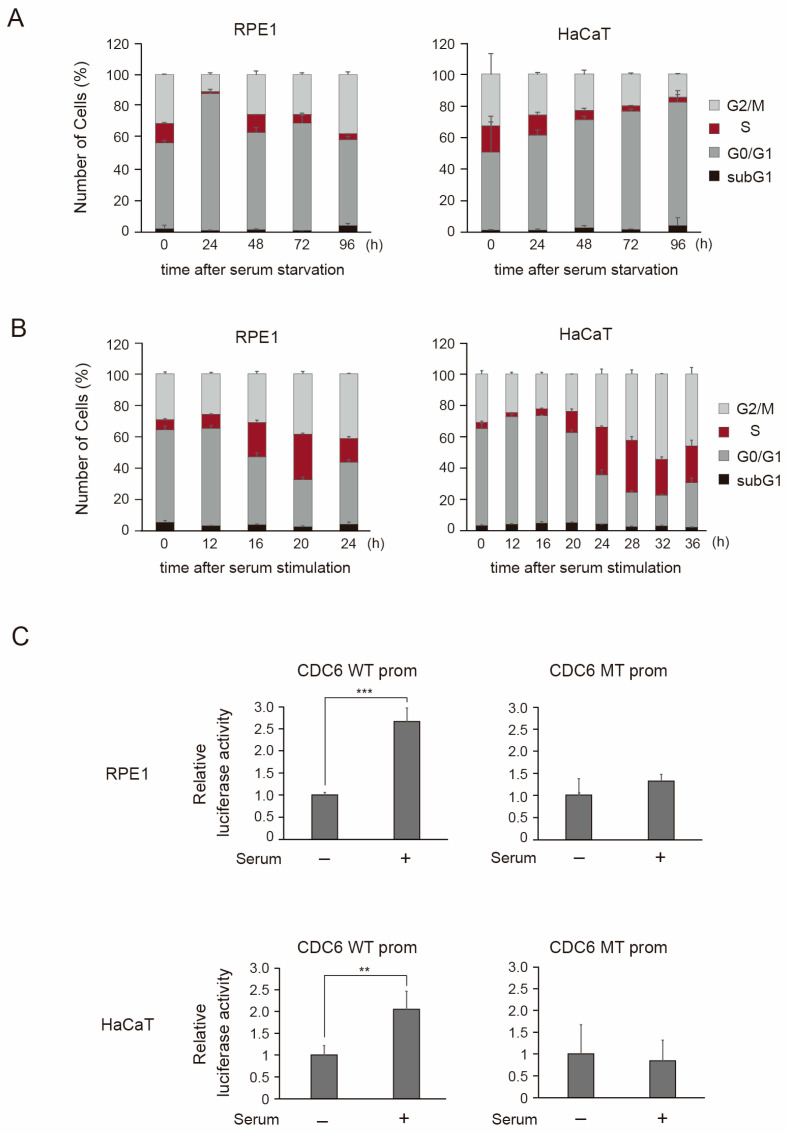 Figure 3
Figure 3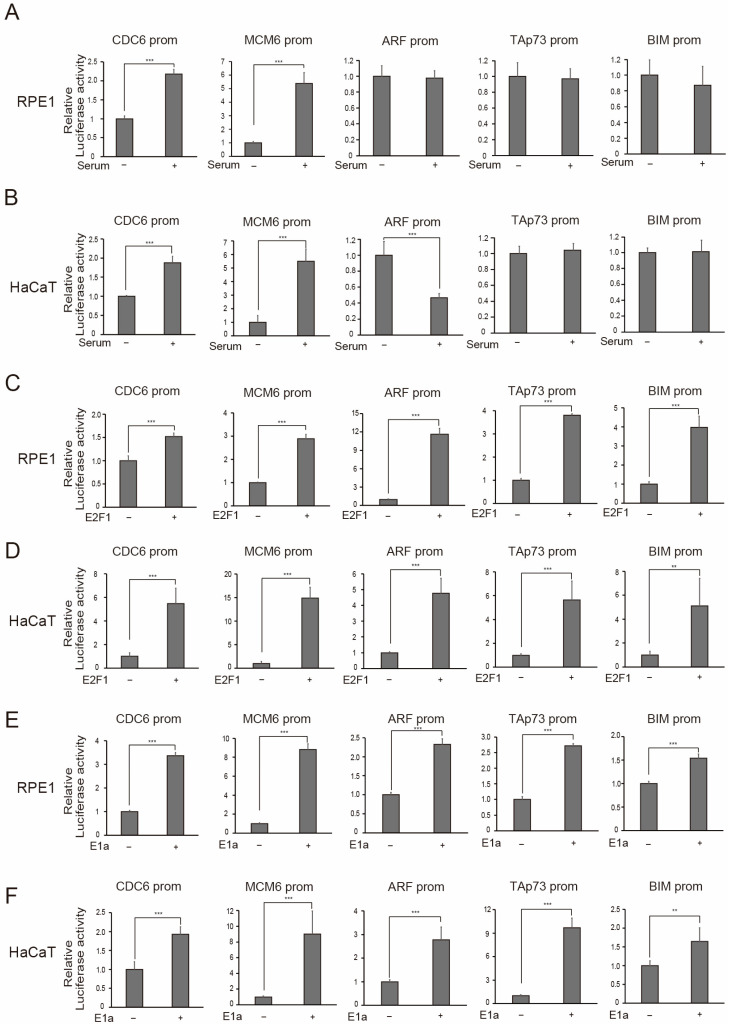 Figure 4
Figure 4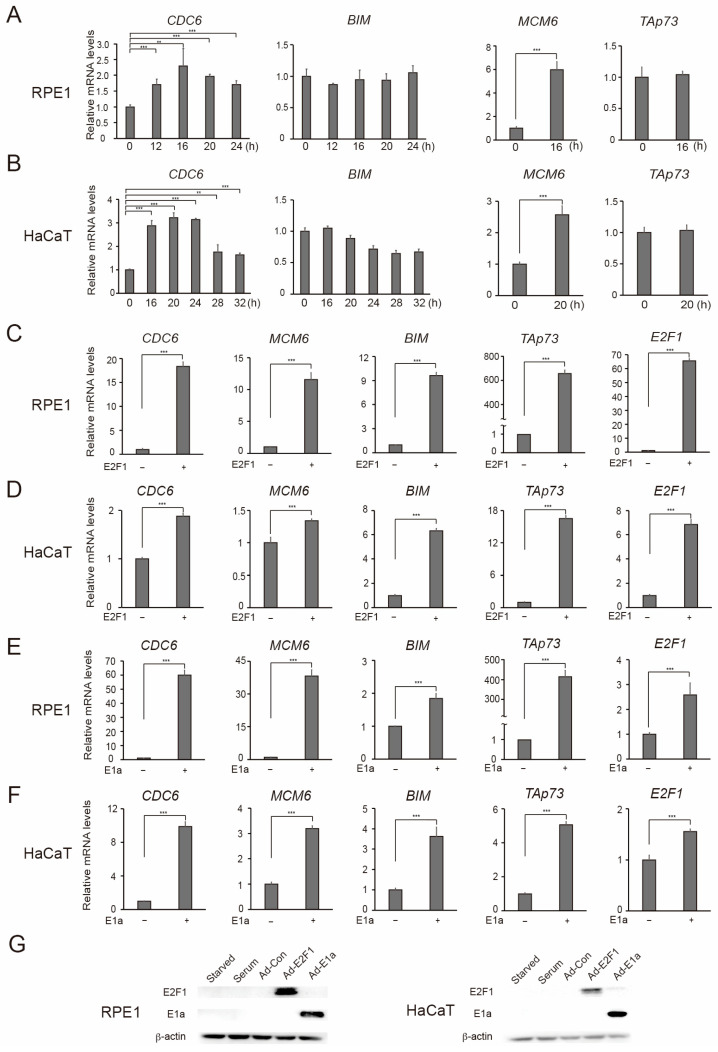 Figure 5
Figure 5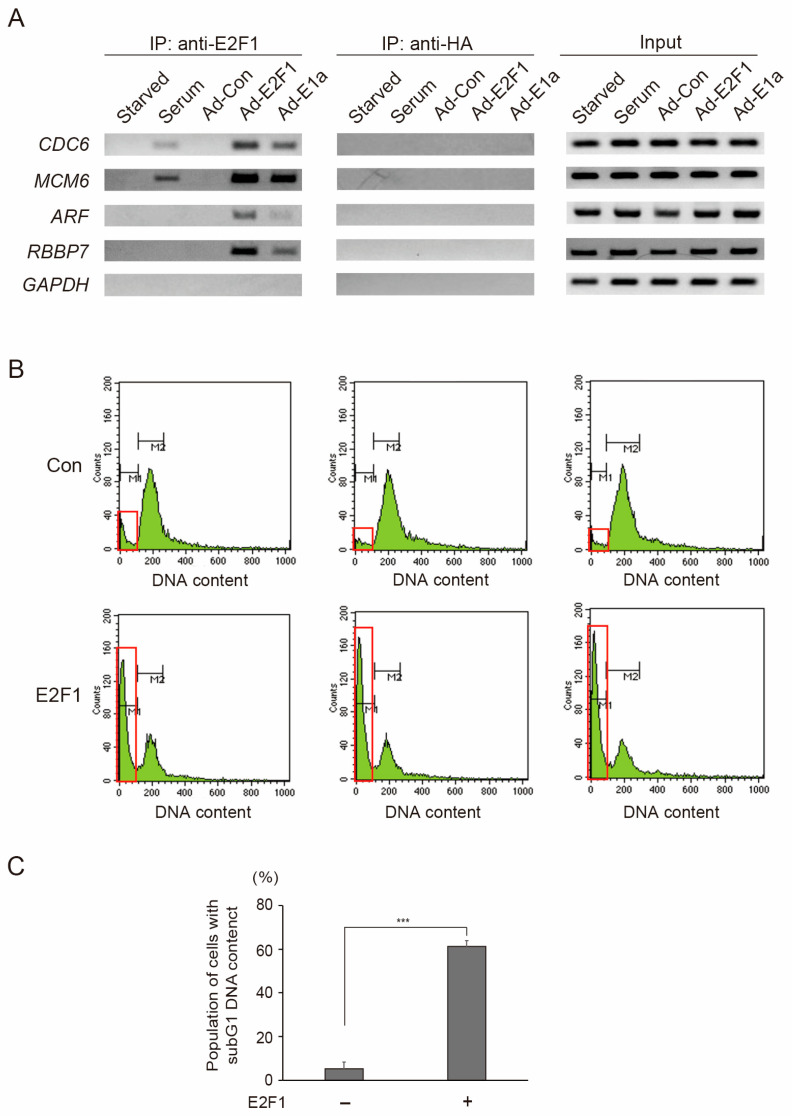 Figure 6
Figure 6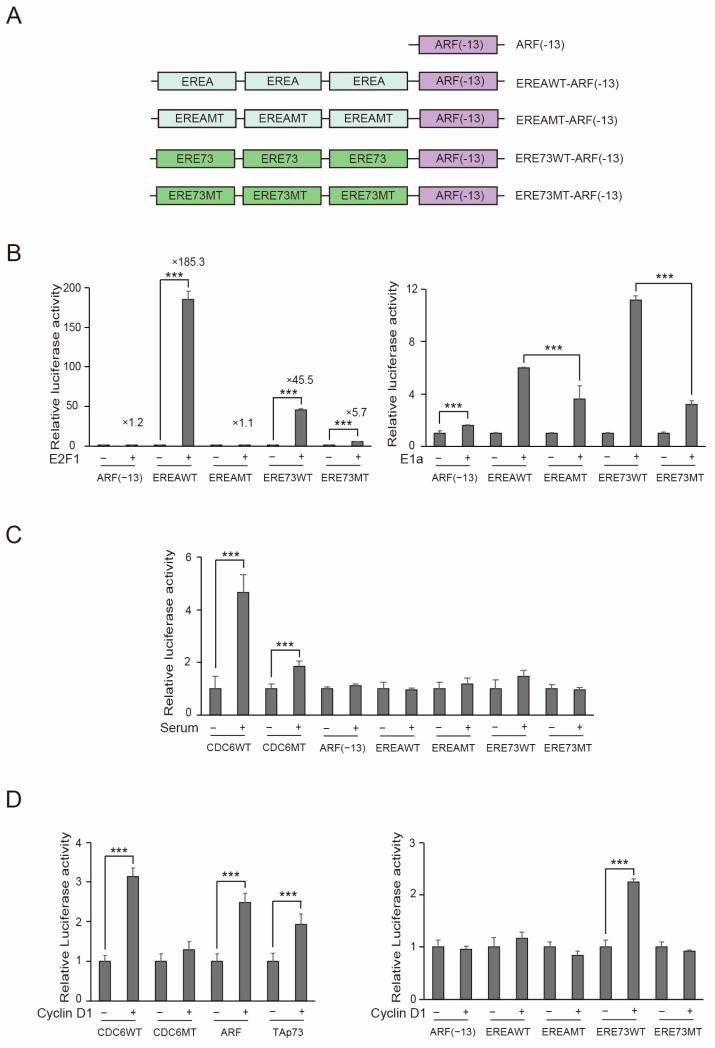 Figure 7
Figure 7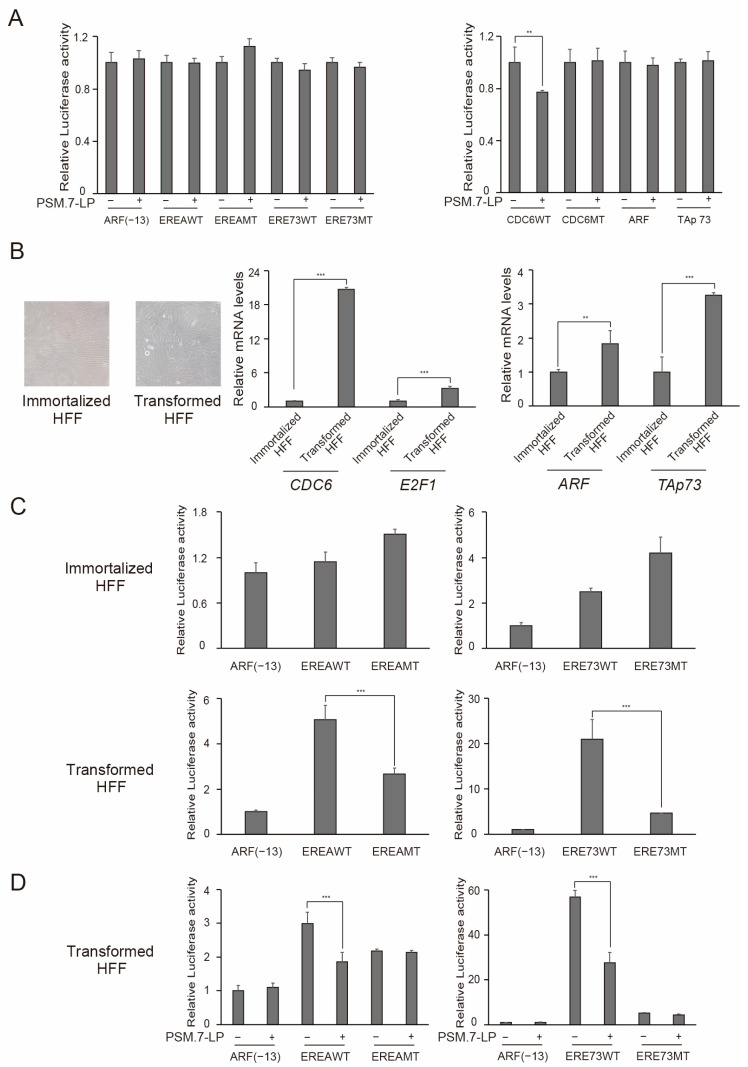 Figure 8
Figure 8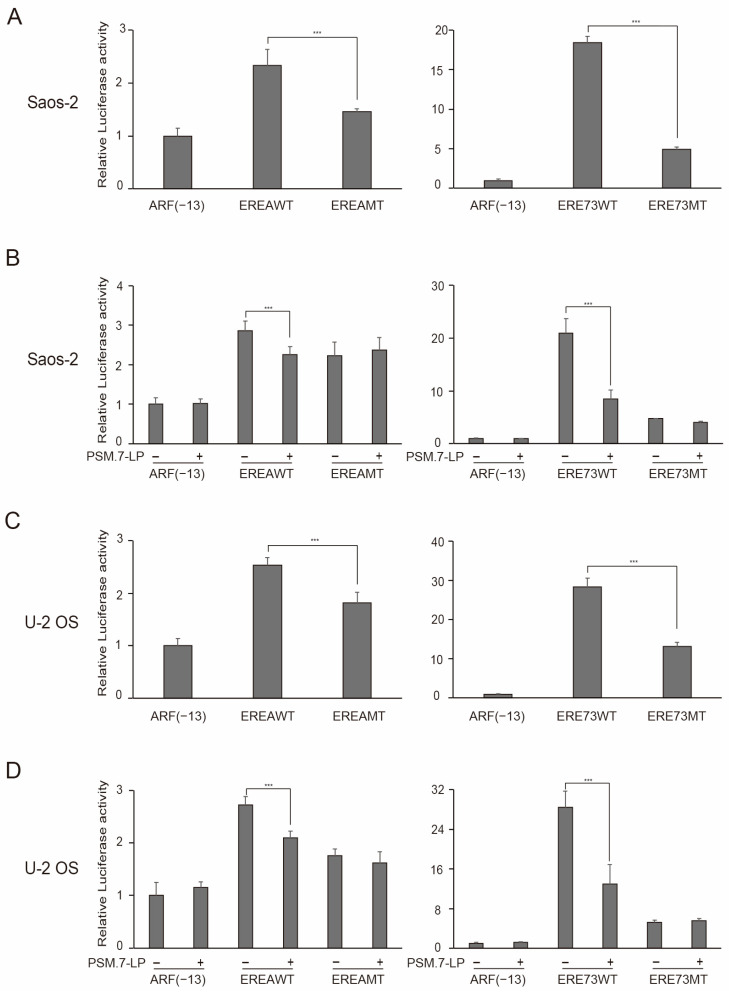 Figure 9
Figure 9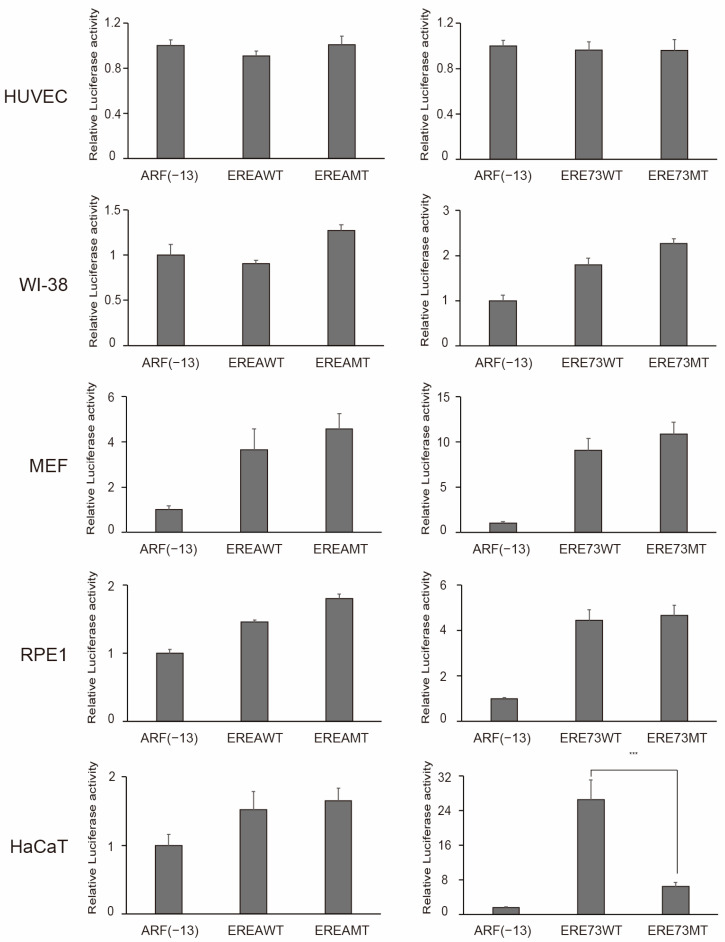 Figure 10
Figure 10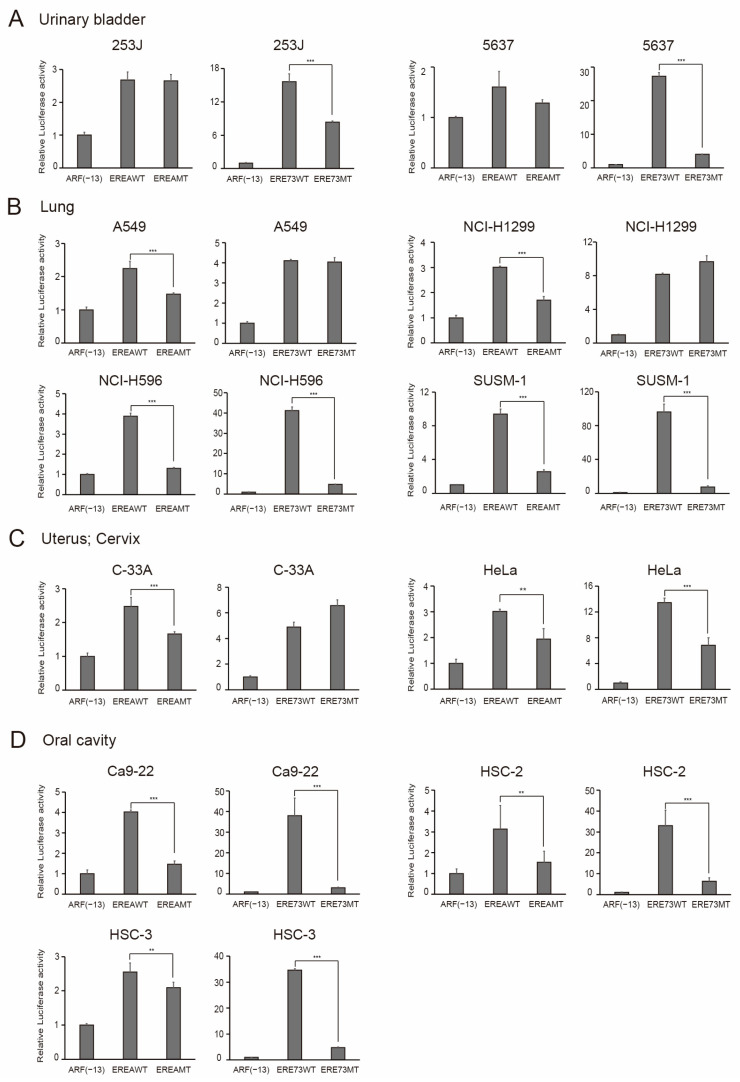 Figure 11
Figure 11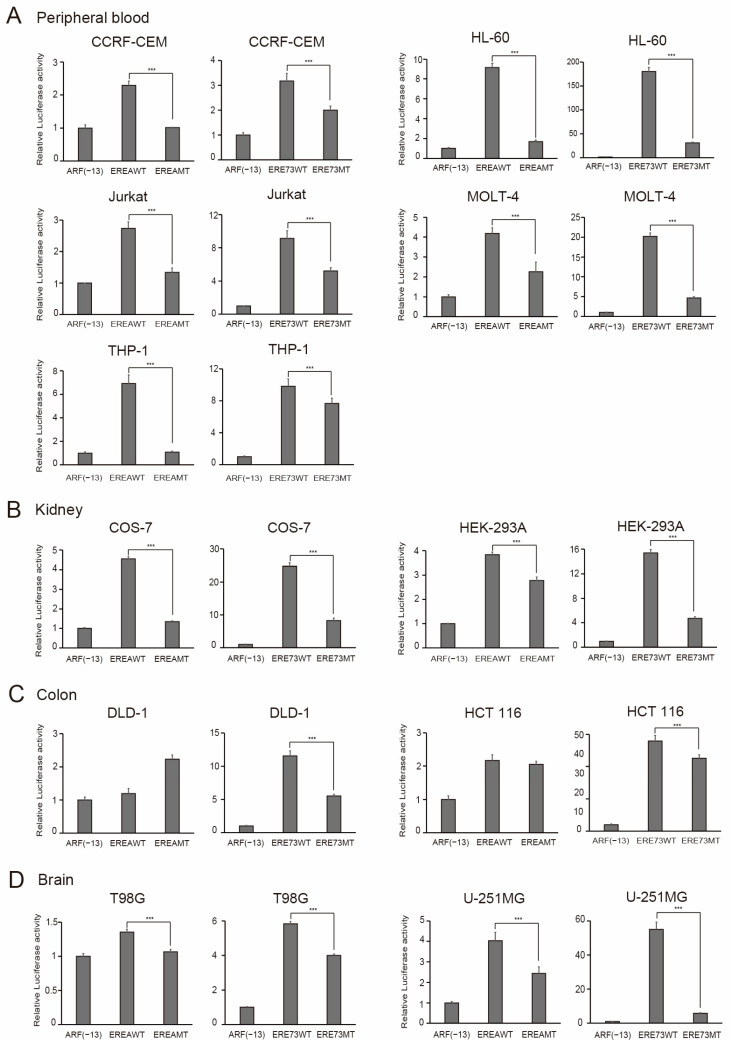 Figure 12
Figure 12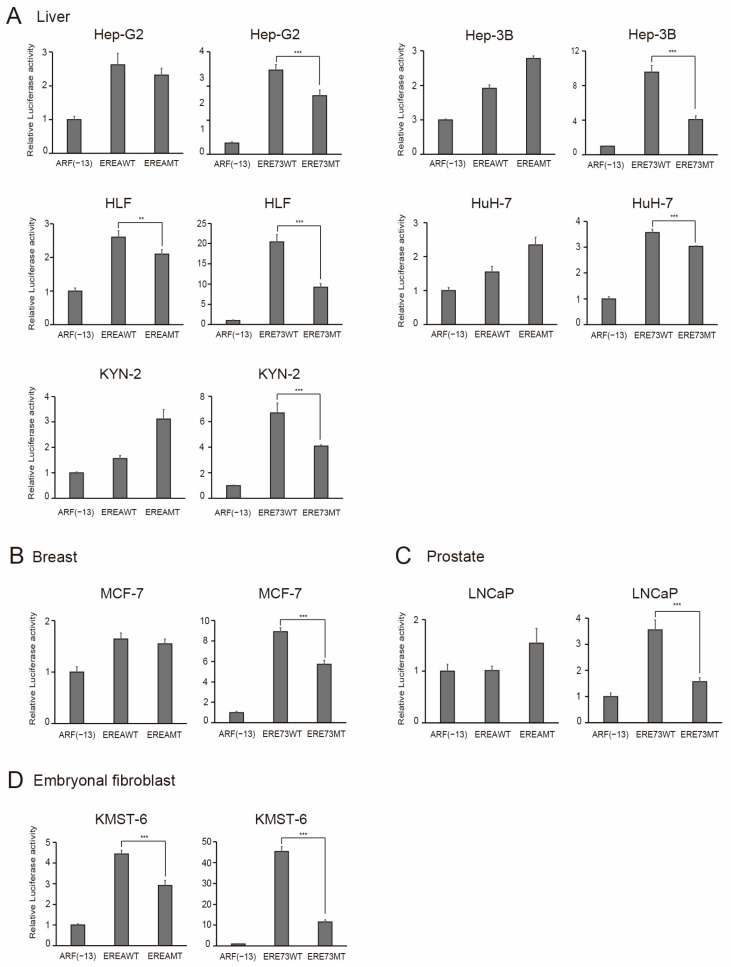 Figure 13
Figure 13- —Ministry of Education, Culture, Sports, Science, and Technology in Japan
- —ANRI fellowship and The Japan Science Society (The Sasagawa Scientific Research Grant)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related Molecular Pathways · Cell death mechanisms and regulation · Microtubule and mitosis dynamics
1. Introduction
The major barrier to cancer treatment lies in the side effects induced by current therapies like radiation and chemotherapy. These therapies preferentially induce death in proliferating cells by damaging the DNA, inhibiting the cellular metabolism, or suppressing mitosis. Hence, they injure not only cancer cells but also normal growing cells, such as epithelial cells and bone marrow cells, thereby leading to side effects that restrict the radical treatment of cancers. To circumvent such side effects and enable radical treatment, cancer cells must be specifically targeted, while preserving normal growing cells. In this context, discrimination of cancer cells from normal growing cells is crucial. However, there is currently no mechanism to universally differentiate the aberrant proliferation of cancer cells from normal cell proliferation.
