Integration of cAMP and TRPV4 Signaling to Optimize Collagen Remodeling for Management of Fibrosis
Connie Di Raimo, Christopher A. McCulloch

TL;DR
The paper explores how cAMP and TRPV4 signaling interact to influence collagen remodeling and fibrosis, suggesting a new approach combining Gαs GPCR activation and TRPV4 inhibition.
Contribution
The study reveals a novel bimodal therapeutic strategy for fibrosis by coordinating Gαs GPCR activation and TRPV4 inhibition.
Findings
Gαs GPCRs generate cAMP, which inhibits fibrotic processes by optimizing collagen degradation.
Dysregulated TRPV4 promotes myofibroblast differentiation and reduces β1 integrin expression.
Coordinated activation of Gαs GPCR and inhibition of TRPV4 may control tissue fibrosis.
Abstract
What are the main findings? Gαs GPCRs generates cAMP, which inhibits fibrotic processes by optimizing intracellular collagen degradation and remodeling.TRPV4 activation regulates collagen remodeling under physiological conditions, but when dysregulated, TRPV4 promotes myofibroblast differentiation and reduces β1 integrin expression. Gαs GPCRs generates cAMP, which inhibits fibrotic processes by optimizing intracellular collagen degradation and remodeling. TRPV4 activation regulates collagen remodeling under physiological conditions, but when dysregulated, TRPV4 promotes myofibroblast differentiation and reduces β1 integrin expression. What are the implications of the main findings? Ca2+ influx through TRP channels modulates cAMP levels by regulating phosphodiesterases and adenylyl cyclases, underpinning the interplay between these signaling systems.Coordinated activation of the Gαs…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
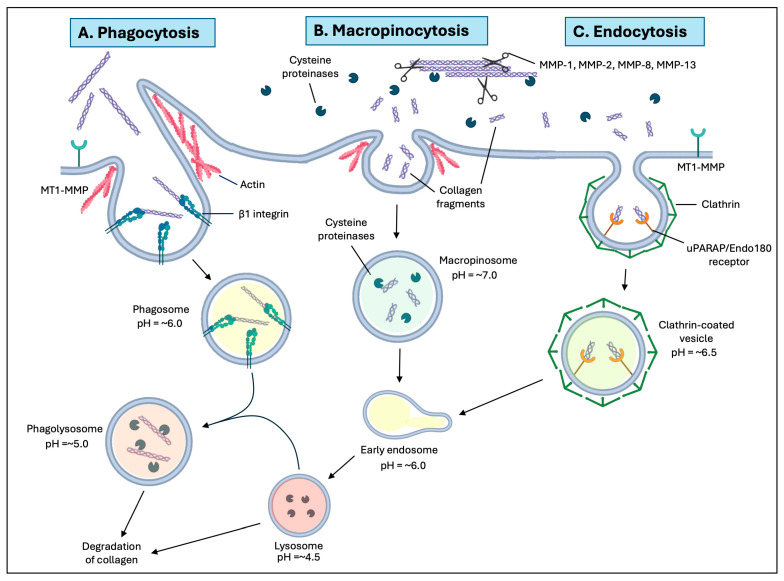 Figure 1
Figure 1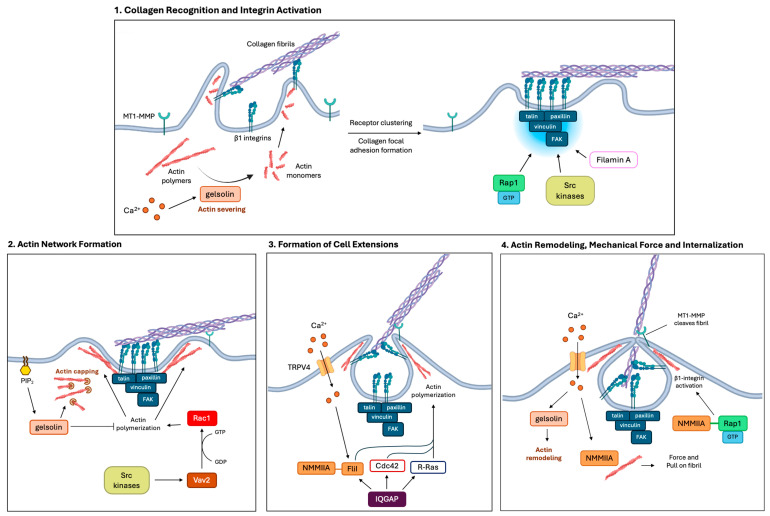 Figure 2
Figure 2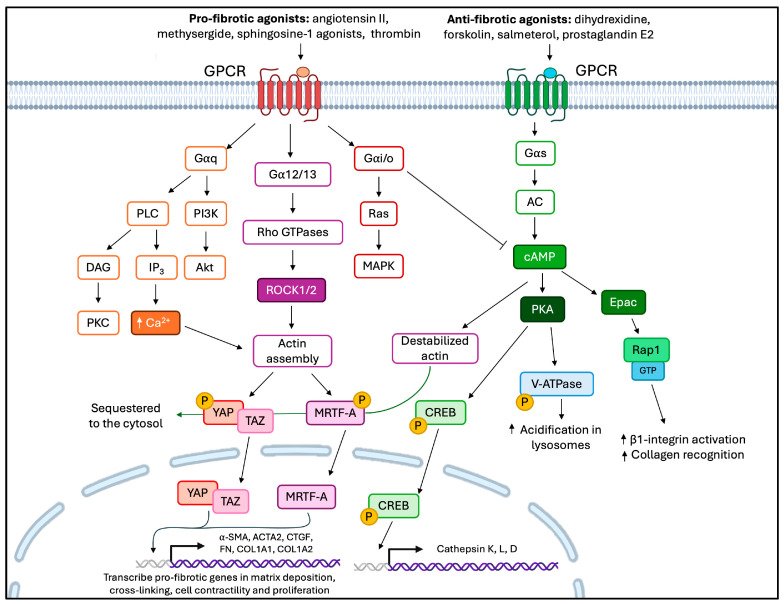 Figure 3
Figure 3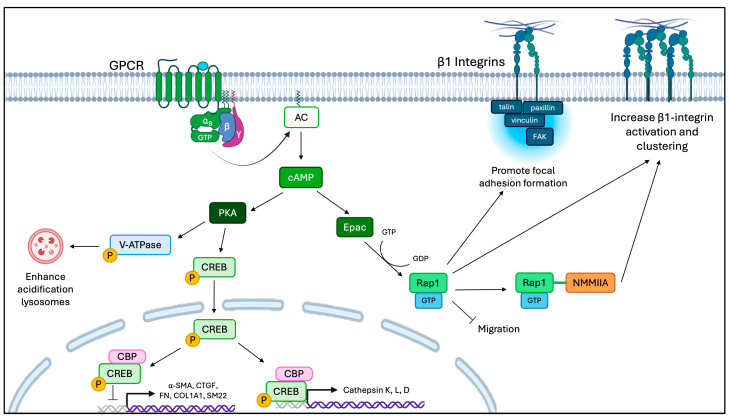 Figure 4
Figure 4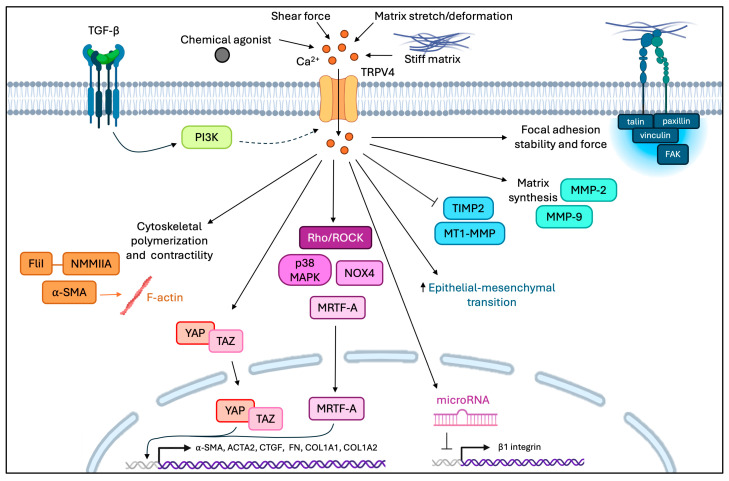 Figure 5
Figure 5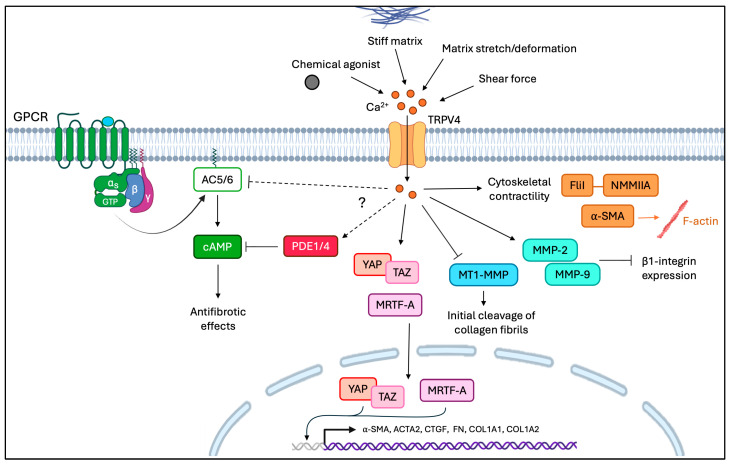 Figure 6
Figure 6- —CIHR operating grant
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsConnective Tissue Growth Factor Research · Interstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Phosphodiesterase function and regulation
1. Introduction
The mechanical and structural properties of the extracellular matrix (ECM) are precisely regulated by complex signaling pathways that enable matrix homeostasis in healthy tissues. The ECM undergoes continuous and controlled remodeling: dysregulation of this process compromises organ structure and function. Fibrosis manifests as the excessive accumulation of stiffened fibrillar collagen that affects multiple organs, such as the liver, lung, skin, cardiac muscle, kidney, eye, and periodontium [1,2]. Multiple stimuli promote the formation of fibrotic lesions, including tissue exposure to chemicals, radiation, injury, and/or as a side effect of certain drugs. High-prevalence diseases, including cardiac hypertrophy, metabolic dysfunction-associated steatohepatitis, idiopathic pulmonary fibrosis, scleroderma, chronic kidney disease, periodontitis, and tumor invasion, manifest with poorly organized, stiffened arrays of fibrillar collagen [1,3,4,5], which reflects a loss of the balance between matrix synthesis and degradation that is perturbed. The fibrotic lesions of these diseases arise in particular from reduced degradation of fibrillar collagen. Accordingly, understanding the control of intracellular collagen degradation is central to our understanding of fibrotic diseases and in the identification of new targets for the development of anti-fibrotic drugs.
