Improvement of Diagnostics in NSCLC Patients with MET Exon 14 Mutations Using Complementary DNA/RNA-NGS and Identification of Two Novel Exonic Splicing Mutations
Edyta Maria Urbanska, Thomas Koed Doktor, Linea Cecilie Melchior, Eva Stampe Petersson, Jens Benn Sørensen, Eric Santoni-Rugiu, Brage Storstein Andresen, Morten Grauslund

TL;DR
This study improves the detection of MET exon 14 mutations in lung cancer patients using combined DNA and RNA sequencing, identifying two new mutations that cause abnormal splicing.
Contribution
The study introduces a new workflow for METex14 diagnostics and identifies two novel exonic splicing mutations previously unrecognized.
Findings
Combined DNA/RNA NGS increased detection of METex14 mutations from 0.2% to 3.5%.
Two novel METex14 mutations were found to cause skipping transcripts despite being outside canonical splice sites.
A new workflow for METex14 detection was proposed based on complementary DNA and RNA sequencing.
Abstract
MET exon 14 (METex14) skipping mutations differ from other non-small cell lung cancer (NSCLC) genomic biomarkers as they result in aberrantly spliced MET transcripts and increased MET-signaling. However, the most accurate method for their detection remains debated. We conducted a retrospective study of previously identified METex14 skipping NSCLC samples by using different, commercially available, diagnostic targeted DNA- /RNA-Next-Generation Sequencing (NGS) panels. We primarily used small DNA-NGS panels covering the 5′ splice site of METex14 and supplemented by targeted RNA sequencing for selected cases. Using this approach, we identified <0.2% patients with METex14 mutations. Due to