Proteasomal Degradation of Mutant Huntingtin Exon1 Regulates Autophagy
Austin Folger, Chuan Chen, Phasin Gonzalez, Sophia L. Owutey, Yanchang Wang

TL;DR
The study shows that proteasomal degradation of mutant huntingtin exon1 is linked to autophagy, revealing a new mechanism of cytotoxicity in Huntington’s disease.
Contribution
The study reveals that proteasomal degradation of mHttEx1 regulates IBophagy, a selective autophagy pathway for misfolded protein aggregates.
Findings
Yeast mutants with defective proteasomal degradation of mHttEx1 show impaired IBophagy.
Cdc48 Ubx cofactors are essential for both proteasomal degradation of mHttEx1 and IBophagy.
Inhibiting the proteasome in yeast cells eliminates IBophagy.
Abstract
What are the main findings? Yeast mutants defective in proteasomal degradation of soluble mutant huntingtin exon1 (mHttEx1) display impaired IBophagy, a selective autophagy pathway for misfolded protein inclusion bodies.Specific Cdc48 Ubx cofactors are required for efficient proteasomal degradation of mHttEx1 and IBophagy.Inhibition of proteasome function in yeast cells abolishes IBophagy. Yeast mutants defective in proteasomal degradation of soluble mutant huntingtin exon1 (mHttEx1) display impaired IBophagy, a selective autophagy pathway for misfolded protein inclusion bodies. Specific Cdc48 Ubx cofactors are required for efficient proteasomal degradation of mHttEx1 and IBophagy. Inhibition of proteasome function in yeast cells abolishes IBophagy. What is the implication of the main finding? The strong correlation between proteasomal degradation of soluble mHttEx1 and IBophagy…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
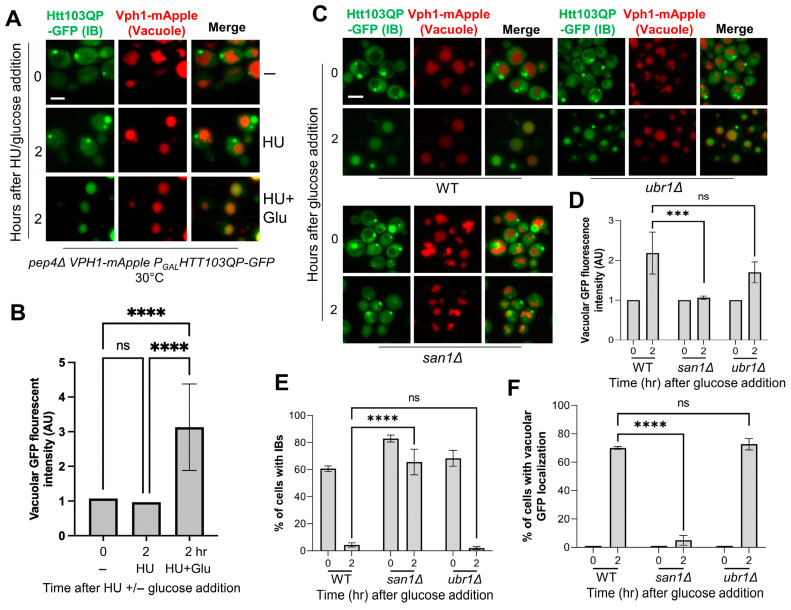 Figure 1
Figure 1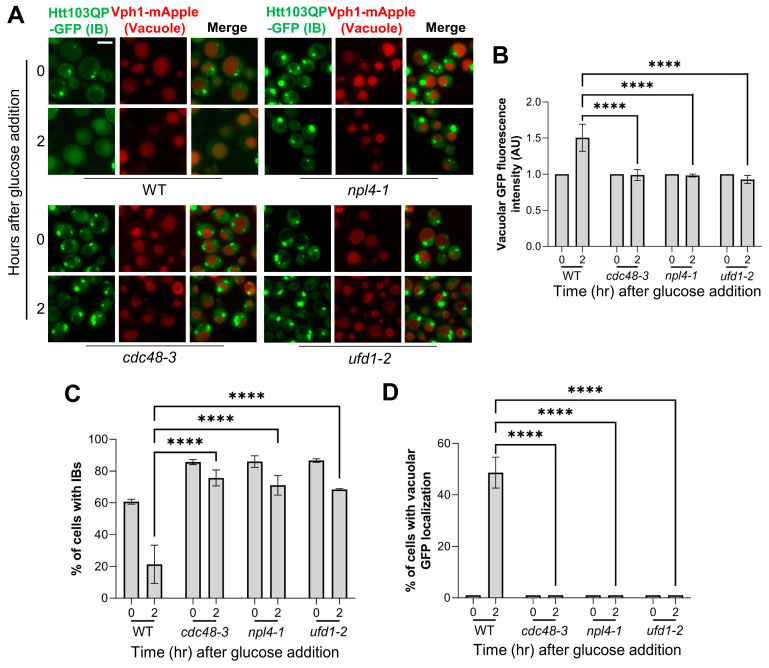 Figure 2