Collagen synthesis and degradation have been extensively characterized in diverse physiological and pathological conditions. In particular, the multi-dimensional aspects of extracellular collagen degradation and uptake continue to be actively explored [6,7]. Collagen is degraded by numerous catabolic pathways that are localized in the extracellular or intracellular space. Extracellular degradation of collagen is directed by matrix metalloproteinases (MMPs) that cleave collagen fibrils into fragments, which are subsequently degraded through intracellular processes [8,9]. Collagen can be degraded intracellularly via three distinct pathways: integrin-mediated phagocytosis, receptor-mediated endocytosis, and macropinocytosis-like uptake. Collagen phagocytosis uses β1 integrin receptors for cell adhesion to fibrils and non-muscle myosin II filaments, and a large cadre of actin-binding proteins to internalize fragmented collagen fibrils [9,10,11]. All three discrete collagen internalization pathways converge on the delivery of collagen to phagolysosomes, vacuolar compartments in which collagen is degraded by lysosomal hydrolases, such as acid-optimal cathepsins. Defining which pathways are involved in enhancing intracellular collagen degradation and in suppressing myofibroblast differentiation are key goals in controlling the pathological accumulation of disorganized collagen that manifests in fibrotic diseases.
G protein-coupled receptors (GPCRs) are implicated in the increase and reduction in fibrosis, depending on which Gα subclass is activated. These proteins are considered promising therapeutic targets for the clinical management of fibrosis. Notably, Gα stimulatory (Gαs) GPCRs stimulate the production of the second messenger cyclic adenosine monophosphate (cAMP), which, in general, slows the progression of fibrosis [3,4,5]. While the potentially anti-fibrotic effect of cAMP could be exploited to increase collagen degradation, we note that ECM remodeling involves multiple, complex processes. These processes must be precisely balanced by multiple signaling systems to maintain matrix homeostasis [1]. Accordingly, therapeutic perturbation of intracellular collagen degradation to reduce fibrosis needs to be carefully managed to avoid unintentional off-target effects, such as excessive ECM degradation.
Anchorage-dependent cells exhibit specialized collagen adhesions (often containing β1 integrins) that act as mechanosensors. These adhesions can “sense” the mechanical properties of the surrounding ECM (e.g., stiffness of collagen fibers). Subsequently, myosin-generated contractile forces enable internalization of individual collagen fibrils. The internalization of collagen fibrils is regulated by multiple processes, including calcium ion (Ca^2+^) conductance through plasma membrane channels [10]. Aberrant function of Ca^2+^-permeable channels and the resultant perturbation of downstream Ca^2+^ signaling pathways are often manifested in fibrotic lesions [12,13,14]. The regulation of Ca^2+^ influx through channels such as transient receptor potential vanilloid type 4 (TRPV4) is important in modulating matrix remodeling. TRPV4 is expressed in multiple cell types and is a polymodal, Ca^2+^-permeable, mechanosensory channel that can detect alterations in the mechanical properties of the ECM. Conversely, TRPV4 affects the mechanical properties of the ECM by its regulation of collagen remodeling [15]. Dysregulation of TRPV4 signaling enhances a pro-fibrotic positive feedback loop in which ECM is pathologically stiffened, which increases matrix stiffness and enhances TRPV4 activation. Ca^2+^ permeation through ion channels like TRPV4 modulates the effects of other second messengers like cAMP. Here, we focus on the functional relationships between the second messengers cAMP and Ca^2+,^ in particular when the balance between matrix synthesis and degradation is perturbed and when ECM structure and function are altered. We discuss the interplay between GPCR and TRPV4 signaling systems in the context of collagen remodeling. We consider how a collective approach to the regulation of cAMP and Ca^2+^ could suggest new treatments for clinical management of fibrotic lesions.
2. Physiological Mechanisms of Collagen Remodeling
The ECM comprises dynamic, three-dimensional, non-cellular structures that are present in most types of tissues and undergo tightly regulated remodeling for tissue homeostasis. ECMs are composed of >300 proteins, referred to as the core matrisome, of which fibrillar collagen is the most abundant component [16,17]. Remodeling of fibrillar collagen maintains the mechanical properties, signaling functions, and structural integrity of different tissues [6,9,18]. Collagen synthesis and secretion are exhibited by numerous cell types, including interstitial fibroblasts, macrophages, and tissue-specific cells (e.g., mesangial cells of the kidney) to maintain tissue health. Fibroblasts in connective tissues comprise heterogeneous subpopulations that have been characterized by transcriptomic and proteomic methods and exhibit tissue-specific remodeling traits [19,20]. In healthy tissues, fibroblasts balance synthesis, degradation, and reorganization of fibrillar collagen to preserve tissue structure and function [21]. Several discrete collagen degradation pathways have been identified in the extracellular space and in certain intracellular compartments (Box 1).
Box 1Collagen degradation pathways.Collagen remodeling is essential for maintaining tissue homeostasis. Collagen can be degraded through extracellular and intracellular pathways. Extracellular degradation of collagen is predominantly performed by matrix metalloproteinases and extracellular cathepsins that cleave collagen fibrils [17]. These collagen fragments can undergo further denaturation in the extracellular space, or the fragments can be internalized by local cells and further degraded by intracellular pathways. The main intracellular pathways for collagen internalization are macropinocytosis, receptor-mediated endocytosis, and phagocytosis, the latter of which is mediated by β1 integrins. Macropinocytosis and receptor-mediated endocytosis primarily internalize cleaved soluble collagen fragments, while β1 integrin-containing adhesion receptors serve as the initial sites where phagocytosis leads to the internalization and eventual degradation of intact collagen fibrils [6]. All three intracellular collagen degradation pathways converge on lysosomes, organelles in which collagen is degraded by acid-optimized cysteine proteases, including cathepsins [6,18].