E2F1-E2F5 of the E2F transcription factor family are the principal targets of the tumor suppressor pRB and its family members p130 and p107 (collectively referred to as RB) [1,2,3]. The E2F transcription factor family exerts essential effects on cell proliferation and tumor suppression and also performs important functions such as apoptosis, differentiation, DNA damage response, and metabolism [4,5,6,7,8,9,10,11,12,13,14,15]. The E2F transcription factor family is composed of eight E2F family members (E2F1–E2F8), which are grouped into activator E2Fs (E2F1–E2F3a) and repressor E2Fs (E2F3b–E2F8) according to their main functions. In the resting state of normal cells, E2F3b, E2F4, and E2F5 suppress target gene expression together with RB family members (E2F3b/pRB, E2F4/p130, and E2F5/p130). Expression of activator E2Fs (E2F1–E2F3a) are induced at the G1/S boundary of the cell cycle by the E2F family itself and contribute to the activation of target genes. When growth stimulation drives the physiological inactivation of pRB through CDK-mediated phosphorylation, activator E2Fs are physiologically activated and induce cell cycle-related genes and promotes cell proliferation. On the other hand, activator E2Fs activated by their exogenous expression or forced inactivation of pRB, such as by adenoviral oncoprotein E1a or shRNA against pRB, which resemble oncogenic changes, activate tumor suppressor genes, such as ARF, an upstream activator of the tumor suppressor p53 [16,17,18]. In this manner, activator E2Fs link two major tumor suppressors pRB and p53, thereby playing crucial roles in tumor suppression. Activator E2Fs also activate the tumor suppressor gene TAp73 coding for TAp73, a family member of p53, which can activate p53 target genes and induce apoptosis in a p53-independent manner [19,20,21,22,23]. Among activator E2Fs, E2F1 has the highest ability to activate pro-apoptotic genes [24,25] and plays a central role in E2F-mediated tumor suppression [26]. However, it is reported that E2F2 and E2F3 can also activate tumor suppressor genes [27]. We previously reported that regulation of these tumor suppressor genes by activator E2Fs is distinct from that of cell cycle-related genes. The ARF and TAp73 genes, unlike cell cycle-related genes, are activated by the over-expression of activator E2Fs and forced inactivation of pRB by adenovirus E1a but not by serum stimulation in human normal fibroblasts [18,22] (Figure 1). This distinct mode of activator E2F regulation of these tumor suppressor genes may serve to permit and preserve normal cell proliferation upon growth stimulation.
Although the mechanism underlying the distinct regulation of the ARF and TAp73 tumor suppressor genes remains to be elucidated, mediators of E2F activity, which activates these genes, appear distinct from E2F signaling induced by growth stimulation. Regulation of cell cycle-related target genes by activator E2Fs depends exclusively on DP as its heterodimeric partner [28,29], since the heterodimerization of activator E2Fs with DP is required for high affinity binding to E2F consensus binding sites (TTT^C^/G^G^/_C_CGC) in cell cycle-related genes [28]. In contrast, E2F1 activation of the ARF gene, whose E2F response element (EREA) lacks T-repeats and is mainly composed of GC-repetitive sequences [18], does not depend on DP, as shown by the knockdown of DP family members DP1 and DP2 [30]. DP knockdown reduced the expression of the cell cycle-related CDC6 gene, indicating that activation of the ARF gene by E2F1 is not due to the increased amount of E2F1/DP [30]. Moreover, EREA does not bind E2F1, induced by growth stimulation or repressor E2F4, and specifically binds exogenously expressed E2F1 or E2F1 activated by forced inactivation of pRB by adenovirus E1a, as shown by the chromatin immunoprecipitation assay [18]. Hence, although the exact molecular nature of E2F1 activity that activates the ARF and TAp73 genes has yet to be elucidated, these observations suggest that E2F1 activity induced by the exogenous expression of E2F1 or forced inactivation of pRB contains functionally and mechanistically distinct E2F1 activity from the E2F1 activity induced by growth stimulation. We refer to this E2F1 activity that activates the ARF and TAp73 genes, which is not induced by growth stimulation, as “distinct E2F1 activity”.
Across most cancers, the RB pathway and p53 pathway are deregulated, leading to the dysfunction of pRB and p53 [31]. If the E2F1-mediated distinct regulation of the ARF and TAp73 tumor suppressor genes applies to cell types other than fibroblasts, it is expected that the dysfunction of pRB in conjunction with the dysfunction of p53 will generate and sustain distinct E2F1 activity in cancer cells. Conversely, since distinct E2F1 activity is not induced by physiological growth stimulation, it should not be present in normal growing cells. Therefore, in theory, the presence of distinct E2F1 activity may serve to discriminate cancer cells from normal growing cells. However, whether the distinct regulation of the tumor suppressor genes by E2F1 also exists in other cell types, especially epithelial cells, from which 90% of cancers arise, has yet to be determined. In addition, it has yet to be elucidated whether the dysfunction of the pathway upstream of pRB generates distinct E2F1 activity, and the extent to which distinct E2F1 activity is present or absent in a variety of cancer cell lines and normal growing cells has not been extensively investigated.
In this study, we characterized the E2F1-mediated distinct regulation of the ARF and TAp73 tumor suppressor genes in multiple cell lines of epithelial origin. We also examined whether over-expression of cyclin D1, an important upstream component of the RB pathway, in human normal fibroblasts generates distinct E2F1 activity. Finally, we examined the presence of distinct E2F1 activity in a panel of 33 cancer cell lines and five normal growing cell types (Table 1), available in our laboratory, utilizing the E2F response elements of the ARF (EREA) and TAp73 (ERE73) genes, which specifically respond to distinct E2F1 activity, and the corresponding E2F1 binding site mutants. Our results show that the tumor suppressor genes were not activated by the physiological E2F1 activity induced by serum stimulation and activated by distinct E2F1 activity in epithelial cells. In addition, over-expression of cyclin D1 generated distinct E2F1 activity in human normal fibroblasts. We also found that the human keratinocyte cell line HaCaT, whose only known defect is the mutation of p53, possessed distinct E2F1 activity. Moreover, all 33 cancer cell lines tested possessed distinct E2F1 activity, while the five normal growing cell types did not. These results suggest that distinct E2F1 activity serves as a universal determinant, based on its fundamental mechanism of tumorigenesis, to distinguish between cancer cells and normal growing cells, and thereby provide means to specifically therapeutically target cancer cells, irrespective of tissue or cell types.
2. Materials and Methods
2.1. Cell Culture
The cell lines used in this study are listed in Table 1. Information about genetic mutations was mainly obtained from Cellosaurus and ATCC. Human umbilical vein endothelial cells (HUVECs, obtained from ATCC, PCS-100-010) were cultured in Vascular Cell Basal Medium (ATCC PCS-100-030) supplemented with 5 ng/mL recombinant human (rh) vascular endothelial growth factor (VEGF), 5 ng/mL rh epidermal growth factor (EGF), 5 ng/mL rh fibroblast growth factor (FGF) basic, 15 ng/mL rh insulin-like growth factor 1 (IGF-1), 10 mM L-glutamine, 0.75 Units/mL heparin sulfate, 1 µg/mL hydrocortisone hemisuccinate, 2% fetal calf serum (FCS), and 50 µg/mL ascorbic acid. To synchronize HUVECs in a resting state, the cells were cultured in Vascular Cell Basal Medium with the supplements without VEGF, EGF, and FGF basic for 24 h. To restimulate growth factor-deprived HUVECs, VEGF, EGF, and FGF basic were added and further cultured for 16 h. Other adherent cell lines including human normal fibroblasts (human foreskin fibroblasts: HFFs, obtained from ATCC, SCRC-1041), human retinal pigment epithelial-1 (RPE-1), and human keratinocyte HaCaT, except 5637 and LNCaP, were cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% FCS. To synchronize HFF, RPE-1, and HaCaT cells in a resting state, the cells were cultured in DMEM containing 0.1% FCS (serum starved condition) for 72 h. To re-stimulate the serum-starved cells, FCS was added at a final concentration of 10%. 5637, LNCaP, and hematopoietic cell lines CCRF-CEM, HL-60, Jurkat, MOLT-4, and THP-1 were cultured in RPMI 1640 medium containing 10% FCS.
2.2. Plasmid
pARF-Luc(−736), pTAp73-Luc(−892), pCMV-β-gal, and pENTR-E2F1 have been described previously [18,22]. pcDNA3-12SE1a(Δ2–11) is an expression vector for the Δ2–11 form of adenovirus E1A, which lacks binding ability to p300/CBP but retains that to RB family members [32]. EREAx3ARF(−13)-Luc, ERE73x3ARF(−13)-Luc, and those E2F binding site mutants were generated by inserting 3 tandem repeats of the E2F responsive element of ARF (EREA) [18] and the E2F responsive element of TAp73 (ERE73(1+2)) [22] into ARF(−13)-Luc [22]. The sequences are shown below. Sequences of the newly constructed plasmids were confirmed by sequencing.
EREA: GGCCCTGAGCCGCCCGCGCGCGCGCCTCC
EREAMT: GGCCCTGAGCCGCCCGATCGATCGCCTCC
ERE73: GGAGCGACGCGCGCCAAAAGGCGGCGGGAAGGA
ERE73MT: GGAGCGACGCGTTCCAAAAGGCGGTTGGAAGGA
pBabe-puromycin-hTERT is a retroviral expression vector, which contains puromycin resistance gene and hTERT cDNA [33]. pBabe-blasticidin-SV40ER contains blasticidin resistance gene and SV40 early region (ER), which expresses SV40 large T and small t [33]. pBabe-neomycin-Ras contains neomycin resistance gene and activated H-Ras (G12V) cDNA [33]. Amphotropic packaging plasmid is derived from the Moloney murine leukemia virus.