Figure 2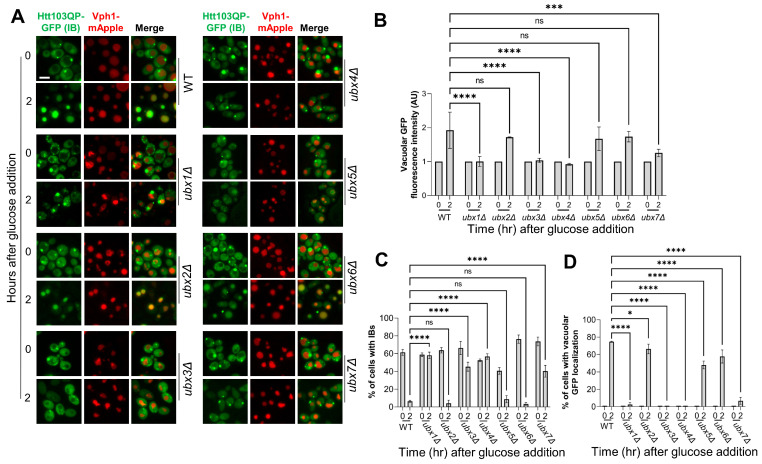 Figure 3
Figure 3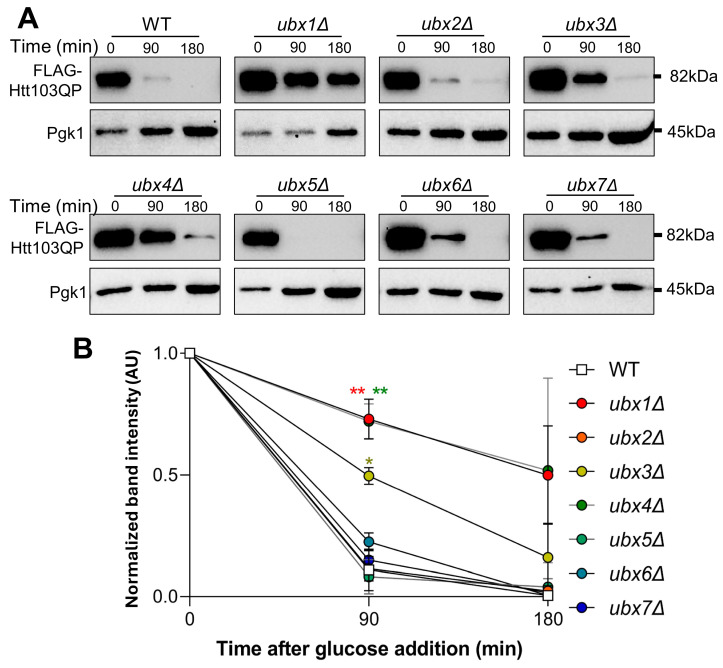 Figure 4
Figure 4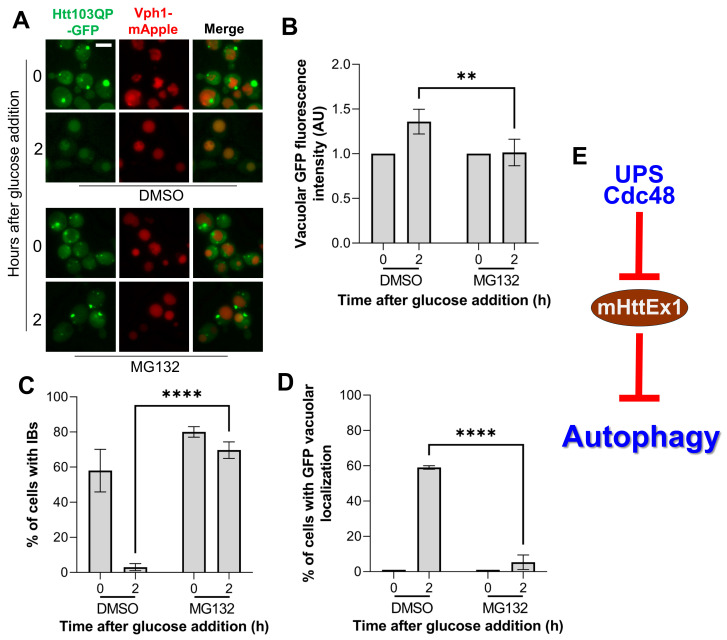 Figure 5
Figure 5- —National Institutes of Health/NIGMS
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Neurodegenerative Diseases · Autophagy in Disease and Therapy · Ubiquitin and proteasome pathways
1. Introduction
Protein folding is a tightly regulated process, and multiple mechanisms ensure correct folding. However, protein misfolding still occurs spontaneously and can be aggravated by mutations, aging, environmental stresses, and translational errors [1]. Misfolded proteins are prone to aggregation, generating small soluble oligomers or large insoluble inclusion bodies (IBs) [2]. This aggregation is primarily driven by exposed hydrophobic surfaces [3,4]. Misfolded protein aggregates show cytotoxicity and are linked to numerous neurodegenerative disorders, such as Huntington’s and Alzheimer’s diseases [5,6]. Cellular mechanisms to combat this toxicity include the degradation of toxic proteins by ubiquitin-proteasome system (UPS) and macroautophagy [7,8,9]. Thus, it is pertinent to understand how these systems respond to the accumulation of misfolded proteins.
Huntington’s disease is a dominant neurodegenerative disorder caused by the expansion of glutamine-encoding trinucleotide (CAG) repeats in exon 1 of the huntingtin gene (HTT). Individuals with greater than 39 CAG repeats in this exon develop Huntington’s disease [10]. The expansion of CAG repeats in HTT results in production of misfolded Htt exon 1 protein with a polyQ track (mHttEx1) that is prone to aggregation [11]. The mHttEx1 fragment includes the 17 N-terminal amino acids of Htt, the expanded polyQ tract, and a proline-rich domain, which is a result of aberrant