Prophage φEr670 and Genomic Island GI_Er147 as Carriers of Resistance Genes in Erysipelothrix rhusiopathiae Strains
Marta Dec, Aldert L. Zomer, Marian J. Broekhuizen-Stins, Renata Urban-Chmiel

TL;DR
This study identifies prophage φEr670 and genomic island GI_Er147 as carriers of antibiotic resistance genes in Erysipelothrix rhusiopathiae, contributing to antimicrobial resistance in Gram-positive bacteria.
Contribution
The study reports novel prophage φEr670 and genomic island GI_Er147 carrying resistance genes in E. rhusiopathiae, expanding understanding of mobile genetic elements in antimicrobial resistance.
Findings
A novel 53 kb prophage φEr670 carrying resistance genes lnuB and lsaE was identified in strain 670.
Genomic island GI_Er147 in strain 147 contains multiple resistance genes including ant(6)-Ia, spw, lnu(J), and vat.
Tn916 transposon and ICEEr1012 are located at genomic hot spots contributing to genome plasticity in E. rhusiopathiae.
Abstract
In this study we employed nanopore whole genome sequencing to analyze the resistance genes, genomic islands and prophage DNA in two multidrug resistant E. rhusiopathiae strains, i.e., 670 and 147, isolated from domestic geese. MLST profiles and core-genome phylogeny were determined to assess strain relatedness. In strain 670 (serotype 8, ST 113), a novel 53 kb prophage φEr670 carrying the lnuB and lsaE resistance genes was identified. Regions highly homologous to the φEr670 prophage were detected in 36 of 586 (6.14%) publicly available E. rhusiopathiae genomes, as well as in some other Gram-positive bacteria, and usually contained resistance genes. E. rhusiopathiae strain 147 (serotype 5, ST 243) was found to contain a composite 98 kb genomic island (GI_Er147) carrying the ant(6)-Ia and spw genes, as well as gene encoding a putative lincosamide nucleotidyltransferase designated lnu(J)…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
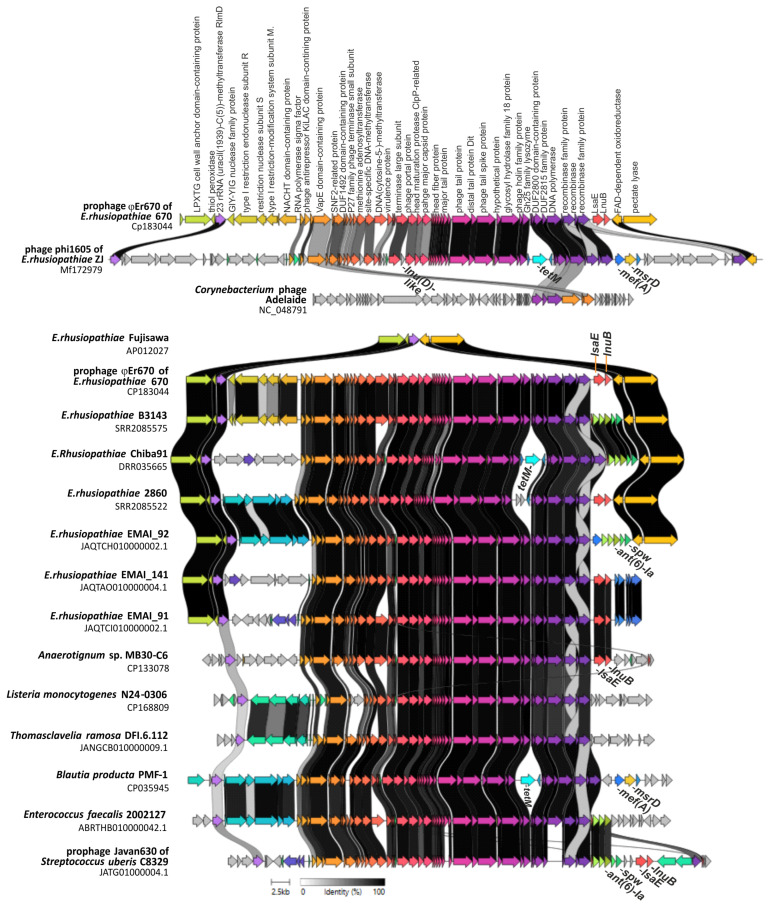 Figure 1
Figure 1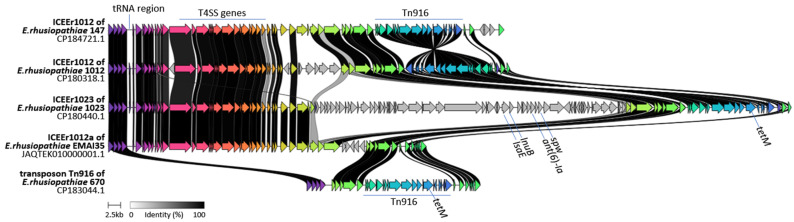 Figure 2
Figure 2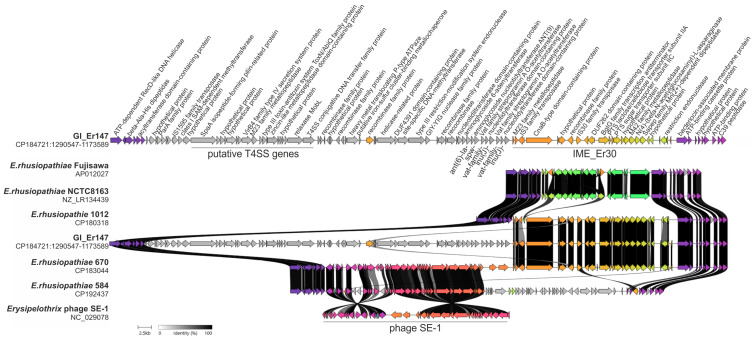 Figure 3
Figure 3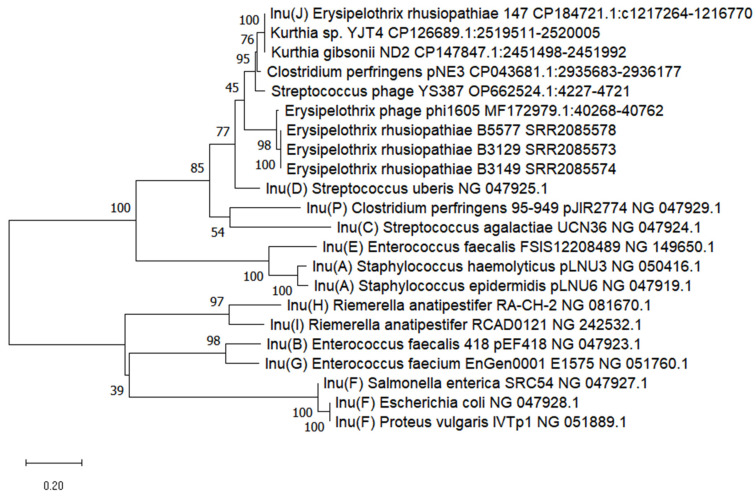 Figure 4
Figure 4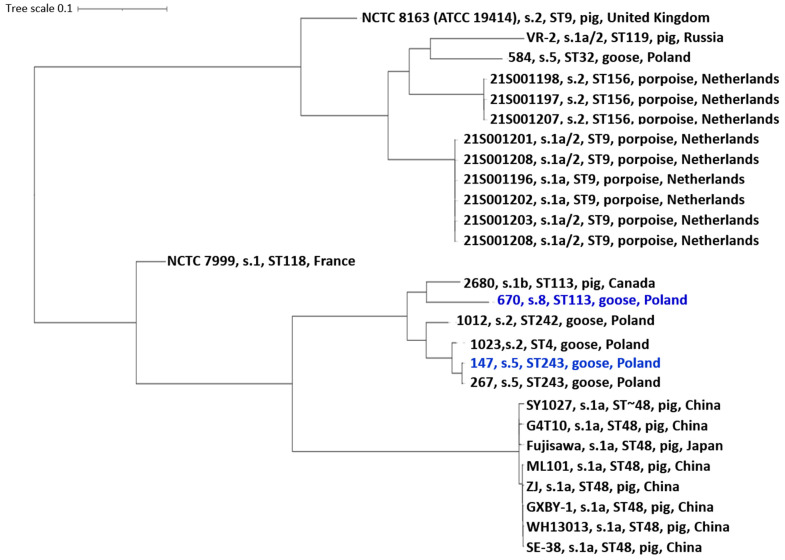 Figure 5
Figure 5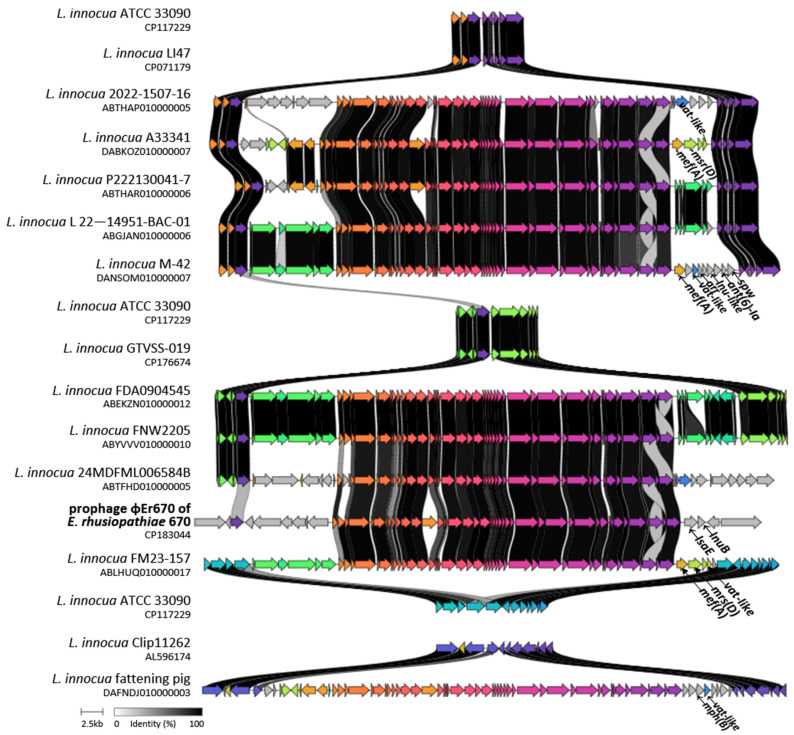 Figure 6
Figure 6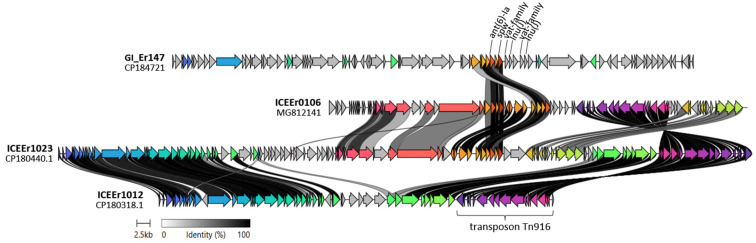 Figure 7
Figure 7 Figure 8
Figure 8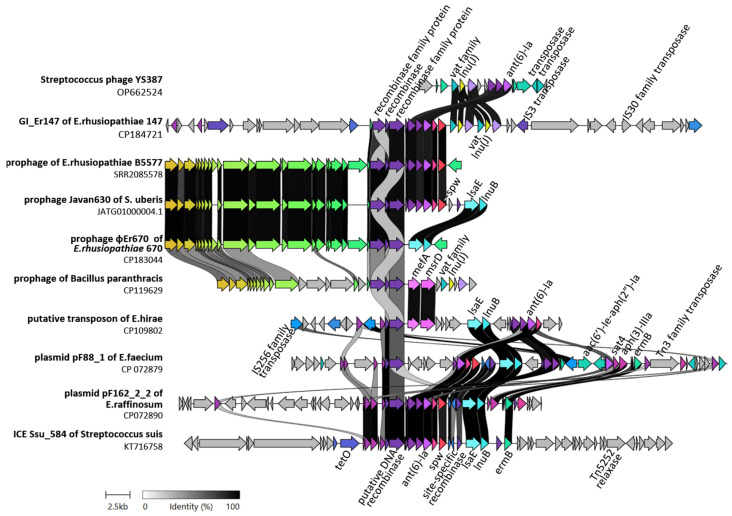 Figure 9
Figure 9| Taxonomic Group | Strain | Prophage Size [kb] | Resistance Genes Located Within Prophage Regions | Homology with Phage φEr670 | Ortho ANI Value [%] | GenBank Acc. No. | Reference | |