3. Pathways of Collagen Degradation
3.1. Extracellular Collagen Degradation
The extracellular collagen degradation pathway is dominated by the catalytic activity of matrix MMPs and, in some cell types (such as fibroblasts), by the lysosomal cysteine protease, cathepsin K [22,23,24]. MMPs are a family of zinc endopeptidases that exhibit collagenolytic activity and have been extensively characterized in the context of ECM degradation in various disease states [25]. Fibrillar collagen is resistant to most proteolytic enzymes and can be cleaved only by a subset of MMPs that include enzymes with triple helicase activity, such as MMP-1, MMP-8, MMP-13, and MMP-14 [8,25]. The extracellular MMP pathway generates collagen fragments that can be further degraded by gelatinases such as MMP-2 or MMP-9; collagen fragments may also be degraded intracellularly. In this review, we focus on the control of intracellular collagen degradation since the extracellular pathway has been examined in considerable depth (see reviews [25,26]).
3.2. Intracellular Pathways of Collagen Degradation
The intracellular pathways of collagen degradation are distinct and can process previously cleaved and intact fibrillar collagen located in the extracellular space. Pre-cleaved collagen fragments can be internalized by macropinocytosis, or for soluble collagen, by receptor-mediated endocytosis (Figure 1). The initial steps of phagocytosis of intact collagen fibrils are largely reliant on β1 integrin-mediated collagen binding; the internalized collagen is subsequently degraded by lysosomal cysteine proteases such as cathepsins (Figure 1) [9]. β1 integrin activation also promotes the expression of the dipeptidase prolidase through the mitogen-activated protein kinase (MAPK) pathway [27]. Prolidase catalyzes the hydrolysis of dipeptides that contain proline or hydroxyproline on the C-terminal [28,29]. Prolidase recycles collagen and is regulated by surface receptor–ECM interactions, as its activity increases with cell growth and density [28,29]. Additionally, nascent collagen can also be degraded by autophagocytosis, in which newly synthesized molecules are destined for destruction prior to secretion and are enclosed in double membrane-bound autophagosomes. These collagen fragments are further degraded by cysteine proteases after the fusion of autophagosomes with lysosomes. Macropinocytosis is an actin-dependent, endocytic uptake pathway by which soluble collagen enters into large vesicles (macropinosomes) [9]. Macropinocytosis is often activated in response to stimulation by growth factors and is also seen in cells with dysregulated signaling (e.g., cancer) and in cells and tissues in which there is rapid ECM remodeling [30,31,32].
In a separate and distinct pathway, pre-cleaved collagen can be internalized via the receptor-mediated endocytic pathway, which involves the urokinase plasminogen activator receptor-associated protein (uPARAP/Endo180) and the formation of clathrin-coated vesicles [33]. uPARAP/Endo180 is a type-1 membrane protein of the mannose receptor family that binds directly to collagen and is expressed in mesenchymal cells, including subpopulations of fibroblasts, osteoblasts, and chondrocytes [33,34,35]. Following internalization of collagen fragments, clathrin-coated vesicles transit to early endosomes and subsequently mature into terminal lysosomes, where collagen is degraded by cysteine proteases [36,37]. Extracellular collagenases and the uPARAP/Endo180 receptor cooperate to efficiently endocytose collagen in fibroblasts [38]. As uPARAP/Endo180-mediated endocytosis occurs independently of the phagocytosis of largely intact fibrils, these pathways are considered to be fundamentally distinct [39,40].
4. Collagen Phagocytosis
4.1. Collagen Recognition, Focal Adhesion Formation, and Actin Network Formation
As described above, intact collagen fibrils are internalized by collagen phagocytosis, which is mediated by β1 integrin-containing collagen receptors [41,42]. The initial cleavage of pericellular collagen fibrils is mediated by membrane-type 1 MMP (MT1-MMP), which is a rate-limiting enzyme for collagen phagocytosis [43]. Fibroblasts recognize collagen fibrils by binding to GFOGER sequences using the cell-surface, heterodimeric transmembrane α2β1 or α11β1 integrins [44,45,46]. Collagens that are not assembled into fibrils preferentially bind to the α1β1 and α10β1 integrins [44,45,46,47]. Integrin binding to collagen fibrils promotes receptor clustering and the activation of Rap1, Src, and talin, which in cultured cells lead to the formation of focal adhesion complexes [48,49]. Cell binding to collagen fibrils also involves Ca^2+^-dependent actin filament severing in focal adhesions by the actin-binding protein, gelsolin (Figure 2) [50]. Collectively, the recognition of collagen and the assembly of signaling-competent adhesion complexes are important rate-limiting steps for phagocytosis of collagen fibrils [42].
Integrin-based focal adhesion complexes contain a large cadre of scaffolding proteins and adaptors that bind actin filaments (i.e., talin, vinculin, paxillin, filamin, α-actinin, zyxin) and signaling proteins such as the focal adhesion kinase (FAK), integrin-linked kinase, and Src [51,52]. Through its interaction with filamin A, talin contributes to β1 integrin activation. The integrin activation step enables high-affinity binding to collagen fibrils [53], which is an important determinant of the initiation of both collagen phagocytosis and synthesis. Focal adhesions are mechanosensing signaling hubs that control how integrins bind to the ECM and integrate with the actin cytoskeleton [10]. The maturation, protein composition, and the regulatory mechanical cues that determine focal adhesion complex function, modify whether fibroblasts favor collagen synthesis or phagocytosis [54]. When ECM polymers are subjected to tensile forces, the formation of focal adhesion complexes is transient and promotes ECM engulfment [10,54]. Meanwhile, mechanosensitive transcription factors remain sequestered in the cytosol [10,54]. Additional regulation of collagen phagocytosis is associated with collagen-bound proteoglycans (e.g., decorin) and glycoproteins (e.g., fibronectin), which can modulate collagen interactions with its cognate receptors. Collagen may also interact with other, non-collagen-binding integrins to promote phagocytosis [55]. Notably, the collagen-binding capacity of β1 integrin-containing adhesions is affected by non-collagenous proteins that coat the surface of the fibrils rather than the core of the collagen molecules [55].
Following collagen recognition and integrin activation, fibroblasts exhibit actomyosin remodeling to enable internalization of collagen fibrils [56,57]. This process involves the small GTPase Rac1 and a constellation of actin-binding proteins (e.g., gelsolin) that contribute to remodeling of sub-cortical actin filaments and engagement of the fibril internalization process [56,57]. The switch from RhoA to Rac1 favors collagen phagocytosis instead of collagen synthesis [56,57]. At early stages of actin network assembly, which is manifest in the phagocytic envelopment of collagen fibrils, Src family kinases phosphorylate the guanine nucleotide exchange factor (GEF) Vav2 [56]. This step is followed by Vav2-mediated activation of the small GTPase Rac1 to promote actin polymerization and the further development of a phagocytic cup to surround the collagen fibril [56]. As the collagen fibril phagocytic cup forms, phosphatidylinositol 4,5-bisphosphate (PIP_2_) accumulates at the site of internalization, which enables gelsolin to uncap the barbed end of actin filaments and mediate rapid actin assembly at the cup [50,58].
4.2. Mechanism of Collagen Internalization
For efficient internalization of collagen, actin-rich cell extensions are generated through the activities of the small GTPase IQGAP1, which interacts with Cdc42, R-ras, and flightless-1 (FliI). These signals promote the formation of extensions that surround the fibril, which is followed by internalization [59,60]. Mechanical force is also required to physically pull the fibril inside the cell [10,50,61,62]. This process relies on Ca^2+^-dependent, actomyosin contractility [10,50,61,62]. The mechanical tension generated by contractile forces applied through integrins to collagen fibrils activates mechanosensitive TRPV4 channels that allow localized influx of extracellular Ca^2+^ at the phagocytic site [61]. Here, Ca^2+^ influx through TRPV4 channels regulates the interaction between non-muscle myosin IIA (NMMIIA) and Flil to generate cell extensions that are important for fibril envelopment [61]. Localized Ca^2+^ bursts activate gelsolin’s actin-severing function, which enhances collagen binding to previously unoccupied β1 integrin-containing adhesions [57]. Gelsolin also binds to full-length NMMIIA at collagen adhesions in a Ca^2+^-dependent manner [10,11]. NMMIIA assembles into filaments that bind to the actin filament network and mediate the generation of localized contractile forces. These activities synergize through the interaction of Rap1 with NMMIIA, which promotes additional β1 integrin activation [10,48]. Collectively, these processes promote cell adhesion to collagen, activate Ca^2+^ entry through plasma membrane channels (e.g., TRPV4), and enhance fibril internalization through actomyosin-dependent membrane remodeling (Figure 2) [10,11,48].