splicing [12]. Cytotoxicity arises when HttEx1 accumulates in the cell. mHttEx1 production in animal models results in symptoms of Huntington’s disease [13]. In addition, the onset of symptoms in aged humans with mHttEx1 is likely attributed to weakened protein quality control systems [14]. Thus, mHttEx1 has been an ideal substrate to study misfolded protein aggregation, clearance, and cytotoxicity.
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved pathway responsible for the degradation and recycling of cellular components [15]. This pathway involves the sequestration of cytoplasmic material into double-membraned autophagosomes, which subsequently fuse with the vacuole or lysosome, where the cargo is degraded and recycled by resident hydrolases [16]. Autophagy can function in either a selective or non-selective manner. Non-selective autophagy is typically induced under nutrient-deprived conditions and involves the bulk, random engulfment of cytosolic constituents for vacuolar or lysosomal degradation [17,18]. Selective autophagy, however, only targets specific proteins/structures for degradation through selective autophagy receptors (SARs) [19,20]. Selective autophagy has been implicated in the degradation of misfolded protein aggregates, including mHttEx1 [8,21]. We recently showed that IB autophagy (IBophagy) utilizes SARs to confer specificity for IBs formed by mHttEx1 in yeast cells [22]. To detect IBophagy, we induced mHttEx1 IB formation in galactose media, but IBophagy was observed only after adding glucose for the shutoff of mHttEx1 production. Therefore, an open question is why IBophagy only occurs after glucose addition. Interestingly, previous studies showed that mHttEx1 itself impairs autophagy [23,24,25], while it is unclear how mHttEx1 affects IBophagy.