2.3. Immortalization and Transformation of HFFs
Immortalization and transformation of HFFs were performed as previously described [33,34]. HEK293T cells cultured in 10 cm dishes were transfected with 5 μg of pBabe-puromycin-hTERT, pBabe-blasticidin-SV40ER, or pBabe-neomycin-Ras along with 5 μg of amphotropic packaging plasmid using PEI Max (Polysciences, Warrington, PA, USA). The next day, the cells were washed with PBS, further cultured for 1 day in fresh media, and the culture media containing the virus were recovered. HFFs were first immortalized by infection with the hTERT expressing virus. HFFs were cultured in a 6-well plate, infected with 2 mL of the supernatant containing the hTERT expressing virus in the presence of polybrene (8 μg/mL) overnight and followed by selection with 0.5 μg/mL puromycin until all of control uninfected HFFs died (3 days). Similarly, the immortalized HFFs were transformed by sequential infection of the SV40ER and activated H-Ras expressing virus, followed by selection with 3 μg/mL blasticidin (7 days) and 400 μg/mL G418 (7 days), respectively.
2.4. Transfection and Reporter Assay
The cells were transfected with a reporter plasmid using PEI Max (Polysciences) with the ratio of DNA:PEI = 1:3. pCMV-β-gal was included as an internal control to monitor the transfection efficiency. For 60 mm dishes, the amounts of reporter plasmid and pCMV-β-gal were 1.7 μg and 0.3 μg, respectively. For 35 mm dishes, the amounts of reporter plasmid and pCMV-β-gal were 0.6 μg and 0.1 μg, respectively. For the co-transfection experiments, the amount of expression vector was 5 ng or 2 ng of pENTR-E2F1 or 100 ng or 30 ng of pcDNA3-12SE1a(Δ2–11) for 60 mm dishes or 35 mm dishes, respectively. The amount of expression vector was optimized for each vector depending on the effects. The next day, the cells were washed with PBS, further cultured for 1 day, and harvested. For serum stimulation experiments, the cells were split (1:15) into 60 mm or 35 mm dishes. The next day, the medium was changed to DMEM containing 0.1% FCS for 2 days to synchronize cells in the quiescent state. The synchronous RPE1 and HaCaT cells were similarly transfected with reporter and internal control plasmids using PEI Max. After 12 h, the cells were washed with PBS, further cultured in DMEM containing 0.1% or 10% FCS, and harvested after 16 h or 20 h, respectively. For hematopoietic cell lines (CCRF-CEM, HL-60, Jurkat, MOLT-4, THP-1), transfection was performed by the DEAE-dextran method. The cells (2.5 × 10^6^) were transfected in 2 mL of RPMI-1640 medium containing 50 mM Tris-HCl (pH 7.4), 200 μg/mL DEAE-dextran (Sigma-Aldrich, St. Louis, MO, USA), 2.5 μg of reporter plasmid, and 2.5 μg of pEF1-LacZ as an internal control at 37 °C for 30 min. After neutralizing DEAE-dextran by adding 5 mL of RPMI 1640 medium containing sodium heparin (5 U/mL), the cells were washed with RPMI 1640 medium, cultured in 10 mL of RPMI 1640 medium containing 10% FCS for 24 h, and harvested. Luciferase activities were assayed using the Luciferase Assay System (Promega, Madison, WI, USA) and normalized to β-galactosidase activities. All assays were performed in biological triplicate, and the results are presented as means ± SD.
2.5. Quantitative Reverse Transcription (qRT)-PCR Analysis
Total RNA was extracted using Isogen II (Nippon Gene) according to the protocol provided by the manufacturer. The quality of RNA was verified by the A260/280 ratio more than 1.8 using the spectrophotometer Smartspec Plus (Bio-Rad, Hercules, CA, USA). First strand cDNA was synthesized using the PrimeScript 1st strand cDNA Synthesis Kit (Takara Bio, San Jose, CA, USA) from 500 ng of total RNA with oligo(dT) primer and random primers in 10 μL reaction volume. The reactions were 37 °C for 15 min, 42 °C for 15 min, 50 °C for 5 min, 85 °C for 15 s, and then kept at 4 °C. After the reaction, the samples were diluted 5 times with TE buffer and stored at −20 °C. Quantitative PCR was performed using 2 μL of each sample, PowerUp SYBR Green Master Mix (applied biosystems, Waltham, MA, USA) and QuantStudio 3 (Applied Biosystems Inc., Carlsbad CA, USA). The amplification program consisted of a hold stage at 95 °C for 60 s, followed by 45 cycles of 95 °C for 10 s, annealing temperature for 30 s, and 75 °C for 30 s. Amplification specificity was confirmed by melting curve analysis (95 °C for 15 s, 60 °C for 30 s, and 95 °C for 15 s). Relative quantification was calculated using the ΔΔCt method, with GAPDH as the internal control gene. All experiments were performed in three biological replicates and three technical replicates, and data are presented as the mean ± standard deviation (SD). Statistical analysis was conducted using Student’s t-test, with p < 0.05 considered statistically significant. The gene specific primer sets for CDC6, ARF, TAp73, BIM, and GAPDH have been described [35]. The gene specific primer sets for E2F1 and MCM6 genes are listed below.
E2F1 (GenBank accession number: NM_005225, annealing temperature: 56 °C, product length: 84 bp)
Fw: 5′-CCTGGAAACTGACCATCAGTACCT-3′ (nucleotide 400–423)
Rv: 5′-GGATTTCACACCTTTTCCTGGAT-3′ (nucleotide 484–452)
MCM6 (GenBank accession number: NM_005915, annealing temperature: 54.9 °C, product length: 160 bp)
Fw: 5′-GAACGGGATCAATGGCTACAATG-3′ (nucleotide 2157–2179)
Rv: 5′-GCTCGCTCCTCTTTAATGCTGACT-3′ (nucleotide 2316–2297)
The results are presented as relative expression levels or fold-induction by serum stimulation, E2F1, or E1a.
2.6. Immunoblot Analysis
The proteins were extracted with 5 times volumes of cell pellets of RIPA buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS)) on ice for 30 min. Protein concentrations were measured using the Protein Assay Dye Reagent Concentrate (Bio-Rad) according to the protocol provided by the manufacturer. Thirty μg of each protein sample was separated on 7.5% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred to Immobilon-P PVDF membrane (Millipore, Burlington, MA, USA) using Transblot SD semi-dry transfer cell (Bio-Rad) according to the protocol provided by the manufacturer. The transferred membrane was blocked with 5% skim milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) at room temperature for 30 min. After washing with TBS-T, the membrane was incubated with first antibody in TBS-T containing 0.25% skim milk at 4 °C overnight. The membrane was washed with TBS-T and incubated with secondary antibody in TBS-T containing 0.25% skim milk at room temperature for 1 h. The membrane was washed with TBS-T, treated with ImmunoStar LD (Fujifilm, Tokyo, Japan), and the luminescent signals were detected using LAS4000 (GE Healthcare, Chicago, IL, USA). The signals were quantified by ImageJ 1.51s (NIH). The antibodies used were anti-E2F1 (sc-251, Santa Cruz Biotechnology, 1:250, Dallas, TX, USA), anti-E1a (554155, BD Biosciences, 1:250, San Jose, CA, USA), anti-β-actin (A1978, SIGMA, 1:2000, Kawasaki, Japan), and anti-mouse IgG-HRP (Jackson ImmunoResearch, 1:1000, West Grove, PA, USA).
2.7. Infection with Recombinant Adenovirus
Recombinant adenovirus expressing E2F1 (Ad-E2F1), the Δ2–11 form of adenovirus 12S E1a (Ad-12SE1a(Δ2–11)), and the control virus (Ad-Con) were described previously [18,22]. The viruses were expanded in HEK293A cells and purified by 2 rounds of ultracentrifuge using a discontinuous CsCl density gradient. The titer of the purified virus was measured by infection of 293A cells with the serially diluted virus followed by immunofluorescence staining using rabbit polyclonal antibodies against the adenovirus E2 gene product. The titers of the viruses were Ad-E2F1; 8.5 × 10^10^/mL, Ad-12SE1a(Δ2–11); 1.5 × 10^11^/mL, and Ad-Con; 4.8 × 10^10^/mL. The cells were infected with the recombinant adenoviruses in 0.5 mL DMEM for 60 mm dishes containing the indicated multiples of infection (MOI) for 1 h. The cells were further cultured in DMEM containing 0.1% or 10% FCS for the indicated times before harvesting.
2.8. FACS Analysis
Cells were fixed with 70% ethanol and stained with propidium iodide (50 μg/mL) containing RNase (50 μg/mL). Cell samples were analyzed with a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA). We set the gates manually for subG0/G1, G0/G1, S, and G2/M phases using a control untreated sample for each cell line. Once the gates were set, all samples of the same cell line were examined with the same gates. For RPE-1 and HaCaT cells, assays were performed in biological triplicate, the % population of cells in each phase was measured, and the values are shown as means ± SD.
2.9. Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was carried out as previously described [18]. PCR amplification was performed with KOD FX Neo (TOYOBO, Osaka, Japan). Gene specific primer sets for the ARF, RBBP7, CDC6, MCM6, and GAPDH genes have been previously described [18,22,36]. The antibodies used for immunoprecipitating protein-DNA complexes were anti-E2F1 (sc-251, Santa Cruz) and anti-HA (sc-7392, Santa Cruz) as a negative control. Input was one 10th of the lysates. The sizes of the PCR products were confirmed by gel electrophoresis.
2.10. Statistical Analysis
Reporter assays, qRT-PCR, and FACS analysis were conducted in biological triplicate. Data are presented as the means ± SD. Statistical comparisons were made using Student’s t-test and Bonferroni correction for multiple comparison. p value < 0.05 was considered significant.