Fibroblasts initially engulf part of the fibril using actin-rich pseudopods at matrix adhesion sites [35]. Once internalized, the membrane-enclosed fibril (in a phagosome) fuses with a lysosome to create a phagolysosome, a vacuolar compartment in which collagen fibrils are degraded by lysosomal enzymes that include cysteine proteases [63,64,65,66]. Certain lysosomal cysteine proteases exhibit efficient and robust collagenolytic activity, including cathepsins L, K, S, and B [23,65,67,68,69]. In theory, deficiencies in the activation, catalytic function, localization, and local pH associated with any cathepsins that degrade collagen will reduce the effectiveness of collagen digestion and contribute to fibrosis [24,70,71,72].
5. Dysregulation of ECM in Fibrotic Lesions
Small perturbations of the signaling modules that control collagen remodeling often contribute to long-standing, chronic disease and to alterations in organ structure and function. Healthy ECM remodeling requires a careful balance between collagen synthesis and degradation, which is dysregulated in fibrotic diseases, connective tissue disorders, muscular dystrophy, and invasive cancers (Box 2) [73,74]. Fibrosis is a major pathological feature of diseases that contributes to ~35% of global deaths (2019) [75].
Box 2Chronic wound healing leads to fibrosis.Fibrosis manifests as an excessive and progressive accumulation of ECM components, such as collagen and fibronectin. These imbalances are frequently found in inflamed or injured tissues [71]. The production of excessive ECM disrupts normal tissue and organ architecture, leading to permanent scarring and/or organ dysfunction [12,71,72]. Aberrant ECM deposition can result ultimately in death, as seen in end-stage liver disease, kidney disease, idiopathic pulmonary fibrosis, and heart failure [72]. Fibrosis affects multiple organ systems, such as skin, lungs, heart, kidneys, pancreas, and periodontal tissues [7,73].
Fibrosis is often associated with chronic inflammation and frequently manifests as excessive accumulation of ECM. The drivers for fibrotic lesions include a wide array of stimuli, including persistent infections, allergic reactions, autoimmune responses, tissue exposure to chemicals, radiation, injury, or side effects of certain drugs (Table 1) [76]. There has been a rise in cases of drug-induced pulmonary fibrosis, which has become increasingly prevalent in respiratory disability [77,78]. Drugs that lead to fibrosis provide a useful method to understand the complex underlying mechanisms of fibrosis. Tissues that sustain repeated injury and/or exhibit chronic inflammation are more likely to manifest fibrosis, partly due to continuous and overactive wound healing processes [79]. For example, in drug-induced periodontal diseases, which are high-prevalence disorders in adults, fibrosis manifests as the formation of dense, stiffened, dysfunctional tissues that fail to effectively seal around the necks of teeth [80].
6. Drivers and Cellular Mediators of Fibrosis
An important, pleiotropic cytokine in wound healing that also promotes the formation of fibrotic lesions is transforming growth factor-β1 (TGF-β1) [1,204,205,206,207]. TGF-β acts through canonical SMAD signaling and non-canonical mitogen-activated protein kinase (MAPK)/JNK/p38 signaling pathways to regulate the expression of pro-fibrotic genes [208]. TGF-β1 potently stimulates the accumulation of the ECM, regulates inflammation, and can enhance α-SMA expression in myofibroblasts; all of these factors promote fibrosis [209,210]. Mechanosensors such as stretch-activated membrane channels (e.g., TRPV4 and Piezo1) can be mechanically activated in myofibroblasts and thereby increase cytosolic Ca^2+^ [211,212]. In addition to the effect of increased cytosolic Ca^+2^ on collagen phagocytosis (see above), increased intracellular Ca^2+^ activates Rho-associated protein kinase (ROCK) signaling, which results in enhanced actomyosin contractility [211,212]. Notably, environmental stimuli such as repeated cyclic strain, shear stress, and enhanced ECM stiffness can activate TRPV4, which in turn, enhances ECM production [208,213]. Further, increased Ca^2+^ influx through TRPV4 has been spatially associated in cells with locally high abundance of the fibrillar collagen-binding discoidin domain receptor 1 (DDR1) [214]. DDR1 increases collagen compaction and alignment driven by its association with Ca^2+^-dependent NMMIIA, which drives localized collagen stiffening at sites of cell attachment [214]. As pathological ECM remodeling is a key characteristic of fibrosis, myofibroblasts are therefore of central importance in the excessive production of stiffened collagen fibrils in these lesions.
7. Role of the Myofibroblast in Fibrotic Lesions
In many fibrotic diseases, a key culprit in fibrotic ECM generation and remodeling is the persistent activation and recruitment of myofibroblasts [215]. These cells exhibit certain prominent features of the mesenchymal lineage, including expression of vimentin intermediate filaments and the synthesis and secretion of collagens I and III at high levels [216]. During normal tissue repair, locally generated signals can promote the conversion of fibroblasts to myofibroblasts, which exhibit specialized ECM contractile machinery that accelerates wound closure of damaged tissues [2]. Indeed, one of the critical phenotypic features of myofibroblasts is their ability to reorganize and condense the ECM, which alters the mechanical properties of tissues (i.e., increased stiffening) during tissue repair.
The phenotypic transition of relatively non-contractile fibroblasts into highly contractile myofibroblasts is temporally associated with increased transcription of muscle-associated genes like ACTA2, which codes for the contraction-associated actin isoform, α-smooth muscle actin (α-SMA) [215]. In fibrotic lesions, excessive synthesis of disorganized collagen fibrils along with the formation of abundant, highly aligned and cross-linked fibrils, fundamentally alters the mechanical properties and function of local tissues [2,217], which is further aggravated by reduced collagen degradation [74]. The formation of a stiff, fibrotic matrix promotes myofibroblast activation, which can generate a positive feedback loop that synergistically increases the deposition of a fibrotic ECM and pathological remodeling [2,215,217,218,219].
Signals That Activate Myofibroblast Formation
Multiple pathways regulate fibroblast-to-myofibroblast conversion. In this context, we note that many downstream mechanosignaling events converge, leading to the activation of the pro-fibrotic transcription factors myocardin-related transcription factor A (MRTF-A), Yes-associated protein 1 (YAP), and transcriptional coactivator with PDZ-binding motif (TAZ) [2,208,211,216]. YAP, TAZ, and MRTF-A translocate to the nucleus in cells that are subjected to increased mechanical cell stress. In the nucleus, YAP and TAZ act as transcriptional co-activators that drive the expression of pro-fibrotic genes like COL1A1, COL1A2, CTGF, and ACTA2, and the upregulation of each other [220,221,222,223]. Following nuclear localization, YAP/TAZ regulates gene transcription through its association with DNA-binding TEA domain family members (TEAD 1–4) [224,225,226]. In unstressed cells, MRTF-A is usually bound to actin monomers and remains in cytosolic sites. However, when the level of actin monomers is reduced because of actin assembly into filaments, MRTF-A enters the nucleus to enhance pro-fibrotic gene transcription, which includes increased expression of α-SMA [211,227]. In contrast with MRTF, YAP/TAZ is mainly regulated through the Hippo pathway (Box 3) by several proteins, including the large tumor suppressor kinase 1 (LATS1/2).
Box 3The Hippo pathway regulates YAP/TAZ localization.The Hippo pathway is a serine/threonine kinase cascade consisting of MST1/2, SAV1, LATS1/2, YAP, and TAZ [89]. When the Hippo pathway is active, YAP/TAZ is sequestered to the cytosol by phosphorylation of LATS1/2 [82]. When YAP/TAZ is localized to the cytosol, it is inactive as a result of its interaction with scaffold protein 14-3-3. It is subsequently degraded in proteosomes [90]. Discrete mechanical and proliferative cues inhibit LATS1/2-mediated phosphorylation of YAP/TAZ, which then enables nuclear localization [91]. YAP/TAZ associates with the TEAD binding domain to promote cell proliferation, stem cell self-renewal, and the expression of pro-fibrotic genes [89].