In addition to autophagy, misfolded proteins can also be degraded by the UPS [26]. This system begins with the ubiquitination of target proteins. For misfolded proteins, the responsible E3 ubiquitin ligases in budding yeast include nuclear-localized San1 and cytoplasmic Ubr1 [27,28]. The efficient degradation of misfolded proteins also requires SUMOylation system and Cdc48 segregase [29,30]. Cdc48 ATPase assembles into a hexamer that translocates polypeptides through its central pore [31]. The yeast Cdc48 complex is composed of the Cdc48 ATPase together with its cofactors Npl4 and Ufd1 [32]. Npl4 recognizes ubiquitinated protein substrates, after which Cdc48 extracts ubiquitinated misfolded proteins from aggregates, unfolds them, and transfers them to the proteasome [31]. Regulators for the Cdc48 complex also include seven Ubx proteins in budding yeast, which contain a ubiquitin-interacting motif (UIM) and a Cdc48 binding motif (UBX domain) to bridge ubiquitinated substrates and Cdc48 [33,34]. We previously showed that proteasomal degradation of mHttEx1 depends on E3 ubiquitin ligase San1 as well as the Cdc48 complex in yeast cells [29]. However, it is unclear if the Cdc48 cofactors are all required for this degradation. Moreover, the role of the UPS pathway in IBophagy remains unknown.
In this work, we investigated the requirements of UPS components as well as Cdc48 and its cofactors in proteasomal degradation of mHttEx1 in yeast cells. We further analyze the role of this degradation in IBophagy. Our work utilizes a mHttEx1 protein with 103 polyQ (Htt103QP) as a model misfolded protein. We previously showed that E3 ligases San1, but not Ubr1, is required for proteasomal degradation of Htt103QP [29]. In this work, we found that San1, rather than Ubr1, is required for Htt103QP IBophagy. Our previous work also showed that the Cdc48 complex is required for proteasomal degradation of Htt103QP, and here we found that all three essential components of the Cdc48 complex, Cdc48, Npl4, and Udf1, are required for Htt103QP IBophagy. We further showed that three of seven Ubx proteins, the nonessential Cdc48 Ubx cofactors, are required for both IBophagy and proteasomal clearance of Htt103QP. In addition, inhibition of proteasomal activity by MG132 also prevents Htt103QP IBophagy. Therefore, our results established a strong correlation between proteasomal degradation and autophagic clearance of Htt103QP. Since Htt103QP presents in two pools: soluble Htt103QP and insoluble IBs, our results indicate that the proteasomal degradation of soluble Htt103QP facilitates IBophagy, likely by alleviating inhibition of autophagy by soluble Htt103QP.
2. Materials and Methods
2.1. Yeast Strains, Growth Conditions, and Plasmids
All yeast strains used in this research are in W303 background. Their relevant genotypes are listed in Table S1. Standard tetrad dissection was used for yest strain construction. The P_GAL_FLAG-Htt103QP-GFP plasmid, originally obtained from the Lindquist lab, is a galactose-inducible construct encoding a FLAG- and GFP-tagged Htt103QP fragment [35]. After linearization, this plasmid was transformed into yeast cells for its insertion into the yeast genome. Yeast cells with the integration were selected using URA3 marker and uracil dropout plates. Yeast peptone dextrose (YPD) was used for the growth of yeast cells. Yeast extract/peptone media (YEP) supplied with galactose was used to induce FLAG-Htt103QP-GFP expression and the subsequent inclusion body (IB) formation.
MG132 (Sigma-Aldrich, St. Louis, MO, USA), is a proteasome inhibitor, but it does not penetrate yeast cell wall and membrane. To enhance permeability of yeast cells, we added 0.1% L-proline and 0.003% SDS into the medium for 3 h prior to MG132 treatment [36]. Our previous studies confirmed that this protocol enables the inhibition of yeast proteasome function by 75 µM MG132 [37,38].
2.2. Detailed Protocol for IBophagy Induction
For the IBophagy assay, saturated yeast cells containing pep4∆ VPH1-mApple P_GAL_-FLAG-Htt103QP-GFP in YPD (yeast extract-peptone-dextrose) were diluted into galactose media (yeast extract, peptone, galactose) at 1:1000 and incubated for 16 h at 30 °C to induce Htt103QP IB formation. Glucose was subsequently added to a final concentration of 2% to repress GAL1 promoter and halt Htt103QP production, thereby inducing IBophagy. To eliminate the potential effect of cell growth on autophagy, 200 mM hydroxyurea (HU) (Ambeed, Buffalo Grove, IL, USA) was also included to arrest yeast cell cycle in S-phase. Cells were harvested prior to glucose addition (time 0) and two hours after glucose treatment. Collected cells were washed with water, resuspended in 1× PBS, and immediately analyzed by fluorescence microscopy.