|---|---|---|---|---|---|---|---|---|
| Query Cover [%] | Similarity [%] | |||||||
| 670 | 53 | NA | NA | 100 | This study | |||
| ZJ | 90 | 63 | 92.7 | 87.8 | [ | |||
|
| EMAI_29 | 51 |
| 71 | 94.2 | 94.9 | JARGDV010000003.1 | [ |
|
| EMAI_31 | ~52 | 76 | 95.2 | ND | JAQTEO010000002.1 | [ | |
|
| EMAI_31 | ~52 |
| 70 | 95.3 | 94.4 | JAQTEO010000011.1 | [ |
|
| EMAI_33 | 51 |
| 70 | 95.3 | 94.4 | JAQTEM010000002.1 | [ |
|
| EMAI_91 | 53 | 82 | 94.4 | 94.5 | JAQTCI010000002.1 | [ | |
|
| EMAI_92 | 54 | 72 | 94.9 | 95.1 | JAQTCH010000002.1 | [ | |
|
| EMAI_141 | 53.5 | 78 | 94.7 | 95.1 | JAQTAO010000004.1 | [ | |
|
| B3129 | 54.5 | 75 | 95.0 | 95.1 | SRR2085573 | UN | |
|
| B3142 | 54.5 | 75 | 95.0 | 95.1 | SRR2085574 | UN | |
|
| B5577 | 54 | 75 | 95.3 | 95.1 | SRR2085578 | UN | |
|
| B3143 | 54 | 93 | 95.0 | 95.3 | SRR2085575 | UN | |
|
| B3144 | 50 | none | 72 | 92.1 | 92.7 | SRR2085576 | UN |
|
| B3159 | 51 | 81 | 91.4 | 91.5 | SRR2085577 | UN | |
|
| G2 | 52.5 | 89 | 94.6 | 94.7 | SRR2085593 | UN | |
|
| 2604 | 51 | 78 | 94.7 | 94.7 | SRR2085518 | UN | |
|
| 2628 | 52 | 82 | 95.2 | 95.4 | SRR2085520 | UN | |
|
| 2860 | 53 | 78 | 94.3 | 94.4 | SRR2085522 | UN | |
|
| 6028 | 53 | 78 | 94.6 | 94.5 | SRR2085524 | UN | |
|
| 6106 | 52 | 80 | 91.9 | 91.8 | SRR2085525 | UN | |
|
| Chiba 91 | 59 | 73 | 92.7 | 92.9 | DRR035665 | [ | |
|
| Saitama 91 | 59 | 73 | 92.7 | 92.9 | DRR035666 | [ | |
|
| Chiba 92A | 59 | 73 | 92.7 | 92.9 | DRR035667 | [ | |
|
| Chiba 92B | 56.5 | 73 | 94.1 | 94.7 | DRR035668 | [ | |
|
| Chiba 93 | 59 | 73 | 92.7 | ND | DRR035669 | [ | |
|
| Kanagawa 95 | 58 | 73 | 92.7 | ND | DRR035671 | [ | |
|
| Nagano 98 | 59 | 73 | 92.7 | 94 | DRR035672 | [ | |
|
| Saitama 01 | 59 | 73 | 92.7 | ND | DRR035675 | [ | |
|
| 16BKT031005 | 55 | 72 | 94.0 | ND | ERR3932976 | [ | |
|
| 16BKT031013 | 51.5 | 82 | 95.7 | 95.3 | ERR3932981 | [ | |
|
| 16BKT31009 | 54 | 77 | 94.8 | ND | ERR3932985 | [ | |
|
| 17MIK0642341 | 52 | none | 73 | 94.6 | 94.7 | ERR3932998 | [ |
|
| 17MIK0642351 | 54.5 | 75 | 95.0 | ND | ERR3932999 | [ | |
|
| 17MIK0642361 | 51.5 | 82 | 95.2 | 95.4 | ERR3933000 | [ | |
|
| 17MIK0642371 | 54.5 | 75 | 95.0 | ND | ERR3933001 | [ | |
|
| swine100 | 52 | 77 | 96.1 | 97.2 | ERR3678831 | [ | |
|
| swine29 | 55 | 72 | 93.4 | ND | ERR3678845 | [ | |
|
| LV19 | ~59 | none | 76 | 94.4 | ND | [ | |
|
| DFI.6.112 | 53 | none | 72 | 94.9 | 94.2 | JANGCB010000009.1 | UN |
|
| DFI.6.30 | 53 | none | 72 | 94.9 | 94.2 | JAJCKK010000013.1 | UN |
| C8329 | 52 | 77 | 95.5 | 94.8 | UN | |||
| MB30-C6 | ND | 81 | 95.6 | 94.6 | [ | |||
|