this low frequency, we validated and introduced complementary NGS testing using combined DNA/RNA-panels. This resulted in an increased number of METex14-positive patients (3.5%) and allowed us to identify…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
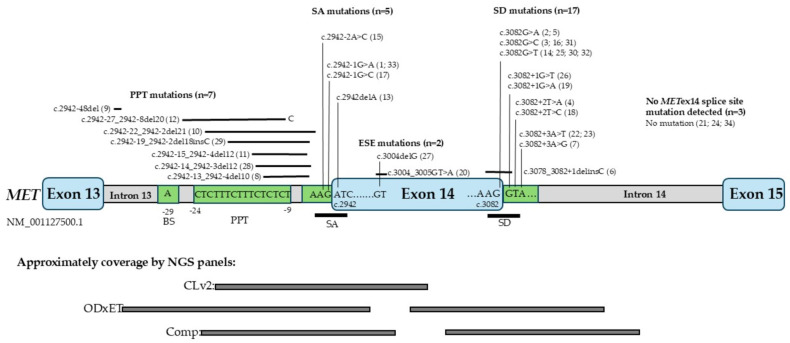 Figure 1
Figure 1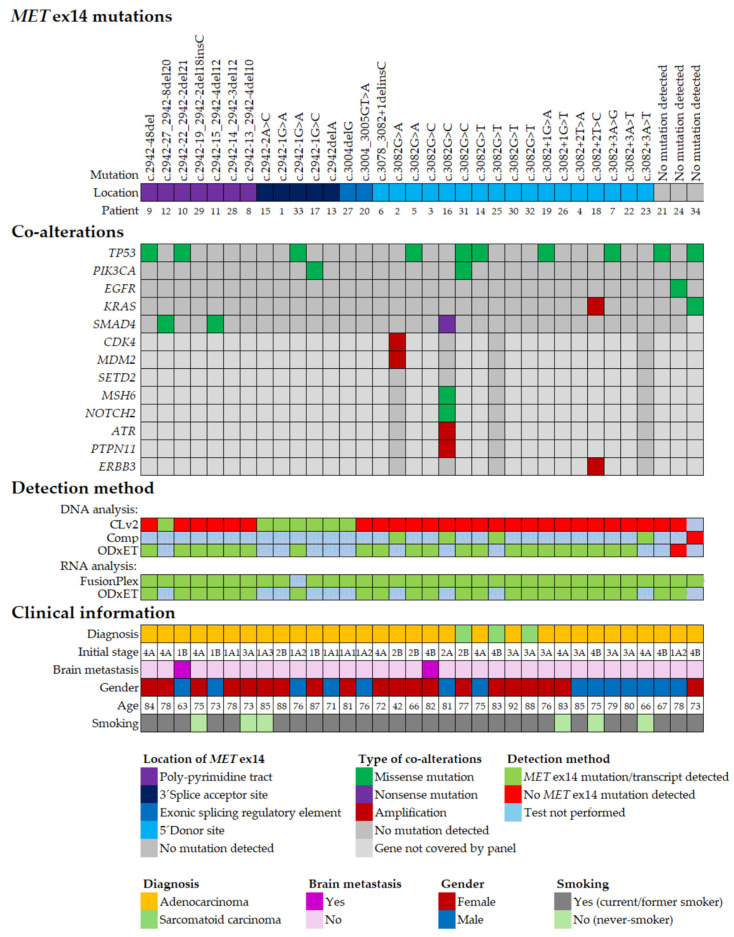 Figure 2
Figure 2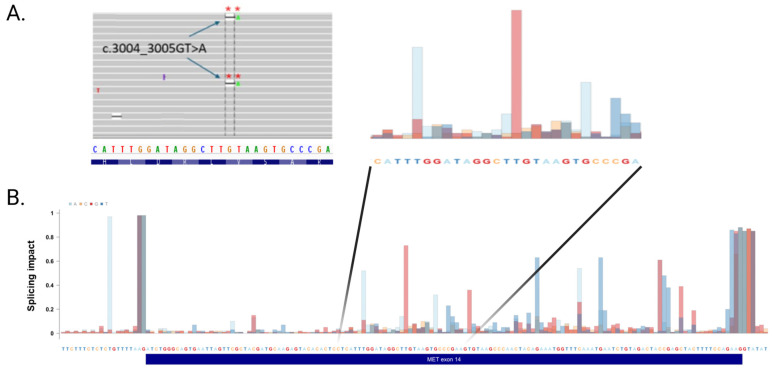 Figure 3
Figure 3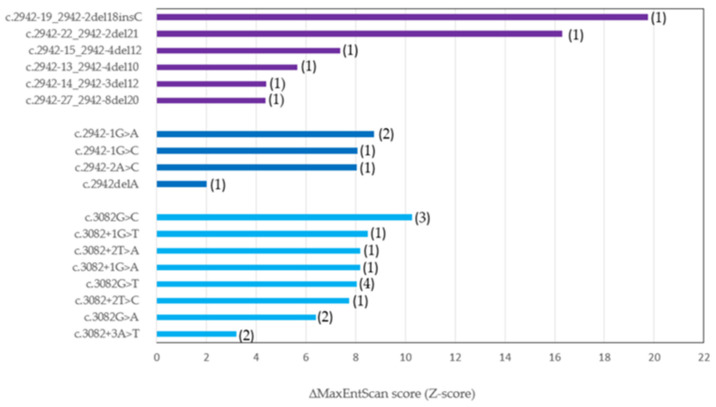 Figure 4
Figure 4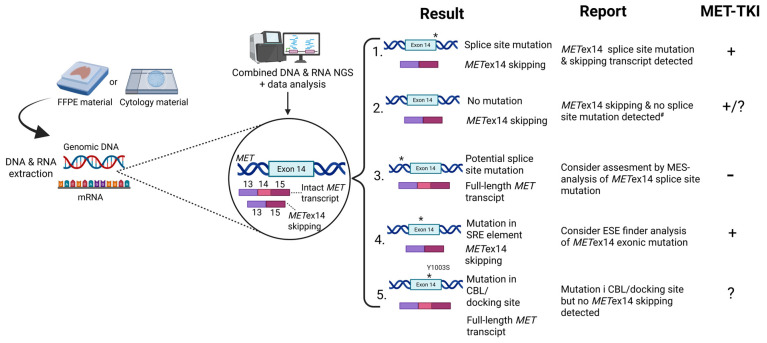 Figure 5
Figure 5- —Merck A/S
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLung Cancer Treatments and Mutations · Lung Cancer Research Studies · Lung Cancer Diagnosis and Treatment
1. Introduction
The N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) transforming gene, MET, encodes the hepatocyte growth factor (HGF) receptor, MET, which plays a fundamental role in regulating cell differentiation, migration, and growth [1]. Pathogenic activation of MET is a well-defined oncogenic driver in many types of cancers [2]. MET exon 14 (METex14) skipping variants occur in 0.6–4% of non-small cell lung cancer (NSCLC), adenocarcinomas [2,3], and belong to the group of molecular biomarkers whose testing is recommended in international guidelines [4,5]. Importantly, NSCLC with METex14 skipping is associated with poor prognosis, as the median overall survival for patients with these mutations when treated with MET tyrosine kinase inhibitors (MET-TKIs) is 24.6 months, while for those not receiving MET-TKIs it is 8.1 months [6]. This demonstrates a strong therapeutic potential in detecting and targeting METex14 skipping mutations.