3. Results
3.1. The ARF Promoter Is Specifically Activated by Distinct E2F1 Activity in HUVECs
To determine whether the ARF and TAp73 tumor suppressor genes are regulated by E2F1 in a distinct manner from cell cycle-related genes in other cell types than fibroblasts, we first used primary human umbilical vein endothelial cells (HUVECs). HUVECs were cultured in Vascular Cell Basal Medium (VCBM) supplemented with vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), fibroblast growth factor (FGF) basic, insulin-like growth factor 1 (IGF-1), and hydrocortisone hemisuccinate, in addition to L-glutamine, heparin sulfate, FCS, and ascorbic acid. We first examined whether HUVECs could be starved of growth factors to arrest in a resting state without apparent cell death and subsequently re-stimulated to re-enter into the cell cycle by re-addition of the growth factors, in order to monitor the physiological E2F activity induced by growth simulation. To identify the conditions to arrest HUVECs in the resting state and re-stimulate the cell cycle, we examined the systematic removal and re-addition of the above growth factors. In this manner, we found that withdrawal of VEGF, EGF, and FGF, while retaining IGF-1, arrested HUVECs in the resting state (population of cell in S phase 3.5%), although the arrest may not be complete, without significant cell death (at 24 h in −GF). Accordingly, subsequent addition of VEGF, EGF, and FGF caused the HUVECs to re-enter the cell cycle (the population of cells in S phase increased from 3.5% to 13.6% at 40 h in −GF → +GF) (Figure 2A). Using this approach, we examined whether growth stimulation activates cell cycle-related and tumor suppressor gene promoters. The CDC6 promoter served as a typical example of a cell cycle-related gene promoter, while the ARF and TAp73 promoters represented tumor suppressor genes. Asynchronously growing HUVECs were transfected with reporter plasmid. The next day, the cells were washed with PBS and further cultured in the media without VEGF, EGF, and FGF for 24 h. VEGF, EGF, and FGF were re-added into the media, and the cells were harvested after 16 h. The addition of growth factors activated the CDC6 promoter about 3.5-fold, but they had no significant effect on a CDC6 promoter construct with mutations in the E2F binding sites (Figure 2B) [37]. These results suggest that growth factor stimulation of HUVECs generated physiological E2F1 activity to activate cell cycle-related genes. Under the same conditions, growth simulation failed to activate the ARF or TAp73 promoter (Figure 2B), suggesting that physiological E2F1 activity, induced by growth simulation, does not activate these tumor suppressor gene promoters in HUVECs. We next examined whether distinct E2F1 activity activates the ARF promoter. The exogenous expression of E2F1 activated the ARF promoter about 3.8-fold and expression of adenovirus E1a, which forcedly inactivates pRB and activates endogenous E2F1, activated the ARF promoter about 1.5-fold (Figure 2C). These results provide further evidence that, in HUVECs, the ARF tumor suppressor promoter is activated by distinct E2F1 activity but not by physiological E2F1 activity, induced by growth stimulation, suggesting that the regulation of tumor suppressor genes by E2F1 is distinct from that of cell cycle-related genes in endothelial cells.
3.2. RPE1 and HaCaT Epithelial Cells Can Be Synchronized in G0/G1 Phase by Serum Starvation and Subsequently Induced to Resume the Cell Cycle Through Serum Addition
The requirement for specialized media, expensive growth factor supplementation, and large-scale culture, precluded the use of the HUVEC model to biochemically analyze the distinct molecular mechanisms underlying E2F1 dependent gene regulation. We thus searched for epithelial cell lines, which could be cultured with addition of FCS alone without additional growth factors. We identified the hTERT-immortalized retinal pigment epithelial cell RPE1 and the immortalized human keratinocyte HaCaT cell line as candidates. RPE1 was successfully serum-starved and re-stimulated by serum in a previous report [38]. HaCaT has been widely used as a human keratinocyte cell line, which can be differentiated to study the differentiation of human keratinocytes, suggesting its normal phenotype. However, HaCaT cells have two mutant p53 alleles, R282Q and H179Y [39], suggesting that they may not be totally normal cells. We first examined whether serum starvation could arrest both cell lines in G0/G1. Asynchronously growing RPE1 and HaCaT cells were starved of serum, and the cells were harvested at the indicated time points to examine the cell cycle phase distribution, by FACS analysis of the DNA content. In RPE1 cells, the population of cells in the S phase almost disappeared after 24 h (Figure 3A). Intriguingly, however, a population of cells in the S phase re-appeared at 48 h and then gradually decreased until 96 h (Figure 3A). In HaCaT cells, the population of cells in the S phase gradually decreased over 96 h (Figure 2B). The population of cells in the S phase was approximately the same at both 72 h and 96 h, and the subG1 population of cells (dead cells) apparent at 96 h was higher than that at 72 h, in both cell lines. These results indicate that RPE1 and HaCaT cells can be arrested by deprivation of FCS for 72 h, without significant cell death.
We next examined whether serum addition induces cell cycle re-entry in serum-starved RPE1 and HaCaT cells. RPE1 and HaCaT cells, starved of serum for 72 h, were treated with serum and collected at the designated time intervals. The cell cycle distribution was examined by FACS analysis. In RPE1 cells, the population of cells in the S phase gradually increased, reaching a maximum (about 29%) at 20 h after serum stimulation and then decreased at 24 h (Figure 3B). In HaCaT cells, the fraction of cells in the S phase gradually increased, although more slowly than RPE1, and reached a maximum (about 33%) at 28 h after serum stimulation, followed by a slight decrease at 32 h (Figure 3B). These results indicated that both serum-starved RPE1 and HaCaT cells can be re-simulated by serum to re-enter the cell cycle, suggesting that physiological E2F activity, induced by growth stimulation, could be effectively monitored using these epithelial cell lines.
To confirm this, we performed reporter assays using the wild type CDC6 promoter and its analogous E2F-binding sites mutant. Serum-starved RPE1 and HaCaT cells were transfected with the reporter plasmids, then re-stimulated with serum or left starved, and harvested after 20 h or 28 h, respectively. Wild type CDC6 promoter activity was increased three- and four-fold, respectively, whereas the E2F-binding sites mutant was not significantly activated (Figure 3C). These results indicate that, in both RPE1 and HaCaT cells, serum stimulation induces physiological E2F activity, which activates the cell cycle-related CDC6 promoter.
3.3. ARF, TAp73, and BIM Tumor Suppressor Gene Promoters Are Specifically Activated by Distinct E2F1 Activity in RPE1 and HaCaT Cells
Using RPE1 and HaCaT cells, we next examined the response of tumor suppressor gene promoters to physiological E2F activity, induced by serum stimulation and the exogenous expression of E2F1, or E2F1 activity induced by the expression of adenovirus E1a. We used ARF, TAp73, and BIM as representatives of tumor suppressor gene promoters and CDC6 and MCM6 as representative cell cycle-related gene promoters.
We first evaluated the influence of serum stimulation on these reporters. In RPE1 cells, serum stimulation activated the cell cycle-related CDC6 and MCM6 promoters about 2.2-fold and 5.4-fold, respectively (Figure 4A). Under the same conditions, serum stimulation did not activate either the ARF, TAp73, or BIM promoters (Figure 4A). Analogous results were observed in HaCaT cells (Figure 4B). The ARF promoter showed rather decreased activity upon serum stimulation. Taken together, these results suggest that the ARF, TAp73, and BIM tumor suppressor gene promoters are not activated by serum stimulation in either RPE1 or HaCaT cells.
We next examined the responsiveness of the reporters to the exogenous expression of E2F1 or E2F1 activity induced by expression of adenovirus E1a. The exogenous expression of E2F1 activated not only the CDC6 and MCM6 promoters but also the ARF, TAp73, and BIM promoters in RPE1 (Figure 4C) and HaCaT cells (Figure 4D). The expression of E1a also activated not only the CDC6 and MCM6 promoters but also the ARF, TAp73, and BIM promoters in both RPE1 (Figure 4E) and HaCaT cells (Figure 4F). These results demonstrate that the tumor suppressor gene promoters are specifically activated by distinct E2F1 activity in RPE1 and HaCaT cells.
3.4. Endogenous Tumor Suppressor Genes Are Specifically Activated by Distinct E2F1 Activity in RPE1 and HaCaT Cells
To verify the results obtained by the reporter assay, we next examined the response of endogenous cell cycle-related and tumor suppressor genes to physiological E2F1 activity induced by serum stimulation and distinct E2F1 activity in RPE1 and HaCaT cells. We first examined the kinetics of cell cycle-related CDC6 and tumor suppressor BIM gene expression after serum stimulation of quiescent RPE1 cells (Figure 5A left panels). Since expression of the ARF gene was not detected in both RPE1 and HaCaT cells, likely due to extremely low expression levels, we examined the BIM and TAp73 genes, which respond to serum simulation and distinct E2F1 activity in a similar manner as the ARF gene in human normal fibroblasts [22,35]. Expression of the CDC6 gene was induced by serum stimulation and reached a peak (about 2.3-fold) at 16 h. In contrast, expression of the BIM gene was not induced throughout this time course. Similarly, in HaCaT cells, expression of the CDC6 gene was induced by serum stimulation, peaking (about 3.3-fold) at 20 h. In contrast, expression of the BIM gene was not significantly increased at this time point (Figure 5B left panels). Coincident with the maximal expression of the CDC6 gene, expression of the cell cycle-related MCM6 gene was also increased, but the tumor suppressor TAp73 gene was not affected in either RPE1 or HaCaT cells (Figure 5A,B right panels). These results suggest that serum simulation induces cell cycle-related E2F target genes, but not BIM or TAp73 tumor suppressor genes, in these epithelial cells. We next examined the expression of these genes in response to the exogenous expression of E2F1 and the expression of adenovirus E1a in RPE1 and HaCaT cells. In both cell lines, expression of not only CDC6 and MCM6 but also BIM and TAp73 genes was induced by exogenous expression of E2F1 (Figure 5C,D) and expression of E1a (Figure 5E,F). We confirmed the expected elevated E2F1 and E1a protein levels in RPE1 and HaCaT cells (Figure 5G). Based on these findings, we conclude that, in RPE1 and HaCaT cells, the BIM and TAp73 tumor suppressor genes are specifically activated by distinct E2F1 activity.