| DSM 2594 | ND | none | 74 | 94.6 | ND | [ | |
|
| DSM 2593 | ND | none | 74 | 94.6 | ND | [ | |
|
| EI1405 | 57 | none | 75 | 93.7 | 94.2 | UN | |
|
| DFI.6.107 | 54 | none | 72 | 93.9 | 94.1 | JAJCLO010000002.1 | UN |
|
| N24-0306 | 56 | none | 69 | 89.9 | 88.3 | [ | |
|
| PMF-1 | ND |
| 82 | 95.2 | ND | UN | |
| HFYG-1003 | ND | 71 | 94.6 | ND | UN | |||
|
| 2002127 | 58 |
| 74 | 92.6 | 90.1 | ABRTHB010000042.1 | UN |
|
| 2022-1507-16 | 54 | none | 72 | 94.8 | ND | ABTHAP010000005.1 | UN |
|
| A33341 | 54 | 76 | 95.6 | 94.8 | DABKOZ010000007.1 | [ | |
|
| 24MDFML00 | ND | none | 72 | 93.6 | ND | ABTFHD010000005.1 | UN |
|
| fattening pig | 54 | 75 | 95.3 | ND | DAFNDJ010000003.1 | [ | |
|
| FDA1205947- | 56 | none | 71 | 89.9 | ND | ABJLEO010000012.1 | UN |
|
| FDA0904545 | 55.5 | none | 71 | 92.9 | 92.9 | ABEKZN010000012.1 | UN |
|
| FM23-157 | 53 | 71 | 95.5 | ND | ABLHUQ010000017.1 | UN | |
|
| FNW2205 | 55 | none | 68 | 92.9 | 93 | ABYVVV010000010.1 | UN |
|
| 22-014951-BAC-01 | 53 | none | 71 | 95.6 | 94.7 | ABGJAN010000006.1 | UN |
|
| P222130041-7 | 51 | none | 78 | 94.7 | 94.3 | ABTHAR010000006.1 | UN |
|
| M-42 | 55.5 | 68 | 91.8 | 92.2 | DANSOM010000007.1 | [ | |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMicrobial infections and disease research · Coccidia and coccidiosis research · Vector-borne infectious diseases
1. Introduction
Erysipelothrix rhusiopathiae is a Gram-positive bacterium responsible for erysipelas in a wide range of hosts, including poultry, swine, and other domestic and wild animals, and it also poses a zoonotic risk to humans [1]. In poultry, infections with E. rhusiopathiae can cause acute septicaemia, sudden mortality, and chronic disease manifestations, leading to significant economic losses in waterfowl and turkey production [2,3,4]. Outbreaks of erysipelas are typically controlled using antimicrobial therapy, with penicillins remaining the treatment of choice [2,5]. However, the increasing detection of antimicrobial resistance (AMR) among E. rhusiopathiae isolates has raised growing concerns regarding the effectiveness of current therapeutic strategies, with potential implications for both animal health and public health [5]. Moreover, the lack of commercially available vaccines specifically approved for the prevention of erysipelas in geese and ducks [5] reinforces the dependence on antimicrobial therapy and emphasizes the need for continuous surveillance of AMR in this pathogen. Accordingly, a deeper understanding of the molecular mechanisms driving AMR and horizontal gene transfer in E. rhusiopathiae is crucial.