The activity of the MET receptor is a result of a dynamic balance between activation by its ligand, HGF, and degradation by the E3 ubiquitin ligase, Casitas B-lineage lymphoma (CBL) protein [7]. METex14 codes a part of the regulatory region in the juxtamembrane domain (JM) that is the docking site for CBL in the MET protein [8]. Consequently, the lack of JM in cells with METex14 leads to an increased steady-state level of MET protein [9]. Additionally, rare mutations in JM, such as amino acid Y1003 (ubiquitin ligase binding site), D1002 (caspase cleavage site), or S985 (phosphorylation site), may reduce degradation of the MET receptor mimicking a METex14 skipping without aberrant splicing [10]. All these alterations enhance the pleiotropic MET signaling, which drives cancer progression via activation of numerous signaling mechanisms like activation of the RAS-MAP kinase pathway, SMAD2/3-signaling independent of TGF-β, and AKT-driven invasive growth [11,12,13].
In eukaryotic cells, mRNA splicing is conducted by the spliceosome, a large RNA-protein complex, which recognizes the splice donor (SD) site at the 5′ end of the intron and the splice acceptor (SA) site at the 3′ end. Near the SA site lies the polypyrimidine tract (PPT), which helps recruit splicing factors. Upstream of this tract is the branch point (BP), an adenosine that initiates the lariat formation during intron removal. Together, these elements ensure precise exon joining and intron excision [14,15]. The SD site consists of the 3 last nucleotides of the exon and the first 8 nucleotides of the intron, and the SA site consists of the last 20 nucleotides of the intron and the first 3 nucleotides of the exon [16,17,18,19,20]. Most exons, including METex14, have their 3′SA and 5′SD with sequence AG (corresponding to the end of an intron) and GT (corresponding to the beginning of an intron), respectively, known as canonical splice sites [19]. Regulation of mRNA splicing leading to alternative splicing is regulated by complex interactions between the spliceosome and splicing regulatory proteins, which bind cis-acting splicing regulatory elements (SREs) like exonic splicing enhancers and silencers (ESEs and ESSs, respectively) and intronic splicing enhancers and silencers (ISEs and ISSs, respectively). An SRE mutation is defined as any nucleotide change within an exon (or sometimes intron) that creates, disrupts, or alters the strength of an SRE motif, thereby influencing splicing. These motifs recruit splicing factors such as SR and hnRNP proteins. The exonic mutations may affect both SREs—ESEs and ESSs. ESEs promote exon inclusion, and ESSs inhibit splicing. However, the activity of SREs depends on position (in one region they may act as ESEs and in another region as ESSs) and gene expression affecting the regulatory effect. Exonic mutations can disrupt ESE/ESS motifs causing mis-splicing: exon skipping or activation the cryptic splice sites. About 25–27% of exons are vulnerable to splicing disruption by exonic mutations [21]. However, the role of exonic mutations has not been described for METex14. In summary, SRE mutations are defined by location (outside the canonical splice sites), motif disruption, and functional consequence. Using in silico prediction (ESEfinder, SpliceTransformer) we can localize the altered binding site.