In fibrotic lesions, YAP/TAZ promotes matrix synthesis, focal adhesion formation, stress fiber assembly, and sustained expression of β1 integrins in collagen adhesions by activated myofibroblasts [2,5,54,223,228,229]. Upstream of MRTF-A and YAP/TAZ, GPCRs regulate the activity of these transcription factors, which depends on the particular type of heterotrimeric G protein [223,230,231,232] that is involved in the specific signaling system. Certain GPCRs and members of the transient receptor potential (TRP) channel family exhibit mutually interactive regulatory relationships [233,234,235]. For example, functional reciprocity is exhibited by TRPV4, which is affected by the Gαq/11-coupled GPCR angiotensin II receptor type 1 (AT1R) and vice versa [236]. In this instance, AT1R modulates TRPV4 by promoting β-arrestin-dependent inhibition and internalization of TRPV4 (which dampens its activity), while TRPV4 inhibits G protein-associated diacylglycerol and inositol-1,4,5-triphosphate accumulation [236]. When GPCRs and TRP channels are co-expressed in the same cells, the expressed channels can affect GPCR desensitization [236]. Collectively, certain GPCRs, and TRPV4 in particular, may regulate one another and thereby play multiple, complex roles in fibrosis by activating YAP/TAZ and MRTF-A.
8. Why Study GPCR and TRPV4 Signaling in the Context of Fibrosis?
In fibrotic lesions, imbalances of collagen remodeling favor the net accumulation of collagen in which synthesis exceeds degradation. Therapeutic interventions that can specifically target collagen degradation and, in a complementary fashion, reduce myofibroblast differentiation have not yet been established. This shortcoming is in part due to the fact that the potential interactions and overlap between their respective signaling pathways are not well-defined. GPCRs are a diverse set of receptors and have been one of the most successfully targeted molecular families for the purpose of drug development [237] (e.g., losartan, an angiotensin receptor blocker [238]; ONO-0260164 a prostaglandin EP4-receptor agonist [239]; and fenoldopam, a selective dopamine D1-receptor agonist [240]). They activate a broad array of signaling pathways, which are dependent on the Gα subclass that is coupled with a specific GPCR of interest. Multiple Gα subclasses have been implicated in reducing fibrosis, which underpins their potential for the development of new therapies for clinical management of fibrotic lesions [223].
Gαs GPCRs generate a key anti-fibrotic second messenger, cAMP [5,241]. Downstream signaling initiated by cAMP suppresses collagen production and fibroblast proliferation, and increases intracellular collagen degradation [3,242,243]. Conversely, and as described above, a prominent mechanosensor, TRPV4, is a pro-fibrotic mediator that regulates myofibroblast differentiation, ECM synthesis, cellular contractility, MMP activity, and cytokine production [244]. Evidently, TRPV4 channels can contribute to the progression of fibrosis through their ability to affect mechanotransduction, TGF-β1 stimulation, and Ca^2+^ signaling [244].
In general, GPCRs have a mutual regulatory relationship with TRP channels. But their interactions that affect downstream signaling pathways are not well-defined [236]. In this review, we consider downstream signaling from Gαs GPCRs and TRPV4 in the context of collagen remodeling and their respective second messengers, cAMP and Ca^2+^. The second messenger cAMP is involved in regulating physiological collagen remodeling, in part by reducing lysosomal pH and enhancing the collagenase activity of cathepsins. In contrast, Ca^2+^ is involved in mediating the interaction between gelsolin and NMMIIA [11,242], a contractile protein that is important for the initial steps in collagen internalization. Ca^2+^ also regulates Ca^2+^/calmodulin-dependent phosphodiesterases (PDE) and Ca^2+^-sensitive adenylyl cyclases (AC) [245]. We posit that Ca^2+^ influx through TRPV4 regulates Ca^2+^-sensitive PDEs and adenyl cyclases that directly affect cAMP levels. Thus, in fibrotic lesions, excessive Ca^2+^ influx through TRPV4 would activate PDEs to reduce cAMP levels [246,247,248,249].
9. GPCRs Are Therapeutic Targets in Fibrosis
GPCRs are the largest class of cell surface receptors that transduce extracellular cues into intracellular signals as part of cellular responses to diverse stimuli [250,251]. Upon activation, GPCRs undergo allosteric modifications that enable them to interact with one or more of the four major and unique trimeric G protein families: Gαi/o, Gαq/11, Gα12/13, and Gαs [252,253]. In healthy connective tissues, GPCR signaling in fibroblasts is involved in tissue growth, maintenance, and repair [250]. As the expression of GPCRs are extremely diverse and tissue-specific, these receptors are potential therapeutic targets in a broad array of diseases, which include fibrosis and cancer [237,250]. G proteins couple external stimuli with multiple intracellular signaling pathways. Depending on which G protein interacts with a specific GPCR, the outcomes of signaling are typically linked to characteristic extracellular stimuli. Further, the expression of GPCRs is often cell-specific in order to generate tissue-appropriate physiological responses [251].
GPCR signaling comprises multiple signaling cascades and pathways that have been extensively reviewed [250,252]. As examples of the complexity of GPCR signaling, we note that Gαs stimulates AC to increase production of the second messenger cAMP, which results in the activation of protein kinase A (PKA) and the exchange protein activated by cAMP (Epac) to enable productive downstream signaling [241]. Gαi/o is antagonistic to Gαs and suppresses AC, thereby decreasing production of cAMP [254]. Gαq/11 stimulates phospholipase C (PLC), which results in the cleavage of PIP_2_ and the production of diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP_3_). Subsequently, IP_3_ triggers the release of Ca^2+^ from the endoplasmic reticulum, and DAG activates protein kinase C (PKC) [255]. Gα12/13 activates the small GTPase Rho, which regulates cytoskeletal remodeling and cell growth [256]. In particular, GPCR signaling has been implicated in the development of fibrotic lesions in which different Gα subclasses inhibit or promote pro-fibrotic feedback signaling (Table 2) [223,250].
9.1. Gαi/o, Gαq/11, Gα12/13 Downstream Pathways Contribute to Fibrosis
GPCR expression can activate different Gα-protein subclasses through the same receptor [232]. Some of the GPCRs that play a role in promoting fibrosis include: protease-activated receptors [265,268,269], lysophospholipid receptors [259,260,261,262], adenosine receptors [257,258], angiotensin II [275,276], endothelin receptor A [277], serotonin receptors 2A and 2B [278], and sphingosine-1-phosphate receptor 1 [263]. As fibrosis can affect many different tissues, certain receptors are more likely to induce pro-fibrotic fibroblast activation based on the specificity of receptor expression in individual tissues [279].
Certain GPCRs can activate Gαi/o, Gαq/11, or Gα12/13 subclasses [279], which we consider here in the context of generating pro-fibrotic signals. For example, the Gαi/o subclass suppresses cAMP production and can regulate Smad2/3 and extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation [280], which in many tissues promotes fibrosis [261,262,280]. The Gαq/11 promotes fibrosis by increasing Ca^2+^ release from the endoplasmic reticulum (via IP_3_ receptors), which promotes collagen secretion and myofibroblast differentiation [13,281]. Pro-fibrotic agonists such as TGF-β1, angiotensin, and endothelin-1 activate robust Ca^2+^ signaling through PLC, which can enhance collagen I gene expression [282,283]. The Gα12/13 subclass stimulates ROCK signaling, which is required for myofibroblast differentiation [284]. Collectively, the activation of these pathways promotes the nuclear entry of YAP/TAZ and MRTF-A, which are important, pro-fibrotic mediators [230,285,286,287]. In addition, GPCR subclasses inhibit LATS1/2 kinase activity through Rho-dependent actin remodeling [231]. Further, as examples of the complex interplay between signaling cascades, GPCR agonists that activate Gαi/o, Gαq/11, Gα12/13 subclasses, increase the abundance of nuclear YAP, which sensitizes the responses of fibroblasts to TGF-β1 [287]. Notably, mechanosensitive processes that involve signaling through integrins and focal adhesion-mediated pathways also promote fibroblast activation through the same convergent pathways [217]. In sum, the GPCRs that selectively stimulate Gαi/o, Gαq/11, Gα12/13 subclasses also favor fibrogenesis and promote myofibroblast differentiation when they are dysregulated (Figure 3).