The acquisition and dissemination of resistance determinants in bacteria are often mediated by mobile genetic elements (MGEs), such as prophages, plasmids, genomic islands, and integrative and conjugative elements (ICEs), including transposons. These elements not only provide a reservoir of AMR determinants but also facilitate their horizontal transfer across strains and even species boundaries [6]. Prophages, in particular, are increasingly recognized as important vehicles of resistance genes in Gram-positive bacteria [7,8], while ICEs represent versatile elements capable of integrating into bacterial chromosomes and mediating conjugative transfer. To date, several MGEs carrying AMR genes have been identified in E. rhusiopathiae, including the bacteriophage φ1605, ICEEr0106 [9], ICEEr1023 and ICEEr1012 [10]. However, the diversity and functional roles of MGEs in this species, and their contribution to shaping its resistome, remain far from fully understood.
In our previous work, we characterized the antimicrobial susceptibility and resistance gene content of E. rhusiopathiae isolates from waterfowl, identifying multidrug-resistant (MDR) strains carrying both resistance genes and phage- or ICE-associated sequences. In serotype 8 isolate 670, the lsaE gene was located adjacent to phage-related genes, whereas serotype 5 isolate 147 harbored conjugation-related genes together with resistance determinants such as ant(6)-Ia and spw [5]. Encouraged by these observations, we carried out the present study to perform detailed genomic analyses of these E. rhusiopathiae isolates (670 and 147). We identified and characterized a novel prophage (φEr670) and a novel composite genomic island (GI_Er147), both associated with resistance genes, as well as the well-known transposon Tn916 carrying tetM. Furthermore, we examined the distribution of φEr670-like prophages in publicly available genomes of E. rhusiopathiae and related Gram-positive bacteria. Collectively, these analyses provide new insights into the contribution of MGEs to resistance gene carriage and dissemination in E. rhusiopathiae, and highlight their broader potential role in the spread of AMR among Gram-positive bacteria.
2. Results
2.1. Basic Genomic Analyses, ST Determination and Detection of Resistance Genes
The genome size of E. rhusiopathiae strains 670 serotype 8 and 147 serotype 5 was 1,874,230 bp and 1,918,116 bp, respectively, and the GC content was 36.3–36.6%. Both genomes contained 55 tRNAs with 3–7 rRNA loci (Table 1). These results are consistent with reports by other authors that the genome size of E. rhusiopathiae strains ranges from 1.6 to 1.9 Mb and the GC content ranges from 36% to 38%; the number of tRNAs ranges from 53 to 57 and the number of rRNA operons from 3 to 9 [10,11,12], NCBI database, The Ribosomal RNA Database (https://rrndb.umms.med.umich.edu, accessed on 15 July 2025).
ST 113 strains such as strain 670 were previously found to express serotype 8 and were isolated from waterfowl and pigs in Poland [5,13]. Globally, this sequence type appears to be rare: among 557 E. rhusiopathiae isolates analyzed, ST113 has been identified only in three serotype 1b strains from Canada [5]. ST 243 identified in strain 147 with serotype 5 was previously detected in only one of 557 E. rhusiopathiae strains, i.e., strain 267 with serotype 5 isolated from geese in Poland [10].
Genotypic resistance profiles obtained from WGS data were consistent with the results of previous PCR-based detection of resistance genes [5]. The multidrug-resistant strain 670 confirmed the presence of tetM, lnuB, and lsaE genes, while strain 147 contained tetM, ant(6)-Ia, and spw genes (Table 1).
2.2. Detection of Prophage Regions
Using the PHASTER tool, three regions containing incomplete prophage DNA were detected in the genome of E. rhusiopathiae strain 147 (PHASTER score < 60), while in strain 670 two prophage regions were detected (PHASTER score 80 and 150). One was highly homologous to the Erysipelothrix phage SE-1 (GB: NC_029078.1; size 33,997 bp) (score 150—intact), the second –designated φEr670—showed some similarity to the Corynebacterium Adelaide phage (GB: NC_048791.1; 44 kb) (score 80—questionable). The sequence of both prophages (SE-1 and φEr670) contained a clearly higher percentage of GC than (>40%) than the whole genome DNA (36.6%), indicating horizontal gene transfer.
The size of the φEr670 prophage was estimated at 53,020 bp (GB: CP183044.1:544708-597727), and further analyses using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 4 June 2025) showed its significant similarity to the sequence of phage Javan630 (48 kb) infecting Streptococcus uberis [14] and to phage φ1605 (90,000 bp, GB: MF172979.1) infecting E. rhusiopathie [9] (Figure 1). Both of these phages represent the class Caudoviricetes [9,14].
Regions highly homologous (≥89.9%) to the φEr670 prophage were detected in the genomes of 36 E. rhusiopathiae strains out of 586 (6.14%) examined, and in 1 out of 30 strains representing other Erysipelothrix species (Table S1). The φEr670-like prophages were also found in two strains of Thomasclavelia ramosa as well as in some genomes of Gram-positive bacteria outside the Erysipelothrixales family, i.e., Listeria, Anaerotignum, Eubacterium, Enterococcus and Blautia (Figure 1, Table 2). A notably high prevalence of φEr670-like prophages was observed in Listeria innocua; BLAST searches identified prophage-positive regions in at least 100 publicly available genomes of this species (Figure A1).