Aberrant splicing is associated with the pathogenesis of different diseases, including NSCLC with skipping mutations in METex14 [2,8].
A growing number of somatic DNA splice site mutations leading to METex14 skipping have been identified [2,22]. The majority of these METex14 splice site mutations are base substitutions/small deletions at the SD, BS, PPT, and SA sites [22]. The mechanism of MET mRNA splicing is also clinically important for explaining the outcome of MET-TKI treatment. Since splice site recognition depends on matching the splicing consensus, alterations in splice sites may result in alternative splicing [17]. In particular, intolerable mismatches due to disrupted sequences lead to aberrant splicing. Exonic mutations may also cause exon skipping by disrupting exonic splicing enhancers or by increasing the strength of exon splicing silencers [16,17,21].
Next-generation sequencing (NGS) is a preferred method for testing molecular driver alterations in NSCLC. Most clinical laboratories use NGS panels of onco- and tumor suppressor genes for detecting DNA single nucleotide variants (SNVs), small deletions, splice site mutations, copy number variants (CNVs), and gene fusions. Both DNA-based and RNA-based NGS can be used to detect METex14 skipping mutations and transcripts. The former detects splice site mutations in the splice site regions that are predicted to lead to METex14 skipping, whereas RNA-NGS detects the direct fusion of MET exon 13 and exon 15 transcripts [6].
In our study, we investigated whether combining DNA-based and RNA-based NGS analyses would improve sensitivity and specificity of METex14 skipping detection in NSCLC samples. Furthermore, we compared targeted DNA-NGS panels commonly used in diagnostics of NSCLC and analyzed how they may perform on the detection of METex14 splice site mutations. We present data based on our own approach to METex14 skipping detection, which supports complementary DNA- and RNA-based NGS in clinical routine.
Since NSCLC is a disease often driven by subclonal alterations, a quantitative approach to METex14 variants might help to better define their impact on the level of protein lacking sequence encoded by exon 14. Therefore, we explored this aspect by using the bioinformatic tool MaxEntScan (MES) to predict the effects of sequence variations on splicing signals [23]. Our cohort was analyzed to investigate the potential relationship between MES scores and genomic localization of detected METex14 splice site variants, alongside clinical features such as age, gender, histological diagnosis, smoking history, and presence of brain metastases.
2. Results
2.1. Identification of METex14 Positive NSCLC Patients
Between January 2018 and August 2023, routine molecular testing of NSCLC using mainly the AmpliSeq Colon and Lung Cancer Research Panel v2 (CLv2) identified seven cases with METex14 splice site mutations among 5000 tested non-squamous NSCLC cases. In three cases, an additional NGS analysis was performed using the Oncomine Comprehensive v3 panel (Comp), and four additional cases were identified with METex14 splice site mutations. During the same period, NGS-RNA testing was conducted with the Archer FusionPlex Lung Panel v1.0 on approximately 350 selected cases (e.g., younger patients, non-smokers, or those with a detected METex14 splice site mutation), identifying 30 METex14 skipping transcript-positive cases with adenocarcinoma histology and three cases with sarcomatoid carcinoma histology The observed frequency of METex14 skipping mutation-positive patients identified by DNA-NGS was much lower (approximately 0.15%) than the reported frequency of 0.6–4% in NSCLC patients [2,3]. A possible explanation for the low incidence of METex14 skipping variants detected with the CLv2 panel is its limited amplicon coverage of METex14, as it includes only the SA site and a part of the PPT region (Figure 1). To improve diagnostic sensitivity, we analyzed 12 cases with the Comp NGS panel with confirmed METex14 skipping transcripts, but no splice site mutations were found by the CLv2 panel. In 10 of these cases, splice site mutations not covered by the CLv2 panel were detected (Figure 1 and Figure 2, Supplementary Tables S1 and S2). In two patients (cases 21 and 24), no METex14 splice site mutation was identified, likely due to larger genomic structural variants [2]. In August 2023, the combined DNA/RNA-NGS panel Oncomine Dx Express Test (ODxET) was clinically implemented at our site as the standard procedure for molecular diagnostic testing of all pulmonary adenocarcinomas. Among the first 519 patient samples analyzed using this method, 19 METex14-positive cases (3.3%) were identified, all showing both a METex14 splice site mutation and exon 14 skipping transcripts.