3.5. ARF and RBBP7 Genes Specifically Bound Exogenously Expressed E2F1 and E2F1 Activated by Expression of E1a in RPE1 Cells
We confirmed that the ARF and RBBP7 tumor suppressor genes specifically bound exogenously expressed E2F1 and E2F1 activated by expression of E1a but not E2F1 physiologically activated by serum stimulation, by chromatin immunoprecipitation (ChIP) assay, using RPE1 cells (Figure 6A). As opposed to the cell cycle-related CDC6 and MCM6 genes, which bound physiological E2F1 induced by serum stimulation, tumor suppressor ARF and RBBP7 genes, which are specifically activated by distinct E2F activity [18,35], did not bind physiological E2F1. Both tumor suppressor (ARF and RBBP7) and cell cycle-related (CDC6 and MCM6) genes bound E2F1, induced by the exogenous expression of E2F1 or expression of adenovirus E1a. These results suggest that the ARF and RBBP7 tumor suppressor genes exhibited specific binding to exogenously expressed E2F1 and E2F1 activated by expression of E1a in epithelial cells.
FACS analysis of the sub G1 DNA content showed that the exogenous expression of E2F1 induced a dramatic increase in cell death in RPE1 cells (Figure 6B,C), confirming that distinct E2F1 activity can also induce apoptosis in epithelial cells.
Collectively, these findings confirm that the tumor suppressor E2F targets are specifically activated by distinct E2F1 activity in epithelial cells, from which 90% of cancers arise. Since growth stimulation did not activate the tumor suppressor genes, E2F1 activity induced by exogenous expression of E2F1 or expression of E1a is thought to contain distinct E2F1 activity that activates the tumor suppressor genes, which is not induced by growth stimulation. We refer to this E2F1 activity that activates the tumor suppressor genes, which is not induced by growth stimulation, as “distinct E2F1 activity”.
3.6. EREA and ERE73 Reporters Specifically Sense Distinct E2F1 Activity
In most cancers, the two key tumor suppressive pathways, the RB and p53 pathways, are deregulated, leading to the dysfunction of pRB and p53 [31]. Dysfunction of pRB is thought to generate distinct E2F1 activity, which is sustained by the concomitant disabling of p53, to facilitate the growth and survival of cancer cells. Since growth stimulation does not generate distinct E2F1 activity, it is hypothesized that distinct E2F1 activity is not present in normal growing cells. Therefore, in theory, the presence of distinct E2F1 activity is a unique feature of cancer cells enabling their discrimination from normal growing cells. To test this possibility, we generated EREA and ERE73 reporters, which specifically sense distinct E2F1 activity. EREA is the E2F-response element of the ARF gene, which specifically responds to distinct E2F1 activity and is unresponsive to the physiological E2F1 activity induced by growth stimulation [18], suggesting that EREA specifically senses distinct E2F1 activity. Similarly, ERE73, in this case ERE73(1+2), is the corresponding E2F-response element of the TAp73 gene [22]. Three tandem repeats of EREA or ERE73 were inserted upstream of the ARF(-13) core promoter, which lacks E2F responsiveness [22] (Figure 7A). As controls, we generated E2F-binding sites mutants of EREA and ERE73, shown to abolish E2F-responsiveness in our previous studies [18,22] (Figure 7A). Theoretically, the presence of distinct E2F1 activity in a cell would be characterized by lower activity of ERE mutant promoters, relative to wild type constructs. Conversely, cells lacking distinct E2F1 activity would exhibit equivalent wild type and mutant promoter activity.
We first characterized the wild type reporters in human normal fibroblasts (HFFs) to see whether they specifically respond to distinct E2F1 activity. EREA and ERE73 reporters were dramatically activated by the over-expression of E2F1, while activation of the point mutants was significantly attenuated, although the ERE73 point mutant retained some response to E2F1 (Figure 7B left panel). Unexpectedly, expression of adenovirus E1a significantly enhanced ARF(−13) core promoter activity, making interpretation of the E1a responsiveness of point mutants difficult (Figure 7B right panel). Activation of the ARF(−13) core promoter by E1a is not thought to be mediated through E2F, since E2F1, the strongest activator of the ARF promoter barely activated the ARF(−13) core promoter (Figure 7B, left panel). Although the exact reason for the enhancement of ARF basal promoter activity is not known, the point mutations did significantly reduce the E1a responsiveness of both EREA and ERE73 reporters. Thus, comparison of wild type and mutant promoters to ascertain the presence of distinct E2F1 activity remains feasible. Serum stimulation activated the CDC6 promoter about 4.7-fold and its E2F binding site mutant about 1.9-fold (Figure 7C). Under the same conditions, EREA and ERE73 reporters were not significantly activated by serum stimulation (Figure 7C). These results indicate that the EREA and ERE73 reporters specifically sense distinct E2F1 activity.
3.7. Exogenous Expression of Cyclin D1 Generates Distinct E2F1 Activity
Approximately 30% of cancers harbor pRB deletions or mutations. We have reported that forced inactivation of pRB by adenovirus E1a generates E2F1 activity that activates the ARF, TAp73, BIM, and RBBP7 genes, which are not activated by growth simulation in human normal fibroblasts [18,22,35], suggesting that the forced inactivation of pRB generates distinct E2F1 activity in human normal fibroblasts. However, it is not known whether the dysfunction of upstream elements of the RB pathway also generates distinct E2F1 activity. To test this possibility, we investigated whether the exogenous expression of cyclin D1, which activates CDK4/6 and inactivates pRB, generates distinct E2F1 activity, using a reporter assay. After introduction of reporter plasmid and cyclin D1 expression vector, HFFs were cultured under the serum-starved condition to suppress endogenous E2F activity. Exogenous expression of cyclin D1 activated not only the cell cycle-related CDC6 promoter but also the ARF and TAp73 promoters (Figure 7D, left panel) and the ERE73 reporter in serum-starved HFFs (Figure 7D, right panel). These results indicate that the deregulation of upstream regulatory factors of pRB has the potential to generate distinct E2F1 activity.
3.8. Transformation of HFFs Generates Endogenous Distinct E2F1 Activity
Using the EREA and ERE73 reporters, we first confirmed that HFFs do not possess distinct E2F1 activity by reporter assay. Introduction of a constitutively active form of pRB, PSM.7-LP, did not significantly reduce EREA or ERE73 reporter activity in growing HFFs (Figure 8A left panel). Moreover, the introduction of PSM.7-LP reduced CDC6 promoter activity but had no significant effect on the ARF or TAp73 promoters (Figure 8A right panel). Together, these results indicate that growing HFFs lack distinct E2F1 activity.
To compare cancer and normal cells from the same source, we immortalized HFFs by introducing hTERT and then transformed immortalized HFFs by viral transduction with SV40 early region (SV40ER), which expresses large T and small t antigens, and the introduction of activated H-Ras. SV40 large T binds to and inactivates pRB and p53. SV40 small t and H-Ras facilitate cell proliferation. The transformed HFFs showed enlarged cell size and nuclei compared to the immortalized HFFs (Figure 8B, left panels). The expression levels of cell cycle-related CDC6 and E2F1 genes were significantly elevated in the transformed HFFs compared to immortalized HFFs (Figure 8B, right panels). Moreover, the expression levels of the tumor suppressor ARF and TAp73 genes were also significantly elevated in the transformed HFFs relative to immortalized HFFs (Figure 8B, right panels), suggesting that the transformation of HFFs generated endogenous distinct E2F1 activity to activate ARF and TAp73 genes. We thus examined the activity of EREA and ERE73 reporters in immortalized and in transformed HFFs. Although wild type of EREA and ERE73 did not show higher activity than analogous mutants in immortalized HFFs (Figure 8C, upper panels), the wild type constructs exhibited significantly higher activity than their corresponding mutants in transformed HFFs (Figure 8C, lower panels), indicating that the transformed HFFs acquired distinct E2F1 activity that activates EREA and ERE73.
To validate that the higher relative activity of wild type EREA and ERE73, compared to corresponding mutants, is due to distinct E2F1 activity, we introduced a constitutively active mutant of pRB, PSM.7-LP, which is the primary regulator of activator E2Fs by directly binding to and suppressing transcriptional activity. Introduction of PSM.7-LP suppressed both wild type EREA and ERE73 reporters, supporting the gain of distinct E2F1 activity in transformed HFFs.
3.9. Cancer Cell Lines but Not Normal Growing Cells Harbor Distinct E2F1 Activity
Finally, we examined a variety of cancer cell lines to determine whether distinct E2F1 activity is a universal property of transformed cells that is absent in normal growing cells, which can be applied to differentiate cancer cells from normal growing cells, based on the presence or absence of distinct E2F1 activity. We first examined this paradigm in the Saos-2 cell line, which lacks both functional pRB and p53, as a representative pRB defective cell line. Both wild type EREA and ERE73 reporters showed higher activity than mutant constructs in Saos-2 cells (Figure 9A), suggesting that this cell line harbors distinct E2F1 activity. Accordingly, the increased activity of wild type ERE promoters in Saos-2 cells was suppressed by introduction of the constitutively active pRB, PSM.7-LP (Figure 9B).
To confirm that the dysfunction of upstream regulatory factors of pRB generates distinct E2F activity, we also examined the U-2 OS cell line, which retains both pRB and p53 but lacks p16^INK4a^ and ARF expression due to DNA methylation of the p16INK4a/ARF locus. Similarly to Saos-2 cells, the wild type EREA and ERE73 reporters showed higher activity than the corresponding mutants (Figure 9C), which was suppressed by the introduction of PSM.7-LP (Figure 9D), implying that U-2 OS cells also harbor distinct E2F1 activity.
We next examined whether normal growing cells possess distinct E2F1 activity. As normal growing cells other than HFF, we used HUVEC, human lung normal fibroblast WI-38 and mouse embryonic fibroblasts (MEF) as primary cells and immortalized cell lines RPE1 and HaCaT, which maintain contact inhibition and do not show transformed phenotype such as anchorage independent growth. These cell lines proliferate with doubling times of about 20 h (RPE1), 24 h (WI-38), 28 h (HaCaT), 32 h (HUVEC), and 38 h (MEF). As expected, HUVEC, WI-38, MEF, and RPE1 did not exhibit distinct E2F1 activity (Figure 10). Unexpectedly, however, HaCaT, which is reported to have mutations in p53, did show distinct E2F1 activity (Figure 10).