9.2. Gαs GPCR/cAMP Pathway Is Anti-Fibrotic
GPCRs that couple to the Gαs subclass activate AC, which subsequently elevates intracellular cAMP levels. Consistent with this notion, in cells stimulated with the AC activator forskolin, elevated cAMP levels block TGF-β1-induced myofibroblast differentiation and collagen synthesis [4,266,267,288]. Likewise, for cells in which GPCRs coupled to Gαs proteins were stimulated, the increased cAMP levels inhibited fibroblast proliferation, collagen production, myofibroblast differentiation, and actin cytoskeletal remodeling [5,266,267,288,289,290]. Further, in pulmonary fibroblasts, increasing cAMP levels with a Gαs GPCR agonist enhanced the degradation of collagen I by lysosomal cathepsin K and reduced ECM stiffness [242].
The second messenger cAMP has two main downstream effector proteins, PKA and Epac, each of which exhibits anti-fibrotic effects independently of the other (Figure 3) [241,291]. PKA is a classical cAMP effector that phosphorylates cAMP-response element-binding protein (CREB; at serine residue 133) and subsequently transits to the nucleus, where it associates with CREB-binding protein (CBP) [4,292,293]. Signaling through cAMP/PKA/CREB/CBP blocks TGF-β1-induced interaction of Smad3 with CBP, which is necessary for myofibroblast differentiation [292,294,295]. Activation of CREB/CBP increases the expression of Smad7, a negative regulator of Smad3 that inhibits collagen expression [296].
Interestingly, PKA agonists can inhibit collagen I expression and revert the phenotype of myofibroblasts to fibroblasts [291,297]. In kidney cells, cAMP mediates PKA activation that increases phosphorylation of the vacuolar-ATPase and subsequent endosomal acidification in kidney cells and astrocytes [298,299]. Additionally, CREB may be involved in modulating the expression of lysosomal cathepsins L and B, possibly enhancing collagen degradation [186,300]. Likewise, when cAMP/PKA signaling in osteoclasts is inhibited, cathepsin K processing and maturation are reduced, which highlights the role of cAMP in lysosomal degradation [274].
In pulmonary fibroblasts, a Gαs GPCR, the dopamine D1 receptor (DRD1), promotes cAMP downstream signaling, which is manifest as increased intracellular collagen degradation [242]. Gαs GPCR signaling also enhances extracellular cleavage, internalization, and lysosomal degradation of collagen I mediated by cathepsin K, and this effect persists even in cells treated with the pro-fibrotic growth factor, TGF-β1 [242,273]. In multiple cell types, Gαs GPCR/cAMP signaling enhances acidification in lysosomal compartments, which provides an acid-optimal environment for cathepsin activation [242,270,271,272]. Indeed, collagenolytic cathepsins can reduce collagen deposition in response to drug-induced fibrosis, which may be upregulated by CREB [23,68,70,186,301]. But in certain types of fibrotic lesions (pulmonary fibrosis), increased extracellular cathepsin K can activate fibroblasts and promote collagen synthesis, demonstrating the complexity of the formation of fibrosis [302].
A separate effector protein that is activated by cAMP is the guanine exchange factor, Epac, which activates the small GTPases Rap1 and Rap2 [303,304]. When cAMP levels increase, Epac is rapidly activated and is redistributed in the cell to promote localized signaling processes [305]. Rap1 is a downstream effector of many pathways, including Ca^2+^-mediated signaling cascades that involve PLC and release of intracellular Ca^2+^ from the endoplasmic reticulum [306,307,308,309]. Rap1 is also an upstream regulator of Ca^2+^ signaling, which exemplifies the importance of “crosstalk” between these signaling platforms in different cell types [309]. In fibroblasts, Rap1 activation reduces migration, decreases proliferation, and interacts with NMMIIA to enhance β1 integrin-mediated collagen phagocytosis [48,310]. A direct link between Epac and collagen phagocytosis has yet to be determined, and an intriguing question is whether Epac increases collagen degradation in the phagocytic pathway by promoting Rap1 activation, which does enhance collagen phagocytosis (Figure 4) [48].
The second messenger cAMP can play a role in regulating focal adhesion function and signaling, as well as actin filament turnover through Rap1-GTP-mediated responses, but this conjecture has not yet been explored. Downregulation of Epac signaling is required for collagen fibrogenesis, as earlier data showed that during pro-fibrotic activation of fibroblasts in multiple tissues, Epac mRNA and protein expression were reduced [310]. Earlier observations in cardiac fibroblasts observed a role of Epac inhibition in collagen synthesis and myofibroblast differentiation [310,311] and suggest a possible anti-fibrotic therapeutic purpose in this pathway. To discern the differential effects of cAMP effectors, cAMP derivatives that selectively activate Epac (8-Me-cAMP) and PKA (N6-cAMP) have been used [241]. Further, cAMP signaling, particularly through Epac, reduces nuclear localization of the pro-fibrotic transcription factors YAP/TAZ and MRTF-A [5,231,273,289,312,313,314]. Since cAMP signaling is a fundamental system for normal cellular function, it is tightly regulated by PDEs, ACs, and by crosstalk with other second messenger systems.
The concentration of cAMP in the cytoplasm of cells is dependent on the abundance, location, and activity of membrane-bound and soluble forms of adenylyl cyclase, and on the rate of cAMP degradation by PDEs [241,245,315]. ACs are the only enzymes that synthesize cAMP, and ACs can integrate signals received from Gαs GPCRs to convert adenosine triphosphate into cAMP [316]. In mammals, there are nine transmembrane isoforms of ACs (AC1-AC9) and one soluble AC that displays varying expression levels in different cells and tissues [317]. Both cardiac and pulmonary fibroblasts mainly express AC3, AC5, and AC6 isoforms, but other isoforms are expressed depending on their subcellular localization [283,318,319]. In pulmonary fibroblasts, overexpression of AC6 reduced the expression of type I collagen and increased the expression of MMP-2 [320], a gelatinase that can reduce the abundance of extracellular collagen. Notably, in comparison to fibroblasts, myofibroblasts produce less cAMP and exhibit decreased expression of AC isoforms and increased expression of PDEs [292].
PDEs convert cAMP into 5′-adenosine monophosphate. There are multiple PDE isoforms in fibroblasts, which include PDE1A [321], PDE4B [247], PDE5 [322], PDE7 [323], and PDE8 [323]. PDE1A belongs to the Ca^2+^/calmodulin-stimulated PDE1 family and promotes ECM synthesis and myofibroblast differentiation in cardiac fibroblasts [323], an observation that underpins the importance of the relative abundance of cAMP in the control of pro-fibrotic processes. The use of experimental approaches that enhance cAMP levels in fibroblasts (e.g., agonists for Gαs GPCRs, PDE inhibitors, PKA enhancers, and Epac activators) provides potential therapeutic targets for clinical management of fibrotic lesions [223,241]. The crosstalk of cAMP with Ca^2+^-dependent signaling pathways contributes to the complexities of understanding how fibrosis is promoted [324,325]. Since Ca^2+^ signaling involves multiple pathways [326], we will provide detailed information on TRPV4 and focus on Ca^2+^ flux through TRPV4 channels because of its implications in fibrosis below.
10. TRPV4 Is a Ca2+ Permeable, Mechanosensing Channel That Promotes Fibrosis
The mammalian Transient Receptor Potential (TRP) channels are conserved, integral membrane proteins that are grouped into six subfamilies based on sequence homology [327]. In particular, the Vanilloid TRP subfamily has six members: TRPV1–4 are non-selective cation conducting pores, while TRPV5 and TRPV6 are highly Ca^2+^ selective [327]. TRPV4 is broadly expressed in several different tissues and by multiple cell types, notably in the peripheral nervous system (root ganglia, trigeminal ganglia, hippocampal pyramidal neurons, and retinal ganglion cells) [328], and importantly, by fibroblasts (Table 3) [61]. Multiple stimuli can activate TRPV4, including heat, osmotic pressure changes, chemical stimulation, and mechanical stress [329,330,331,332,333]. Activation of TRPV4 in fibrotic tissues promotes fibroblast differentiation into contractile myofibroblasts and enhances collagen deposition [208,329,334].
Two recent reviews have considered several mechanisms that link TRPV4 mechanotransduction and Ca^2+^ influx in the context of fibrosis [21,208]. Here, we will focus on Ca^2+^ conductance and mechanosensing by TRPV4, as it has been linked to the pathophysiology of multiple diseases, including fibrosis, cancer, and neurodegenerative diseases [208,329].