The size of φEr670-like prophage regions detected in E. rhusiopathiae strains (n = 36/586) and other Gram-positive bacteria ranged from 50 to 59 kb (excluding the larger 90 kb φ1605 phage) (Table 2), which is consistent with their classification as medium-sized dsDNA phages [15].
Within the φEr670 prophage, two AMR genes, i.e., lsaE and lnuB, were identified. Similarly, in most of the 36 E. rhusiopathiae strains carrying φEr670-like prophages, prophage-associated DNA also contained resistance determinants, including lsaE and lnuB (n = 12/36). In 17 genomes the prophage contained the ant(6)-Ia and spw genes, while in 11 genomes the tetM or mefA gene was additionally present. The tetM, mefA, msrD and lnuD-like genes were present in the φ1605 prophage of E. rhusiopathiae ZJ, and strain EMAI_31 had two homologous prophage regions, one containing the msrD and ermG resistance genes and the other the mphB gene (Table 2 and Table S1). Various resistance genes (lnuB, lsaE, ant6-Ia, spw mefA, msrD, mphB, tetM, arr, and putative resistance genes vat-like and lnu-like) were present within φEr670-like prophages detected in the genomes of Gram-positive bacteria outside the Erysipelothrix genus (Table 2, Figure 1).
Comparative genomic analysis revealed a high degree of structural conservation across φEr670 and its homologous prophages identified in E. rhusiopathiae and other Gram-positive bacteria. As shown in Figure 2, extensive synteny blocks span the major functional modules, including those involved in DNA replication, genome packaging (terminase subunits), virion assembly (capsid and tail proteins), and host–cell lysis. They correspond to the 38–46 core genes (out of 51 identified in φEr670) that were consistently present across the 36 prophage–positive E. rhusiopathiae genomes (Table S1). In contrast, genomic variation is largely confined to the accessory regions near the prophage termini, which exhibit substantial differences in gene content. Such divergence likely reflects intra-lineage modular exchange, including the acquisition of AMR genes, and is consistent with the natural genetic variability commonly observed among closely related prophages [15,16]. BLAST-based comparisons demonstrated 63–93% query coverage and 89.9–95.7% nucleotide identity relative to φEr670 (Table 2), and the corresponding orthoANI values ranged from 87.8 to 97.2% (or 92.7–97.2% for E. rhusiopathiae prophages when the divergent φ1605 phage is excluded). Together, these metrics indicate that the φEr670-like prophages form a cohesive evolutionary group, with most elements falling within or just below the ~95% ANI threshold often used to delineate species in dsDNA phage taxonomy [16].
The φEr670 prophage and φEr670-like prophages were characterized by a specific integration site (att). In E. rhusiopathiae strain 670 and other E. rhusiopathiae strains (except strains EMI_91 and EMAI_141), the prophage DNA was flanked by a gene encoding 23S rRNA (uracil(1939-(C5)) methyltransferase RlmD [its product catalyzes the formation of 5-methyl-uridine at position 1939 (m5U1939) in 23S rRNA] and a gene encoding FAD-dependent oxidoreductase. The RlmD methyltransferase gene also flanked prophage DNA in the genomes of other Gram-positive bacteria, indicating a key role of this gene in prophage DNA integration. (Figure 1 and Figure A1).
In our previous study, we confirmed the presence of three genes of prophage φEr670 (encoding the major capsid protein, a minor tail protein, and a site-specific recombinase) as well as the adjacent region encompassing the phage recombinase and lsaE gene in 7 out of 60 E. rhusiopathiae isolates recovered from waterfowl and in 1 out of 14 isolates from pigs. Notably, these three prophage genes were found exclusively in multidrug-resistant strains (lsaE-lnuB-tetM) of serotype 8 and ST113 [5]. In contrast, isolates from geographically distant regions that carried φEr670-like prophages represented other serotypes, most often 1a (10/36), 1b (11/36) and 2 (11/36), (Table S1). These findings suggest that the infection of E. rhusiopathiae strains with φEr670 phage is not dependent on serotype-specific receptor recognition. Instead, the occurrence of φEr670-positive strains of serotype 8 and ST113 on several poultry and swine farms in Poland may reflect clonal expansion and passive maintenance of integrated prophage DNA, rather than recent active transduction events.
The results of our analyses suggest that prophages of the φEr670 lineage play an important evolutionary role in shaping the resistome of E. rhusiopathiae and may contribute more broadly to the dissemination of antimicrobial resistance across Gram-positive bacteria. The phenomenon of AMR gene transfer via bacteriophage-mediated transduction is increasingly well documented and understood [26,27]. In Erysipelothrix rhusiopathiae, this mechanism was demonstrated by Gu et al. [9], who showed mitomycin C–induced transfer of the φ1605 prophage carrying the mef(A), msr(D), and tetM resistance genes from strain ZJ to strain G4T10. Similarly, future studies may assess whether the φEr670 prophage is capable of producing progeny phages that could infect E. rhusiopathiae strains and mediate the transfer of the lnuB and lsaE genes. To date, the occurrence of these resistance determinants within prophage-associated DNA has been reported only in Streptococcus pyogenes [28]. The present study therefore provides the first evidence of lnuB and lsaE being located within prophage sequences in E. rhusiopathiae as well as in Streptococcus uberis and Anaerotignum sp. (Table 1).
The detection of prophages of φEr670 family across distinct Gram-positive bacterial species is an intriguing finding that warrants further investigation. Bacteriophages have traditionally been considered narrow-host-range viruses, often restricted to specific strains of single species [29]. However, increasing evidence points to the existence of broad host range phages that contribute to interspecies gene transfer. For example, Listeria phage A511 targets a wide range of Listeria species [30] while Staphylococcus phage K infects multiple coagulase-positive and coagulase-negative staphylococci [31]. Similarly, wastewater-derived staphylococcal phages were shown to infect multiple Staphylococcus species [32], and phage Φm46.1 is able to transfer between S. suis and S. pyogenes [33]. Mechanistically, such broadened host tropism may result from the utilization of conserved surface receptors (e.g., wall teichoic acids), structural adaptability of tail fiber or baseplate proteins, and the deployment of anti-restriction or anti-CRISPR mechanisms that allow phages to circumvent taxon-specific bacterial defense systems [34,35]. Together, these observations—combined with the presence of φEr670-like prophages not only in E. rhusiopathiae but also across multiple Gram-positive bacterial species—strongly support the hypothesis that this phage lineage is capable of cross-species transfer. Such interspecies mobility positions φEr670-related prophages as important evolutionary drivers shaping the resistome of E. rhusiopathiae and potentially contributing to the broader dissemination of AMR among Gram-positive bacteria.