2.2. Subtypes of METex14 Mutations
To gain a deeper understanding of the diversity of METex14 splice site mutations leading to METex14-skipping transcript, we analyzed the subtypes of MET mutations based on their location in the gene. In total, using these three different DNA-NGS panels, we identified 17 samples with MET mutations causing aberrant splicing, which were localized in the 5′ SD site, seven cases with mutations in the PPT region located in intron 13, and five patients with mutations of the SA site. Lastly, in two cases, a potential ESE was affected by previously unreported mutations localized in the middle of exon 14 (Figure 1 and Figure 2).
The two most frequently identified splice site mutations were c.3082G>C and c.3082G>T, present in three and four samples, respectively. These two mutations were also among the most common splice site mutations identified by Kim et al. [22]. All mutations identified in the SA- and SD sites were described in previous studies [2,22]. As formerly reported [8,22], we also observed a wide distribution of deletions within the PPT region in seven affected cases. In one instance (case 9), we identified a 1 bp deletion located 48 base pairs upstream of the SA element. The impact of this short deletion on METex14 splicing is unclear.
2.3. Co-Occurring Genomic Alterations in METex14 Positive NSCLC
We analyzed whether the METex14 positive samples in our cohort harbored alterations in other relevant genes. We identified co-existing genetic alterations in 53% (18/34) of METex14-skipping positive patients (Figure 2, Supplementary Table S3). Co-mutations of the tumor-suppressor gene TP53 were the most frequent co-alteration, occurring in 10/34 patients (29%). Two patients with METex14 alterations (case 24 and 34) also harbored concurrent mutations in the oncogenic drivers, EGFR and KRAS, respectively. Notably, in both cases, METex14 mutations were detectable only at the RNA transcript level, with no corresponding DNA splice site mutations identified. The patient with the EGFR co-mutation exhibited the well-known pathogenic p.L858R substitution in the tyrosine kinase domain of EGFR and, because of stage II disease, underwent curative surgery. In this case, METex14 transcripts were present at low levels, as confirmed by two independent RNA-based assays (FusionPlex and OdxET). Since no METex14 DNA splice site mutations were detected, we cannot rule out that the observed METex14 transcript results from background splicing [14,16]. On the other hand, the patient with KRAS p.G12V mutation had a high level of METex14 transcripts. Other identified pathogenic co-alterations were mutations in SMAD4, PIK3CA, SETD2, MSH6, and NOTCH2, as well as amplification of CDK4, MDM2, ATR, and PTPN11. In two patients (cases 31 and 34), co-mutation of TP53 and PIK3CA were found, while in one patient (case 2) the NSCLC cells harbored co-amplification of CDK4 and MDM2, and in another case (no. 16) co-existing mutations in MSH6, NOTCH2, and SMAD4 together with amplification of ATR and PTPN11 genes were detected (Figure 2, Supplementary Table S3). Interestingly, no MET amplification was observed, which in approximately 15% of cases co-occurs with the METex14 skipping mutation; this may be explained by the small size of the patient group [8,22]. Since the gene content differs in the used NGS panels (Supplementary Table S1), not all genes were analyzed across all samples.
2.4. Clinical Characteristics of METex14 Positive NSCLC
Most of the patients were ≥70 years old (30/34), with co-existing comorbidities (30/34), in performance status (PS) 0–1 (32/34), females (20/34), with no spread to the central nervous system (32/34), in advanced/metastatic stage (18/34), and previous smokers (28/34) (Supplementary Table S4). The clinical characteristics of our cohort are presented in Figure 2 and are consistent with previous observations in the literature and larger cohorts [6,8,20,24,25]. There were only 5 patients treated with MET-TKI, which makes our cohort too small for reliable statistical analysis of the clinical effect of MET-TKI. Patients no 3, 4, and 18 are still being treated with MET-TKI (November 2025) and all had mutations in the SD site (Figure 2).