Fibrotic disease progression can be promoted by dysregulated TRPV4 signaling. TRPV4-induced Ca^2+^ influx is required for TGF-β1-induced myofibroblast differentiation in numerous tissues, including the heart, lung, airways, and skin [208,329,334]. In mice subjected to bleomycin-induced fibrosis, TRPV4 expression is upregulated [329]. In TRPV4-deficient mice, the animals are protected from bleomycin-induced pulmonary fibrosis and exhibit reduced myofibroblast differentiation. These processes are reversed when TRPV4 is expressed [329]. Likewise, following suppression of TRPV4 function by chemical antagonists, siRNA-mediated knockdown, or loss-of-function genetic manipulation, TGF-β1-mediated type 1 collagen and fibronectin production are reduced, and force-induced cellular contractility, epithelial-to-mesenchymal transition, and α-SMA expression and incorporation into stress fibers are all inhibited [208,354,357,362].
TGF-β1, which is one of the most potent pro-fibrotic agonists that have been described, activates phosphoinositide 3-kinase (PI3K), NADPH oxidase 4 (NOX4), ROCK, and p38 MAPK during fibroblast differentiation and increases nuclear localization of MRTF-A and YAP/TAZ (Figure 5) [208,334,354]. In cells treated with the TRPV4 inhibitor RN1734, the activation of Rho, p38 MAPK, and MRTF-A nuclear localization was all inhibited [334]. Interestingly, PI3K was not altered by TRPV4 inhibition, indicating that PI3K is upstream of TRPV4 and regulates TRPV4 in response to TGF-β1 in fibroblasts [334].
TRPV4 activation enhances matrix synthesis and strongly increases the expression of α-SMA [334]. Evidently, TRPV4 regulates the pro-fibrotic functions of TGF-β1 by modulating both the non-canonical SMAD-independent and the SMAD-dependent downstream pathways [329,334,357,363]. Independent of TGF-β1, TRPV4 independently upregulates the expression of α-SMA, the formation of highly aligned collagen fibrils, and the activation of MRTF-A, as shown in cultured cells treated with the small molecule TRPV4 agonists GSK1016790A and 4α-PDD (Figure 5) [208,364]. In response to increased matrix stiffness and TGF-β1, the expression of TRPV4 is needed for nuclear translocation of YAP/TAZ, myofibroblast differentiation, and for the regulation of epithelial-to-mesenchymal transition [357].
In matrix mechanosensing, TRPV4 can impact ECM remodeling, which is dependent on Ca^2+^ conductance through TRPV4 channels [329,337,365]. TRPV4 is enriched in focal adhesions and is activated by mechanical force transfer from the β1 integrin cytoplasmic binding protein, CD98 [366]. Higher TRPV4 expression is associated with reduced β1 integrin abundance, adhesion to collagen, focal adhesion size, alignment, and the compaction of extracellular collagen fibers [367]. The expression of TRPV4 enhances microRNAs that promote downregulation of β1 integrin mRNA, which directly impacts the ability of cells to recognize and bind tightly to the underlying ECM [367]. Subsequently, after activation, TRPV4 initiates Ca^2+^ oscillations that trigger signaling, which are involved in multiple cellular processes like tissue repair and ECM remodeling [368]. In response to pro-fibrotic stimulation, elevations of Ca^2+^ are solely dependent on TRPV4 and are not affected by TGF-β1 [337]. In collagen remodeling, TRPV4-mediated Ca^2+^ influx regulates the functions of the actin-binding protein Flil and NMMIIA to enable the generation of cell extensions and enhanced contractility [61]. TRPV4-dependent Ca^2+^ influx is also associated with the collagen receptor DDR1, which influences processes that mediate collagen compaction and alignment [214]. In human mesenchymal stem cells, TRPV4 Ca^2+^ signaling is critical for the initial stages of collagen matrix assembly and can regulate force generation at cell–matrix adhesions [364].
TRPV4 activity influences the abundance of matrix-degrading MMPs in specific cell types and microenvironments. For example, in human respiratory epithelial cells, Gi/o GPCRs, PLC, and PI3K activate TRPV4 Ca^2+^ influx, which subsequently promotes MAPK-mediated MMP-1 production and secretion [338]. In whole mouse lungs, TRPV4 activation increases the abundance and activation of the gelatinases, MMP-2 and MMP-9, while reducing the expression of the MMP inhibitor, tissue inhibitor of metalloproteinases 2 (TIMP-2) and MT1-MMP [369]. MMP-2 and MMP-9 associate and cleave β1 integrins in colorectal carcinoma and in corneal epithelial cells [370,371]. TRPV4 increases the expression of MMP2 and MMP9, which reduces β1 integrin expression [369] and possibly affects the recognition of collagen fibrils at focal adhesions. TRPV4 also reduces MT1-MMP expression [369], which may affect collagen phagocytosis and the intracellular degradation of collagen. Indeed, MT1-MMP plays a crucial role in facilitating collagen phagocytosis by preparing collagen fibrils for subsequent internalization [43]. TRPV4-induced MT1-MMP downregulation has not been explored in the context of collagen phagocytosis but may contribute to reduced intracellular collagen degradation. In summary, TRPV4 is evidently involved in collagen remodeling. When dysregulated, TRPV4 contributes to the development and progression of fibrotic lesions, thereby indicating that TRPV4 is an attractive target for therapeutic inhibition.
11. Gαs GPCR and TRPV4 Signaling Are Interconnected Through the Second Messengers, cAMP and
Ca2+
As described above, impaired collagen remodeling is one of the main characteristics of fibrosis, in which the accumulation of poorly organized, stiff collagen in the ECM is favored. In order to reduce the “burden” of excessive fibrillar collagen in the ECM, it will be important to discover which pathways specifically enhance intracellular collagen degradation. One pathway that is being explored as a potential therapeutic for reducing fibrillar collagen in the ECM is the Gαs GPCR-mediated pathway, as it is clearly involved in resolving fibrotic lesions through the involvement of cAMP downstream effectors [186,241,314,320]. The cAMP effector Epac mediates activation of Rap1, which interacts with NMMIIA to enhance collagen internalization and degradation in the phagocytic pathway [48,241]. Gelsolin also interacts with NMMIIA; however, this interaction is Ca^2+^-dependent [10], which may be mediated through TRPV4 or possibly by other Ca^2+^-permeable channels or from store-operated Ca^2+^ entry (Box 4).
Box 4Ca^2+^ signaling involves external and internal stores.Many plasma membrane, Ca^2+^-permeable channels control Ca^2+^ influx from the external environment in response to diverse stimuli, including mechanical forces, decreased cytosolic Ca^2+^, membrane depolarization, extracellular agonists, intracellular messengers, and depletion of intracellular stores [200]. Many stimuli activate the release of Ca^2+^ from intracellular stores (through PLC). Gαq/11 GPCRs generate IP_3_ [110,200], which binds to its cognate receptor on the endoplasmic reticulum to trigger Ca^2+^ release and oscillations [110,200]. Ca^2+^ controls Ca^2+^ release from intracellular stores, which are also affected by a large group of messengers (e.g., IP_3_, sphingosine-1-phosphate) [200].
In human pulmonary fibroblasts, Ca^2+^ oscillations contribute to the pro-fibrotic effects of TGF-β1, while prostaglandin E_2_ (PGE_2_), EP_2_ and EP_4_ receptors (which regulate AC), disrupt pro-fibrotic signaling through cAMP/PKA [372]. TGF-β1 triggers Ca^2+^ oscillations in human pulmonary fibroblasts [282,372] and in human ventricular fibroblasts, TRPV4-dependent Ca^2+^ influx mediated TGF-β1-induced myofibroblast differentiation: in these experiments, TRPV4 was the only TRP channel upregulated at mRNA and protein levels [373].
Ca^2+^ oscillations can modulate the expression of ECM genes in a frequency-dependent manner, either through TGF-β1 or independent of TGF-β1 [182,282]. The Ca^2+^ oscillations that are modulated by TGF-β1 are generated by the activation of Ca^2+^/calmodulin-dependent protein kinase-II (CaMK-II) and by TRPV4-activated pro-fibrotic signaling processes [373,374,375]. The anti-fibrotic effect of PGE_2_ is thought to be mainly explained by the interference of the EP_2_ receptor with Ca^2+^ signals. These signals inhibit CaMK-II and Akt activation in TGF-β1-treated pulmonary fibroblasts [372].