2.3. Detection of ICEs and Genomic Islands
In Erysipelothrix rhusiopathiae strains 670 and 147, an 18 kb tetM-carrying Tn916 transposon was identified. It showed >99.7% sequence similarity to Tn916 elements described in other Gram-positive bacteria, including E. rhusiopathiae [9,10,36,37,38]. In strain 670, the Tn916 transposon occurred as an independent element, whereas in strain 147, it was located within the 80 kb ICEEr1012 element, previously identified in E. rhusiopathiae strain 1012 isolated from geese in Poland [10]. The ICE_Er1012 element containing Tn916 was additionally detected in E. rhusiopathiae strain EMAI_31, while a variant lacking Tn916 (designated ICEEr1012a) was found in several E. rhusiopathiae isolates of swine origin from Australia (EMAI_35, 159, 130, 131, 133, 134, 135, 136, 171, 172, and 180) [17] (Figure 2). The presence of ICEEr1012a and Tn916 in association with a resistance gene cluster was also previously confirmed on the large (130 kb) genomic island ICEEr1023, identified previously in E. rhusiopathiae strain 1023 [10]. All of these MGEs (Tn916, ICEEr1012a, ICEEr1012, and ICEEr1023) were integrated at a specific chromosomal site located in the vicinity of tRNA-encoding genes (Figure 2), suggesting that this region may represent a genomic “hot spot” for the acquisition and recombination of MGEs in E. rhusiopathiae.
A 98 kb genomic island was detected in E. rhusiopathiae strain 147 (CP184721.1: 1185333-1283798) and designed as GI_Er147. It carries resistant genes, i.e., ant(6)-Ia, spw, vat-like gene and lnu-like gene as well as genes involved in bacterial conjugation, i.e., genes encoding recombinases, transposases, relaxase MobL and the genes found on type IV secretion systems (T4SS) (Figure 2). However, it is not known whether this is the complete set of genes enabling the formation of protein machinery for DNA transfer between bacterial cells. Therefore, this element was classified as a genomic island and not as an ICE (Figure 3).
GI_Er147 showed only little similarity to the genomic islands previously found in E. rhusiopathiae, i.e., ICEEr1023, ICEEr1012 [10] and ICEEr0106 [9] (Figure A2).
Within GI_Er147, a 30 kb integrative mobile element was identified, harboring two transposase genes (belonging to the IS3 and IS30 families) and a recombinase-family gene, which together suggest a potential capacity for mobility. This element, designated IME_Er30, was detected also in strain 670 and 3 other E. rhusiopathiae strains from geese in Poland (1023, and 267), in several dozen Australian isolates [17], and in E. amsterdamensis strain A18Y020d (GB: OW659496.1). In contrast, IME_Er30 was not found in the reference strains E. rhusiopathiae Fujisawa and NCTC 8163 (ATCC 19414), nor in isolates originating from China (ZJ, WH13013, ML101, SY1027, GAT10, SE38, and GXBY-1). The functions of IME_Er30 remain unknown, but this element may contribute to the acquisition of additional adaptive traits in E. rhusiopathiae. In the genomes of E. rhusiopathiae strains 670, EMAI_5, and EMAI_51, IME_Er30 is located adjacent to prophage SE-31, forming a hybrid IME–prophage element (Figure 3). In E. amsterdamensis strain A18Y020d, as well as in E. rhusiopathiae strain 147, IME_Er30 is positioned next to a large integrative element carrying Type IV Secretion System (T4SS) genes. IME_Er30, the SE-1 prophage, and hybrid elements formed through the combination of IME_Er30 with other MGEs such as GI_Er147, were all integrated at the same chromosomal locus in E. rhusiopathiae, located near the gene encoding tRNA 2-thiouridine(34) synthase MnmA. This integration site, similar to the insertion site of the Tn916 transposon and larger Tn916-containing ICEs, may represent a genomic “hot spot” for horizontal gene transfer in E. rhusiopathiae (Figure 3). Similar preferential integration near tRNA or tRNA-related genes has been widely reported for both prophages and integrative elements, as these loci contain short, conserved sequence motifs (att sites) that are efficiently recognized by integrases [39,40]. The coexistence of prophages and IMEs at the same chromosomal position also supports the concept of mosaic or hybrid genomic islands, which arise through sequential integration and recombination events between different MGEs [41].
Within GI_Er147, the presence of several resistance genes was demonstrated, i.e., ant(6)-Ia (aadE, coding for aminoglycoside nucleotidyltransferase ANT(6)-Ia), spw (encoding for ANT(9) spectinomycin adenyltransferase), vat family gene encoding putative streptogramin A O-acetyltransferase (2 copies), and gene encoding nucelotidyltransferase domain-containing protein (2 copies). The coexistence of ant(6)-Ia and spw genes noted in these studies is common in Gram-positive bacteria [10]. In E. rhusiopathiae they can be located on large genomic islands (e.g., ICEEr1023 and Er0106) [9,10,37], including prophages of φEr670 family [this study] (Figure 1 and Figure A2).
The gene encoding nucelotidyltransferase domain-containing protein (GB: CP184721.1 locus_tag AC-PCEN_09575 and ACPCEN_09590), designed as lnu(J) (495 bp, GB: PV882487), showed similarity to the lnu family genes encoding lincosamide nucleotidyltransferase, with the highest homology, i.e., 83.6% (100% coverage), with the lnu(D) gene (GB: NG_047925.1, 513 bp) (Figure 4). Genes homologous to lnu(J) gene (83–100% homology) were found in strains of various Gram-positive bacteria, i.e., Kurthia, Streptococcus, Anaerocolumna, Clostridium, and in phages infecting streptococci, as well as in several E. rhusiopathiae strains, i.e., ZJ (within prophage φ1605), and B3129, B3142 and B5577 (Figure 4). Unfortunately, susceptibility of these strains to lincosamides is unknown.
PCR analysis of 60 wild-type isolates of E. rhusiopathiae from waterfowl [5], including strains 147 and 670, showed that the lnu(J) gene was present only in two strains representing serotype 5, i.e., strain 147 and 136. Both strains were characterized by intermediate resistance to lincomycin, i.e., MIC 4–8 µg/mL and were susceptible to clindamycin (Table 3) [5].