2.3. Fluorescence Imaging and Analysis
Htt103QP IB formation and autophagic clearance were analyzed using a fluorescence microscope (Keyence BZ-X700; Keyence of America, Itasca, IL, USA). Prepared cells were imaged with a 60× objective using appropriate filter sets for mApple and GFP. Z-stack images were acquired at 0.2-μm intervals and processed using BZ-X800 software (1.1.2.4.) to generate composites images. Autophagic activity was evaluated by assessing average GFP density inside vacuoles through automated hybrid cell counting with BZ-X800 software. Briefly, vacuolar regions were defined by extracting the mApple signal from the composite image, and the average integrated GFP intensity within mApple-labelled areas was measured in more than 50 cells per condition.
2.4. Western Blotting
Protein extracts were prepared using an alkaline extraction protocol and separated by 10% SDS–PAGE. Anti-FLAG antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA); anti-Pgk1 antibody was purchased from Invitrogen (Waltham, MA, USA). Horseradish peroxidase–conjugated goat anti-mouse IgG secondary antibodies were from Cell Signaling Technology (Danvers, MA, USA). Following enhanced chemiluminescence (ECL; PerkinElmer, Waltham, MA, USA), membranes were imaged using a Bio-Rad ChemiDoc system (Hercules, CA, USA).
2.5. Statistical Analysis
Data are presented as mean ± standard error of the mean (SEM). For each strain, fluorescence average integrated density was calculated by quantifying vacuolar GFP intensity using BZ-X800 software. Statistical analyses were performed using GraphPad software (10.0.1). One-way or two-way ANOVAs was applied as appropriate to determine p-values, with the specific test indicated in the figure legends. Statistical significance was defined as p < 0.05 (*).
3. Results
3.1. Loss of E3 Ubiquitin Ligase San1 Blocks IBophagy of Htt103QP
As a model misfolded protein, Htt103QP forms IBs in yeast cells after overnight induction of Htt103QP expression in galactose media. Interestingly, IBophagy occurs only after glucose is added to shut off Htt103QP production [22]. We previously showed that San1 E3 ligase is essential for the ubiquitination and proteasomal degradation of Htt103QP [29]. Thus, we were curious whether San1 is required for Htt103QP IBophagy. To examine Htt103QP IBophagy, we utilized a previously constructed integrating plasmid P_GAL_-FLAG-Htt103QP-GFP, which contains a galactose-inducible promotor GAL and the first exon of HTT gene with 103 CAG expansion in fusion with FLAG and GFP gene. Yeast strains harboring this integrating plasmid produce FLAG-Htt103QP-GFP (hereafter Htt103QP) in the presence of galactose, forming Htt103QP IBs [37]. To analyze IBophagy, yeast cells used in this study also produce Vph1-mApple as a vacuolar marker, and are lacking the Pep4 protease (pep4Δ) to avoid protein degradation inside the vacuole [39,40].
To exclude the effect of cell cycle on autophagy, we added hydroxyurea (HU) to block cell cycle in our previous studies [22]. However, the HU-induced DNA replication stress may induce autophagy [41,42,43]. Therefore, we first examined the effect of HU on IBophagy in yeast cells. pep4Δ VPH1-mApple yeast strains with integrated P_GAL_-FLAG-Htt103QP-GFP plasmid were cultured in galactose-containing media for 16 h at 30 °C to induce IB formation. Then, HU alone or in combination with glucose was added to the medium. The appearance of GFP signal in the vacuole, which is marked by Vph1-mApple, was used as an indicator of successful IBophagy. The addition of HU alone did not cause any noticeable change in vacuolar GFP signal, but addition of both HU and glucose increased vacuolar GFP signal significantly (Figure 1A,B). The result suggests that treatment with HU alone does not induce IBophagy under our experimental conditions, likely because short-term HU exposure is insufficient to activate autophagy in yeast. We therefore applied this protocol to identify additional IBophagy regulators.