In certain cell systems, Ca^2+^ signaling directly regulates cAMP by modulating AC and PDE function [283,315,317,321]. For example, in osteocyte mechanotransduction, TRPV4- mediated Ca^2+^ influx disrupts AC6 function [376]. It is unknown whether this process also occurs in fibroblasts, but conceivably, TRPV4 Ca^2+^ signaling can reduce cAMP levels by adjusting the catalytic activities of ACs and PDEs. Ca^2+^ inhibits AC5 and AC6 isoforms and reduces cAMP generation in neuronal cells and cardiac myocytes [377,378,379]. Ca^2+^ may inhibit the production of cAMP by inhibiting AC and thereby reducing collagen degradation in fibrotic lesions. Likewise, Ca^2+^ can regulate cAMP by promoting the activity of PDE1 and PDE4, a process which depletes cAMP from cells [245,249,321,362,380]. In cardiac fibroblasts, PDE1A activity reduces cAMP-Epac-Rap1 signaling, which leads to increased myofibroblast formation and ECM synthesis [321]. Similarly, PDE4 promotes signaling through the MAPK, p38α, which generates a pro-fibrotic response in skin and lung fibroblasts [380].
In a therapeutic context, PDE4 inhibitors in combination with PGE_2_ agonists can alleviate organ fibrosis by enhancing the “dedifferentiation” of myofibroblasts into fibroblasts [380]. Likewise, PDE1 and PDE4 inhibitors increase cAMP to promote an anti-fibrotic response [247,249,323]. TRPV4-mediated Ca^2+^ influx may also be involved in activating PDEs and inhibiting AC5/6, which decreases cAMP and promotes pro-fibrotic responses (Figure 6). Notably, Ca^2+^ signaling not only upregulates pro-fibrotic responses: increased Ca^2+^ signaling can also induce AC3 to produce cAMP and thereby attenuate collagen synthesis in cardiac fibroblasts [283]. Further, cAMP can modulate Ca^2+^ influx by reducing Ca^2+^ channel activities in vascular smooth muscle cells, although the potential clinical implications for the management of fibrosis are not understood [381]. Collectively, the complex interactions between cAMP and Ca^2+^ signaling may be important contributing mechanisms that alter the balance between collagen synthesis and degradation. Further, in-depth examination of the interplay between TRPV4 and Gαs GPCR cAMP pathways may provide new insights into potential anti-fibrotic therapeutics.
12. Conclusions and Future Perspectives
Small perturbations in TGF-β1, TRPV4, and Ca^2+^-mediated signaling can create positive feedback loops that promote fibrosis and drive aberrant collagen remodeling. Accordingly, understanding the fundamental mechanisms that drive tissue-specific fibrotic lesions by analyzing relevant signaling pathways may be useful in developing rational treatment options. We have reviewed above which GPCR pathways enhance or reduce fibrosis progression, based on the specific Gα proteins that are activated. Specifically, we focused on Gαs GPCRs, which generate cAMP and signal through PKA and Epac to elicit an anti-fibrotic response. Both of these cAMP downstream pathways (i.e., Epac and PKA) decrease the activation of pro-fibrotic signaling systems by decreasing myofibroblast activation, enhancing collagen degradation, and reducing pro-fibrotic gene expression [3,242,295,306,310,380]. An important gap in our understanding of pro-fibrotic processes is that the role of cAMP signaling in regulating focal adhesions and cytoskeletal dynamics through Rap1 is not defined. In the context of collagen remodeling, focusing on Gαs GPCR pathways and their relationships with pro-fibrotic signaling pathways would be beneficial to provide more focused targets for potential treatments.
We considered here the role of TRPV4 in collagen remodeling and mechanosensing. Notably, TRPV4 function and activation are integrated with β1 integrin-mediated adhesion to collagen. But with stiff matrices, TRPV4 becomes dysfunctional and promotes pro-fibrotic signaling [367]. Understanding the role of TRPV4 Ca^2+^ signaling in fibrosis is complicated because it is also involved in a large cadre of processes, including wound healing, cytoskeletal remodeling, epithelial–mesenchymal transformation, and matrix synthesis and turnover [208]. Currently, while it is not known whether TRPV4 influences collagen internalization directly, recent data indicates that TRPV4 expression decreases β1 integrin expression through microRNAs and reduces MT1-MMP expression [367,369].
Maintaining a tight balance between collagen synthesis and degradation is important for healthy organ structure and function. Accordingly, we considered here how Ca^2+^ and cAMP signaling are connected in physiological collagen remodeling and what processes occur when these pathways are disrupted in fibrosis. Based on current analyses, the interplay between TRPV4 and GPCR is not fully defined. Although their downstream pathways and associated second messengers exhibit extensive crosstalk, our understanding of the underlying regulatory mechanisms, compensatory systems, and interactions is incomplete. For example, in osteocytes, Ca^2+^ flux through TRPV4 inactivates ACs [376], but it is unknown if this same effect also occurs in fibroblasts. Ca^2+^ signaling is known to regulate PDEs that are expressed in fibroblasts [245,372,380], but the role of TRPV4 channels in these processes is not understood. Using mouse models with fibroblast-specific deletion of TRPV4 fibroblasts could help to distinguish whether Ca^2+^ signaling in PDE regulation is mediated by TRPV4 or other channels. More defined data sets describing how TRPV4-mediated Ca^2+^ signaling regulates AC and PDE function are needed to understand the functional interplay between cAMP and TRPV4 in fibrotic lesions.
Exploring whether Gαs GPCR agonists can rescue TRPV4-driven pro-fibrotic phenotypes is functionally relevant in new treatments for fibrosis and for preservation of physiological collagen remodeling. Since the Gαs GPCR pathway increases intracellular collagen degradation and decreases myofibroblast differentiation in response to TGF-β1 signals [4,242,289], we anticipate cAMP stimulators may reverse TRPV4-mediated, pro-fibrotic collagen remodeling. In the context of ECM health, defining the relationship between TRPV4 and Gαs GPCRs signaling provides an opportunity to discover new drug targets for the development of new therapies for clinical management of fibrosis.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Rockey D.C. Bell P.D. Hill J.A. Fibrosis—A Common Pathway to Organ Injury and Failure N. Engl. J. Med.20153721138114910.1056/NEJ Mra 130057525785971 · doi ↗ · pubmed ↗
- 2Hinz B. Mc Culloch C.A. Coelho N.M. Mechanical Regulation of Myofibroblast Phenoconversion and Collagen Contraction Exp. Cell Res.201937911912810.1016/j.yexcr.2019.03.02730910400 · doi ↗ · pubmed ↗
- 3Andrabi K.I. Kaul N. Mudassar S. Dilawari J.B. Ganguly N.K. Intracellular c AMP Determines the Extent of Degradation and Not the Synthesis of Collagen by Rat Hepatocytes Mol. Cell Biochem.1992109899410.1007/BF 002308781319551 · doi ↗ · pubmed ↗
- 4Liu X. Sun S.Q. Hassid A. Ostrom R.S. c AMP Inhibits Transforming Growth Factor-Beta-Stimulated Collagen Synthesis via Inhibition of Extracellular Signal-Regulated Kinase 1/2 and Smad Signaling in Cardiac Fibroblasts Mol. Pharmacol.2006701992200310.1124/mol.106.02895116959941 · doi ↗ · pubmed ↗
- 5Haak A.J. Kostallari E. Sicard D. Ligresti G. Choi K.M. Caporarello N. Jones D.L. Tan Q. Meridew J. Diaz Espinosa A.M. Selective YAP/TAZ Inhibition in Fibroblasts via Dopamine Receptor D 1 Agonism Reverses Fibrosis Sci. Transl. Med.201911 eaau 629610.1126/scitranslmed.aau 629631666402 PMC 7066514 · doi ↗ · pubmed ↗
- 6Gelse K. Pöschl E. Aigner T. Collagens—Structure, Function, and Biosynthesis Adv. Drug Deliv. Rev.2003551531154610.1016/j.addr.2003.08.00214623400 · doi ↗ · pubmed ↗
- 7Sodek J. Brunette D.M. Feng J. Heersche J.N.M. Limeback H.F. Melcher A.H. Ng B. Collagen Synthesis Is a Major Component of Protein Synthesis in the Periodontal Ligament in Various Species Arch. Oral Biol.19772264765310.1016/0003-9969(77)90094-2414699 · doi ↗ · pubmed ↗
- 8Song F. Wisithphrom K. Zhou J. Windsor L.J. Matrix Metalloproteinase Dependent and Independent Collagen Degradation Front. Biosci.2006113100312010.2741/203616720379 · doi ↗ · pubmed ↗