The MIC for lincomycin of the E. rhusiopathiae strains in which neither the lnu(J) nor the lnu(B) gene was detected (n = 49/61, including 60 wild-type strains and reference strain E. rhusiopathiae ATCC 19414) was in the range 0.25–1 µg/mL, while the lnu(B)-positive E. rhusiopathiae strains (n = 9/61) are characterized by very high lincomycin MIC values, i.e., >64 µg/mL (with a simultaneous clindamycin MIC of 1–4 µg/mL) (Table 3) [5]. Despite these indications of the involvement of the lnu(J) gene in the reduction in the susceptibility of E. rhusiopathiae strains to lincomycin, the unambiguous definition of the role of the lnu(J) gene requires additional analyses.
It was previously shown that the lnu(D) gene, to which the lnu(J) gene shows 83.6% homology, was responsible for the moderate sensitivity of S. uberis UCN 42 strain to lincomycin (MIC = 2 µg/mL) (MIC for lnuD-negative S. uberis 72 strain was 0.06 µg/mL). Introduction of the recombinant pUC18 plasmid with the cloned lnu(D) gene into E. coli resulted in a change in the lincomycin MIC from 32 µg/mL to 128 µg/mL [42].
The gene encoding putative streptogramin A O-acetyltransferase, located within GI_Er147 showed 71.2% (89% coverage) similarity to the vat(B) gene (GB: NG_048538.1) and lower homology to other genes of the vat family (Figure A3). This gene was also detected in the genomes of other Gram-positive bacteria (deposited in the GenBank database), and in some strains (Kurthia and Bacillus, and within the phage YS387 detected in Streptococcus suis where it was located next to the lnu(J) gene. However, our studies did not confirm that the vat family gene of GI_Er147 confers resistance to streptogramins. Phenotypic testing using E-tests showed no difference in the sensitivity of strain 147 and vat-negative strains to quinupristin/dalfopristin (mixed streptogramins type A and B) (Table 3). It is also possible that the gene could also not be expressed as its promoter is not well recognized or because it encodes a different functionality as it is only 71.2% similar to known vat genes.
It is worth emphasizing that the serine recombinase encoding gene within GI_Er147 shows high homology to the recombinase of prophage φEr670 and related phages in other Erysipelothrix strains, as well as to recombinases carried by diverse MGEs (phages, plasmids, transposons and other ICEs) found in other Gram-positive bacteria (Figure A4). This pattern reflects the strong evolutionary conservation of serine recombinases, which mediate site-specific integration and excision. Unlike tyrosine recombinases, which use a stepwise cleavage–religation mechanism, serine recombinases catalyze coordinated double-strand breaks and strand exchange in a single concerted reaction [43], making them particularly efficient modules for horizontal movement across MGEs.
2.4. Core Genome Phylogeny
The phylogenetic tree based on the core-genome alignment clearly separates E. rhusiopathiae isolates into several distinct clades corresponding to their sequence types (STs) and geographic origins. All Polish isolates, except for strain 584 (ST32), form a single, well-supported branch, indicating a close genomic relationship. Strains 147 (ST243) and 670 (ST113) show the greatest similarity to isolates from Poland and Canada sharing the same STs. This clustering pattern suggests a common evolutionary background of Polish goose isolates, distinct from pig-derived strains from China and Japan and from porpoise isolates originating from the Netherlands. The reference strains NCTC 8163 (serotype 2, ST9, United Kingdom) and NCTC 7999 (serotype 1, ST118, France) form separate branches, while isolates from Asia (e.g., Fujisawa, ZJ, ML101) group together within the ST48 lineage (Figure 5).
3. Materials and Methods
3.1. Isolation, Identification, and Phenotypic Characterization of E. rhusiopathiae Strains
The two isolates of E. rhusiopathiae included in this study, i.e., 670 serotype 8 and 147 serotype 5, were isolated in 2020 from internal organs of domestic geese that were delivered dead to the laboratory for diagnostic purposes [2].
Strains E. rhusiopathiae 670 and 147 were selected from a pool of 60 E. rhusiopathiae isolates from waterfowl based on phenotypic and genotypic resistance profiles as well as detection of putative phage genes (coding for major capsid protein and minor tail protein) and ICEs related genes (mobL, int-Tn). The serotype of the strains was previously determined using multiplex PCR [5].
3.2. Whole Genome Sequencing
Nanopore sequencing was performed according to protocol SQK-RBK110.96 with Flow Cell ver. R10 on a MinION device (FLO-MIN106D; Oxford Nanopore, Oxford, UK), using the super-accurate base-calling method in MinKNOW v22.12.7. Reads were trimmed and down-sampled to 200× coverage using Filtlong (https://github.com/rrwick/Filtlong, accessed on 12 May 2025) and assembled into circular contigs using Flye v2.9.1 [44]. Genomes were polished using Medaka (https://github.com/nanoporetech/medaka, accessed on 4 June 2025) and Homopolish [45] and annotated using NCBI Prokaryotic Genome Annotation Pipeline (PGAP) [46]. The genome assemblies of both strains were deposited in the GenBank database (NCBI) under accession numbers CP183044.1 (strain 670) and CP184721.1 (strain 147).
3.3. Detection of Resistance Genes, Genomic Islands and Prophage DNA
Resistance genes were previously identified using PCR in E. rhusiopathiae strains 147 and 670 [5]. In this work, more detailed detection of resistance genes was performed based on WGS analysis using Resfinder 4.1 [47] and Resistance Gene Identifier (RGI) ver. 6.0.3 [48]. Synonymous mutations in the gyrA gene were determined by aligning amino acid sequences predicted using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/, accessed on 12 June 2025). Genomic islands of E. rhusiopathiae strains 670 and 147 were identified by aligning the whole genome sequences of these strains with those of the reference strains E. rhusiopathiae Fujisawa and NCTC 8163 using the BLAST tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 1 October 2025). Prophage DNA was detected in the genomes of the E. rhusiopathiae strain 147 and 670 using the PHAge Search Tool—Enhanced Release (PHASTER) [49]. Regions homologous to the φEr670 prophage were detected in the genomes of E. rhusiopathiae strains (n = 586) and other Erysipelothrix species (n = 30) retrieved from the GenBank database (Table S1) using Kaptive v3 and a custom database containing the annotated sequence of φEr670 [50].
3.4. Determination of Susceptibility to Streptogramins and Lincosamides
The susceptibility of strain 147 (vat-family positive) and several other E. rhusiopathiae strains with known genomic sequence to streptogramins was determined using E-tests containing quinupristin/dalfopristin (Liofilchem, Roseto degli Abruzzi, Italy). The test was performed on blood agar with a bacterial inoculum of 0.5 McFarland density. The plates were incubated for 24 h at 37 °C, 5% CO_2_. The susceptibility of the bacteria to lincomycin and clindamycin was determined using the broth microdilution method [5]. The antimicrobial susceptibility test was performed in duplicate.