Because San1 E3 ubiquitin ligase is required for Htt103QP proteasomal degradation [29], we first tested whether this degradation is required Htt103QP IBophagy. For this purpose, wild-type (WT) and san1Δ cells were grown overnight in galactose media to induce Htt103QP IB formation. Glucose was then added to halt Htt103QP production and induce IBophagy. HU was also added to 200 mM to arrest the cells in S-phase. Two hours after glucose addition, most WT cells exhibited vacuolar localization of GFP, indicating successful Htt103QP IBophagy. However, minimal GFP vacuolar localization was observed in san1Δ cells (Figure 1C). Ubr1, the cytoplasmic E3 ligase for misfolded proteins, is dispensable for proteasomal degradation of Htt103QP [29]. Interestingly, IBophagy occurred normally in ubr1Δ cells as in WT cells. We measured the intensity of vacuolar GFP before and after glucose addition and found a significant increase in WT and ubr1Δ cells, indicating IBophagy, but no significant increase was observed in san1Δ cells (Figure 1D). Consistently, cells with IBs sharply declined in WT and ubr1Δ cells after glucose addition (Figure 1E), but we observed increased number of cells displaying vacuolar GFP signal (Figure 1F), consistent with successful IBophagy. In clear contrast, these changes were absent in san1Δ cells, where the number of cells with an IB or vacuolar GFP remains consistent. Collectively, these findings demonstrate that San1, but not Ubr1, is required for both proteasomal degradation of Htt103QP and IBophagy.
3.2. The Cdc48 Complex Is Required for IBophagy of Htt103QP
Our lab has shown that the Cdc48 complex promotes the proteasomal degradation of Htt103QP [29]. Thus, we tested if the Cdc48 complex is also required for Htt103QP IBophagy. Because the Cdc48 complex is essential in budding yeast, we used three temperature-sensitive mutants: cdc48-3, npl4-1, and ufd1-2. These mutations were introduced into pep4Δ cells producing Htt103QP and Vph1-mApple. We then grew cells in galactose media overnight at 25 °C to induce Htt103QP IB formation and shifted to 37 °C for one hour to inactivate the Cdc48 complex. Glucose and HU were then added to halt Htt103QP production and arrest the cell cycle while inducing IBophagy. We found that all mutants exhibited persistent presence of Htt103QP IBs and minimal GFP vacuolar localization, indicating defective IBophagy in these mutants (Figure 2A). Although WT cells show greater vacuolar GFP intensity and fewer IBs after glucose addition, the cells with defective Cdc48 complex showed no significant change in vacuolar GFP intensity and maintained a relatively consistent proportion of cells with either IBs or vacuolar GFP (Figure 2B–D). Therefore, the Cdc48 complex is required for both proteasomal degradation and IBophagy of Htt103QP.
3.3. The Proteasomal Degradation and IBophagy of Htt103QP Depends on Specific Ubx Proteins
In budding yeast, the Cdc48 complex uses seven Ubx domain-containing cofactors to facilitate the recruitment of ubiquitinated proteins [44,45]. Thus, some Ubx proteins are likely required for Htt103QP proteasomal degradation and IBophagy. To test this, we examined their requirements in Htt103QP IBophagy. For this purpose, we deleted UBX1–7 genes in yeast cells with pep4Δ VPH1-mApple P_GAL_-FLAG-Htt103QP-GFP. Using the described IBophagy protocol, we found that vacuolar Htt103QP-GFP localization was deficient in ubx1Δ, ubx3Δ, ubx4Δ. A slight increase in vacuolar GFP was observed in ubx7Δ cells after IBophagy induction, while the increase in vacuolar GFP signal in ubx2Δ, ubx5Δ, and ubx6Δ mutant cells was similar to WT cells (Figure 3A,B). In addition, ubx1Δ, ubx3Δ, ubx4Δ mutants displayed a relatively consistent proportion of cells with either IBs or vacuolar GFP after glucose addition (Figure 3C,D). These results indicate that specific Ubx cofactors, Ubx1, Ubx3, and Ubx4, are required for the efficient Htt103QP IBophagy.