Finally, we extended the characterization of distinct E2F1 activity to all of the cancer cell lines available in our laboratory (Table 1). The results are shown in Figure 11, Figure 12 and Figure 13, according to the organ of origin. Essentially, all cancer cell lines tested possessed distinct E2F1 activity, based on the activation of EREA or ERE73 compared to the corresponding mutant. These results strongly suggest that the presence of distinct E2F1 activity is a widespread, perhaps universal characteristic of cancer cell lines, originating from multiple tissues, which enables their discrimination from normal growing cells.
4. Discussion
Discrimination of cancer cells from normal growing cells is crucial to specifically target cancer therapy. We showed here that the presence of distinct E2F1 activity, which activates EREA and ERE73, enables the discrimination of cancer cell lines from normal growing cells. We previously reported, using human normal fibroblasts of mesenchymal origin, that E2F regulation of ARF and TAp73 genes is distinct from that of cell cycle-related genes, in that these genes are activated by E2F activity induced by exogenous expression of E2F1 or expression of E1a but not by physiological E2F activity induced by growth stimulation [18,22], indicating that E2F activity induced by exogenous expression of E2F1 or expression of E1a contains distinct E2F1 activity that activates ARF and TAp73 genes, which is not induced by growth simulation. We showed here that this unique mode of E2F1 regulation of the tumor suppressor genes also applies to epithelial cells (Figure 4 and Figure 5), from which 90% of cancers arise. We also demonstrated that the exogenous expression of cyclin D1 generated distinct E2F1 activity (Figure 7D), suggesting that alterations in the upstream regulatory factors of pRB can also potentially generate distinct E2F1 activity. Using the E2F responsive elements of ARF (EREA) and TAp73 (ERE73) [18,22], we tested whether cancer cell lines possess distinct E2F1 activity. We showed that transformation of human normal fibroblasts (HFFs) by SV40ER and H-Ras generated distinct E2F1 activity. Furthermore, all the cancer cell lines tested exhibited distinct E2F1 activity, whereas normal growing cells, such as WI-38, HUVEC, MEF, and RPE1, did not. These results raise the possibility that the existence of distinct E2F1 activity has the potential to discriminate cancer cells from normal growing cells.
EREA and ERE73 contain extra sequences in addition to minimal E2F responsive elements (GC repetitive sequences). Hence, the enhancement of reporter activity by addition of EREA or ERE73 elements does not necessarily indicate that the cell line assayed possesses distinct E2F1 activity. We thus confirmed the presence of distinct E2F1 activity by examining whether mutation of these GC repetitive sequences in EREA or ERE73 reduced the reporter activity compared to wild type constructs. Intriguingly, we found in some cell lines, mutation of EREA or ERE73 did not reduce reporter activity. In some cases, mutation increased reporter activity. Although the exact reason remains poorly understood, we speculate that the mutation of EREA or ERE73 inadvertently generated a binding sequence for another transcription factor, which is activated in that cancer cell line. Thus, despite these limitations, if either EREA or ERE73 reporter activity was higher than the corresponding mutant, the cell line was judged to possesses distinct E2F1 activity.
HaCaT is a spontaneously immortalized human epithelial cell line, which is widely used as a model system to study human keratinocyte differentiation. We thus chose HaCaT as a candidate normal epithelial cell line. However, we discerned subsequently that this cell line possesses mutations of the tumor suppressor p53 (His179Tyr, Arg282Trp) [39]. Since this mutation resides in the DNA binding domain of p53, it is likely to compromise p53 functions. Accordingly, we found that HaCaT cells possess distinct E2F1 activity. These observations suggest that the loss of p53 function may indirectly generate distinct E2F activity, likely due to the reduced expression of the CDK inhibitor p21^Cip1^, which is an upstream regulatory factor of pRB. Consistent with this, it is reported that in MCF7 and HCT116 cells, knockdown of p53 increased TAp73 gene expression through E2F1-dependent transcription, mediated by p21^Cip1^ [40]. These observations suggest that the loss of p53 function has the potential to generate distinct E2F1 activity via reduced expression of p21^Cip1^. However, the possibility that HaCaT cells acquired additional defects in the RB pathway cannot be formerly excluded.
In most cancers, pRB function is disabled with a consequent increase in E2F activity in cancer cells [31]. This elevated E2F activity has been utilized to drive expression of cytotoxic genes (suicide gene therapy) or genes required for viral replication (oncolytic virotherapy), preferentially in cancer cells. For this purpose, the E2F1 gene promoter has been widely utilized, such as in the oncolytic adenovirus CG0070 [41,42,43,44,45,46,47,48,49,50,51,52,53] and ONYX-411 [54,55,56,57,58,59]. However, E2F1 is a cell cycle-related E2F target gene, and its promoter is also activated by physiological E2F activity, induced by growth stimulation, and hence exhibits high activity in normal proliferating cells [54]. Thus, utilizing the E2F1 promoter to drive cytotoxic gene expression has the potential to injure normal proliferating cells. This is also true for promoters of other cell cycle-related E2F target genes such as CDC6 [37]. The promoters of these cell cycle-related E2F target genes harbor a typical E2F binding consensus sequence (TTT^C^/G^G^/_C_CGC), which exhibits responsiveness to physiological E2F activity triggered by growth stimulation [35]. This indicates that the cell cycle-related E2F target promoters have the potential to drive gene expression in normal growing cells. However, E2F-responsive elements of the ARF gene (EREA) and the TAp73 gene (ERE73), whose sequences diverge from the typical E2F binding consensus and are mainly comprised of GC repeats, specifically respond to distinct E2F1 activity, yet fail to respond to physiological E2F1 activity triggered by growth stimulation in fibroblasts [18,22] and also in epithelial cells, as shown here. We have shown that all the cancer cell lines tested exhibit distinct E2F1 activity, whereas this activity is undetectable in normal growing cells. Thus, utilizing distinct E2F1 activity acting on tumor suppressor promoter elements, such as EREA and ERE73, would represent a universal means to specifically induce gene expression in cancer cells while preserving normal growing cells [60].
One possible application of the utilization of distinct E2F1 activity by tumor suppressor gene promoter elements would be to drive cytotoxic gene in suicide gene therapy or to drive genes required for viral replication in oncolytic virotherapy specifically in cancer cells, minimizing off-target toxicity to normal cells. For diagnostics, EREA or ERE73 could be integrated into biosensor platforms such as fluorescent reporters to detect distinct E2F activity in clinical samples, serving as a novel diagnostic biomarker for early cancer detection. Although the utilization of distinct E2F1 activity by EREA or ERE73 is expected to be more cancer specific than using promoters of cell cycle-related E2F target genes, there may be some limitations. One possible limitation would be basal promoter activity, which drives certain level of gene expression in normal growing cells, although weaker than cell cycle-related promoters. A potential trade-off of this approach would be the promoter activity of EREA or ERE73 constructs in cancer cells, whether they exhibit enough activity to obtain the expected level of gene expression. Another possible limitation would be a rare situation where normal cells may express distinct E2F1 activity. For example, it is reported that the inflammatory region of ulcerative colitis exhibits highly active CDK2 [61]. Since CDK2 is involved in the inactivation of pRB by phosphorylation, the cells in the inflammatory region may express distinct E2F1 activity by the forced inactivation of pRB by hyperactive CDK2. In such a case, use of distinct E2F1 activity may be limited. The potential utility and limitations are issues to be addressed in utilizing distinct E2F1 activity for cancer specific therapy.
5. Conclusions
The findings suggest that distinct E2F1 activity is a widespread and robust marker for the discrimination of cancer cells and highlight its promise for the selective delivery of cytotoxic therapies, minimizing harm to normal proliferating cells.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nevins J.R. The Rb/E 2F pathway and cancer Hum. Mol. Genet.20011069970310.1093/hmg/10.7.69911257102 · doi ↗ · pubmed ↗
- 2Stevaux O. Dyson N.J. A revised picture of the E 2F transcriptional network and RB function Curr. Opin. Cell Biol.20021468469110.1016/S 0955-0674(02)00388-512473340 · doi ↗ · pubmed ↗
- 3Frolov M.V. Dyson N.J. Molecular mechanisms of E 2F-dependent activation and p RB-mediated repression J. Cell Sci.20041172173218110.1242/jcs.0122715126619 · doi ↗ · pubmed ↗
- 4Nevins J.R. Leone G. De Gregori J. Jakoi L. Role of the Rb/E 2F pathway in cell growth control J. Cell. Physiol.199717323323610.1002/(SICI)1097-4652(199711)173:2<233::AID-JCP 27>3.0.CO;2-F 9365528 · doi ↗ · pubmed ↗
- 5Phillips A.C. Vousden K.H. E 2F-1 induced apoptosis Apoptosis 2001617318210.1023/A:101133262574011388666 · doi ↗ · pubmed ↗
- 6Trimarchi J.M. Lees J.A. Sibling rivalry in the E 2F family Nat. Rev. Mol. Cell Biol.20023112010.1038/nrm 71411823794 · doi ↗ · pubmed ↗
- 7Rogoff H.A. Kowalik T.F. Life, death and E 2F: Linking proliferation control and DNA damage signaling via E 2F 1Cell Cycle 2004384584610.4161/cc.3.7.97515190206 · doi ↗ · pubmed ↗
- 8Stevens C. La Thangue N.B. The emerging role of E 2F-1 in the DNA damage response and checkpoint control DNA Repair 200431071107910.1016/j.dnarep.2004.03.03415279795 · doi ↗ · pubmed ↗