3.5. Detection of lnu(J) Gene
The lnu(J) gene identified in the genome of strain 147 was detected by PCR in 60 wild-type E. rhusiopathiae isolates listed in our previous paper [5]. The negative control was the reference strain E. rhusiopathiae ATCC 19414 (NCTC 8163), whose genome is deposited in the GenBank database (NZ_LR134439.1). Amplification of lnu(J) gene was performed using the following primers: forward 5’-TTGGATAGATGGCGGTTGGG-3’ and reverse 5’-ACTTGTATTCACTCGGAACAGGAA-3’. The PCR product size was 431 bp. For 5 wild type isolates whose WGSs are publicly available (nos. 1023, 1012, 584, 267, 670), detection was based also on in silico analysis.
3.6. Determination of Homology Between DNA Sequences
The similarity of the MGEs detected in strains 670 and 147 to the sequences found in the genomes of other E. rhusiopathiae strains as well as other bacterial species was determined using BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 13 September 2025) and Clinker [51].
Average nucleotide identity (ANI) between prophage φEr670 and homologous prophages was calculated with OrthoANI [52] using online calculator (www.ezbiocloud.net/tools/ani, accessed on 6 December 2025).
The phylogenetic relationships between the lnu(J) and vat-family genes identified in E. rhusiopathiae strain 147 and reference lnu and vat genes were inferred using the Maximum Likelihood method implemented in MEGA ver. 11 [53].
3.7. Multilocus Sequence Typing
Multilocus sequence typing (MLST) of E. rhusiopathiae strains was performed according to the scheme developed by Webster et al. [17] using mlst v.2.19.0 (https://github.com/tseemann/mlst, accessed on 18 June 2025).
3.8. Phylogenetic Inference
The phylogenetic analysis included the E. rhusiopathiae strain 670 and 147 from geese and 25 other E. rhusiopathiae strains, including several MDR strains which, like strain 670, contained the lsaE, lnuB and tetM genes. Two of these strains, i.e., ZJ and 2860, contained regions homologous to the φEr670 prophage, and strain 2860, like strain 670, represented ST113. In turn, strain 267 represents the same sequence type as strain 147, i.e., 243. WGSs of 25 E. rhusiopathiae strains used for comparative analysis were downloaded from the GenBank database (accession numbers in Table S1). Whole genome alignments were performed using parsnp v2.0 [54]. Phylogenetic trees were constructed using Fasttree v 2.1.11 [55] and visualized using iTOL v5 [56].
4. Conclusions
In this study, we provide a comprehensive whole-genome characterization of MGEs associated with AMR in two E. rhusiopathiae strains, i.e., multidrug-resistant serotype 8 strain 670 and the serotype 5 strain 147, isolated from domestic geese. We report the identification of a previously undescribed prophage, φEr670 (53 kb), carrying the lnuB and lsaE resistance genes, as well as a novel hybrid genomic island, GI_Er147 (98 kb), harboring several resistance determinants and T4SS genes. Together with the detection of Tn916 (18 kb), ICEEr1012 (80 kb), and two chromosomal “hotspots” that promote the integration and recombination of diverse MGEs, these findings reveal a previously underappreciated complexity of the E. rhusiopathiae mobilome.
The widespread distribution of φEr670-like prophages—most of which also carry AMR genes—strongly suggests that this phage lineage plays an important role in shaping the resistome of E. rhusiopathiae and other Gram-positive bacteria.
Overall, this study provides new insights into the diversity and organization of MGEs in E. rhusiopathiae, thereby advancing current understanding of the genomic mechanisms underlying the evolution and dissemination of AMR in this species and potentially across Gram-positive bacteria. Further studies are warranted to elucidate the mobility of the newly identified genetic elements and the functional significance of the lnu(J) gene encoding a putative lincosamide nucleotidyltransferase.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stackebrandt E. Erysipelothrix Bergey’s Manual of Systematics of Archaea and Bacteria 1st ed. Whitman W.B. John Wiley & Sons, Inc.Hoboken, NJ, USA Bergey’s Manual Trust Glasgow, UK 201511510.1002/9781118960608.gbm 00763 · doi ↗
- 2Nowak T. Wódz K. Kwiecieński P. Kwieciński A. Dec M. Incidence of erysipelas in waterfowl in Poland—Clinical & pathological investigations Br. Poult. Sci.20246576276810.1080/00071668.2024.240633139373393 · doi ↗ · pubmed ↗
- 3Bobrek K. Nowak M. Borkowska J. Bobusia K. GawełA. An outbreak of erysipelas in commercial geese Pak. Vet. J.201636372374
- 4Jordan F.T.W. Bisgaard M. “Erysipelothrix rhusiopathiae—Różyca” [Erysipelothrix rhusiopathiae—Erysipelas]Choroby Drobiu [Poultry Diseases]1st ed. Wieliczko A. Edra Urban & Partner Wrocław, Poland 2011257261
- 5Dec M. Nowak T. Webster J. Wódz K. Serotypes, Antimicrobial Susceptibility, and Potential Mechanisms of Resistance Gene Transfer in Erysipelothrix rhusiopathiae Strains from Waterfowl in Poland Int. J. Mol. Sci.2024251219210.3390/ijms 25221219239596258 PMC 11595068 · doi ↗ · pubmed ↗
- 6Han B. Feng C. Jiang Y. Ye C. Wei Y. Liu J. Zeng Z. Mobile genetic elements encoding antibiotic resistance genes and virulence genes in Klebsiella pneumoniae: Important pathways for the acquisition of virulence and resistance Front. Microbiol.202516152915710.3389/fmicb.2025.152915740066273 PMC 11891212 · doi ↗ · pubmed ↗
- 7Huang J. Dai X. Wu Z. Hu X. Sun J. Tang Y. Zhang W. Han P. Zhao J. Liu G. Conjugative transfer of streptococcal prophages harboring antibiotic resistance and virulence genes ISME J.2023171467148110.1038/s 41396-023-01463-437369704 PMC 10432423 · doi ↗ · pubmed ↗
- 8Quirós P. Colomer-Lluch M. Martínez-Castillo A. MiróE. Argente M. Jofre J. Navarro F. Muniesa M. Antibiotic resistance genes in the bacteriophage DNA fraction of human fecal samples Antimicrob. Agents Chemother.20145860660910.1128/AAC.01684-1324165177 PMC 3910789 · doi ↗ · pubmed ↗