Because the Ubx protein family bridges ubiquitinated proteins to the Cdc48 complex for extraction/unfolding and eventual proteasomal degradation, we investigated their requirement for Htt103QP proteasomal degradation. The strains used for this experiment contain pep4Δ, which reduces autophagic degradation of Htt103QP. Our previous studies using expression shut-off assay show proteasome-dependent Htt103QP degradation, since proteasome inhibitor MG132 blocks this degradation [29]. We applied the same protocol to analyze the role of Ubx proteins in proteasomal degradation of Htt103QP. For this experiment, we grew WT and ubxΔ mutant cells in raffinose media to log phase before inducing HTT103QP expression with galactose for one hour. Expression was shut off by adding glucose, and we then examined Htt103QP degradation kinetics over time. As shown previously, Htt103QP was readily degraded in WT cells over 180 min [29], but delayed Htt103QP protein degradation was observed in ubx1Δ, ubx3Δ, and ubx4Δ cells. In contrast, the delay was not obvious in ubx2Δ, ubx5Δ, ubx6Δ, and ubx7Δ cells, indicating only certain Ubx proteins are required for proteasomal degradation of Htt103QP (Figure 4A,B). Strikingly, all three ubxΔ mutants exhibiting impaired proteasomal Htt103QP degradation, ubx1Δ, ubx3Δ, and ubx4Δ, also showed defective IBophagy of Htt103QP (Figure 3). This strong correlation suggests that proteasomal degradation of Htt103QP or some other protein is required for IBophagy.
3.4. Proteasome Function Is Required for Htt103QP IBophagy
Our results so far reveal the strong correlation between proteasomal Htt103QP degradation and IBophagy. Thus, we examined whether proteasome activity is required for IBophagy. To this end, we grew cells overnight in galactose media to induce IB formation. After treatment of the cells with L-proline and SDS to increase cell permeability, MG132 was added to 75 μM for 30 min to inhibit proteasome activity [36,37]. Then, glucose was added to stop Htt103QP production and induce IBophagy. We found that the vacuolar localization of Htt103QP-GFP as well as the disappearance of Htt103QP IBs was blocked in cells treated with MG132 (Figure 5A–D), indicating defective IBophagy. Previous studies showed that yeast cells treated with MG132 triggers selective autophagy of proteasomes [46], indicating that inhibition of proteasome function per se does not affect autophagy. Therefore, this result indicates that proteasomal degradation of Htt103QP or some other protein is likely required for IBophagy.
4. Discussion
UPS and Cdc48 segregase contribute to the clearance of soluble mHttEx1 [9,29], but constant expression of mHttEx1 or other misfolded proteins results in the formation of large insoluble IBs. We previously demonstrated autophagic clearance of mHttEx1 IBs through IBophagy [22]. However, IBophagy began only when Htt103QP production was shut off with glucose. To determine the underlying mechanism, we examined whether the proteasomal degradation of soluble Htt103QP affects Htt103QP IBophagy. E3 ubiquitin ligase San1 and the core Cdc48 subunits are shown to be essential for proteasomal degradation of Htt103QP [29]. In this study, we found they are also required for Htt103QP IBophagy. In addition, we identified several Cdc48 Ubx cofactors required for Htt103QP IBophagy. Strikingly, only the ubxΔ mutants exhibiting defective Htt103QP IBophagy showed compromised proteasomal Htt103QP degradation, thereby establishing a strong correlation between proteasomal and autophagic degradation of Htt103QP. In addition, the inhibition of proteasome activity by MG132 also blocks Htt103QP IBophagy. Together, these results support the possibility that proteasomal degradation of soluble Htt103QP is a prerequisite for IBophagy (Figure 5E).
We and others found that mHttEx1 aggregates and IBs are cleared by autophagy [8,22]. Interestingly, mHttEx1 has been shown to impair autophagy [23,24,25]. For the IBophagy protocol in this research, Htt103QP IB formation was induced by growing yeast cells in galactose medium overnight. Under this condition, we speculate the presence of both soluble Htt103QP monomers or oligomers and insoluble Htt103QP IBs. Therefore, it is likely that soluble Htt103QP inhibits IBophagy until their proteasomal degradation. This notion can explain why IBophagy does not occur for yeast cells in galactose medium. However, the addition of glucose shuts off Htt103QP production, and the degradation of soluble Htt103QP by UPS alleviates its inhibition on IBophagy (Figure 5E). The strong correlation between proteasomal degradation of Htt103QP and IBophagy revealed in this study supports this possibility.
The Cdc48 complex extracts ubiquitinated misfolded proteins from aggregates and unfolds them for proteasomal degradation [47]. In our model, the Cdc48 complex likely promotes IBophagy by facilitating the proteasomal degradation of soluble Htt103QP. Previous studies indicate that the Cdc48 complex is also involved in the autophagy of stress granules and ribosomes in budding yeast and mammalian cells [48,49]. Moreover, the Cdc48 complex promotes autophagy by facilitating autophagosome closure by extracting Atg8 with the assistance of cofactor Ubx1 [50]. Although our results support the possibility that the Cdc48 complex promotes IBophagy by facilitating the degradation of soluble Htt103QP, the Cdc48 complex may also promote IBophagy through other mechanisms.
mHttEx1 interacts with cellular membranes and disrupts lipid bilayers [51,52]. In addition, mHttEx1 IBs interact with ER membrane, with portions of ER membrane being incorporated into IBs during their formation [53,54]. As the transmembrane protein in the autophagy machinery, Atg9 scramblase collaborates with Atg2 bulk lipid transporter, which together promote autophagosome growth and formation [55,56]. Therefore, one untested possibility is that the interactions of mHttEx1 with ER membrane may compromise Atg9/Atg2-dependent vesicle transport and subsequent autophagosome formation. However, more research is needed to further define the negative role of mHttEx1 in autophagy.
UPS and autophagy are conserved protein degradation pathways, and they are critical to combat the cytotoxicity of misfolded proteins. Deficiencies in autophagy and proteasomal degradation are linked to human diseases, including neurodegenerative disorders, cancer, diabetes, and cardiovascular disease [57,58]. Therefore, enhanced clearance of misfolded proteins should be a strategy to combat the cytotoxicity of misfolded proteins. Indeed, promoting autophagic and proteasomal turnover of mHttEx1 is shown to ameliorate mHttEx1-induced toxicity in neurons [9,59]. Our results established an interesting correlation between the proteasomal degradation of mHttEx1 and autophagy. Because previous studies indicate the negative role of mHttEx1 in autophagy, it is likely that UPS-dependent degradation of soluble mHttEx1 alleviates its negative effect on autophagy. Therefore, our observation provides a new angle to understand the cytotoxicity of soluble mHttEx1 and the role of UPS in combating this toxicity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Tyedmers J. Mogk A. Bukau B. Cellular strategies for controlling protein aggregation Nat. Rev. Mol. Cell Biol.20101177778810.1038/nrm 299320944667 · doi ↗ · pubmed ↗
- 2Fink A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid Fold. Des.19983 R 9R 2310.1016/S 1359-0278(98)00002-99502314 · doi ↗ · pubmed ↗
- 3Chiti F. Taddei N. Baroni F. Capanni C. Stefani M. Ramponi G. Dobson C.M. Kinetic partitioning of protein folding and aggregation Nat. Struct. Biol.2002913714310.1038/nsb 75211799398 · doi ↗ · pubmed ↗
- 4Zaman M. Khan A.N. Wahiduzzaman Zakariya S.M. Khan R.H. Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: An overview and therapeutic strategies to inhibit aggregation Int. J. Biol. Macromol.20191341022103710.1016/j.ijbiomac.2019.05.10931128177 · doi ↗ · pubmed ↗
- 5Folger A. Wang Y. The Cytotoxicity and Clearance of Mutant Huntingtin and Other Misfolded Proteins Cells 202110283510.3390/cells 1011283534831058 PMC 8616338 · doi ↗ · pubmed ↗
- 6Ross C.A. Poirier M.A. Protein aggregation and neurodegenerative disease Nat. Med.200410 S 10S 1710.1038/nm 106615272267 · doi ↗ · pubmed ↗
- 7Saitoh Y. Fujikake N. Okamoto Y. Popiel H.A. Hatanaka Y. Ueyama M. Suzuki M. Gaumer S. Murata M. Wada K. p 62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies J. Biol. Chem.20152901442145310.1074/jbc.M 114.59028125480790 PMC 4340391 · doi ↗ · pubmed ↗
- 8Lu K. Psakhye I. Jentsch S. Autophagic clearance of poly Q proteins mediated by ubiquitin-Atg 8 adaptors of the conserved CUET protein family Cell 201415854956310.1016/j.cell.2014.05.04825042851 · doi ↗ · pubmed ↗