2.5. Mutations in an Exonic Splicing Enhancer in METex14 Dependent for Splicing
Interestingly, two of the identified METex14 skipping transcript positive cases had exonic METex14 mutations (case 27: c.3004delG and case 20: c.3004_3005GT>A) involving the same position. Because these mutations both lead to a reading frame shift, which would cause nonsense-mediated decay, it was surprising that METex14 transcripts were identified by RNA-NGS testing (Figure 2), although with a reduced number of METex14 transcripts (114 and 515 reads, respectively). To investigate these two exonic mutations, we performed an analysis using the ESE finder tool (version 3.0), which identified a potential altered binding site for the serine/arginine-rich splicing factor 2 (SRSF2) (Supplementary Table S5). Furthermore, we analyzed the effect of mutation in each nucleotide in METex14 using the newly developed SpliceTransformer (SpTransformer) algorithm [26]. SpTransformer identified in silico that the SD and SA sites were most affected by point mutations (Figure 3) [27]. Furthermore, several potential METex14 exonic SREs, including nucleotide c.3003 which is located in the potential SRSF2-binding site and neighboring nucleotide c.3004 which is deleted in cases 20 and 27, were identified using ESE finder. These findings suggest that exonic mutations within METex14 may influence its splicing by disrupting SREs and thus be important in the choice of MET-TKI therapy.
2.6. In Silico Prediction of METex14 Splice Site Mutations
MES relies on the “Maximum Entropy Principle” and extends beyond many earlier probabilistic models of sequence motifs, including weight matrix models and inhomogeneous Markov models [23]. Analysis of the METex14 splice sites using MES showed that METex14 contains a relatively strong SA splice site with a score of 10.86 and a relatively weak SD splice site with a score of 7.84. Two patients with the strongest effect of METex14 mutations had relatively large deletions of the 5’ parts of the PPT element (case 10 and 29). The lowest MES score difference was observed in the SA mutation c.2942delA (score: 2.02) and the SD mutation c.3082+3A>T (score: 3.20), which might be less severe and only partially result in the expression of METex14 transcript. The MES analysis of the MET c.2942-48del (case 9) mutation could not be performed due to the mutation’s location too distant to the SA and SD sites (Figure 4, Supplementary Table S6).
Our MES analysis showed the highest Z-scores for identified splice site mutations such as c.2942-1G>A and c.3082+2T>A and the lowest Z-scores for mutations in less conserved nucleotide positions like c.2942delA and c.3082+3A>T.
3. Discussion
Reliable detection of potentially pathogenic mutations in METex14 is critical for identifying NSCLC patients who will benefit from MET-TKIs. NGS is a frequently used method for detecting genetic alterations in NSCLC, including METex14 skipping variants. Advantages of utilizing NGS-technology include the ability to analyze multiple biomarkers simultaneously within a single assay with acceptable sensitivity (approximately 5% mutated allele). This approach conserves both time and tumor tissue when compared to sequential testing of individual biomarkers [28]. The phenomenon of METex14 skipping was first identified in 1994 as an alternative splicing variant in cDNA from normal mouse embryos, without any associated mutations affecting splicing consensus sequences [29]. METex14 skipping was first identified in NSCLC tissue in 2005, resulting from a somatic 141 bp deletion that led to the skipping of exon 14 [30]. METex14 splice site variants occurring in NSCLC are now counted in hundreds [22,24,25]. Certain METex14 splice sites variants such as c.3082+1G>T and c.3082+1G>A are already well-defined in genomic databases like the Catalogue of Somatic Mutations in Cancer (COSMIC), the Oncology Knowledge Base (OncoKB), and cBio Cancer Genomics Portal (cBioPortal), in terms of their impact on splicing [31,32,33].
In our small cohort (n = 34), the clinical and molecular data are also comparable with the largest to date reported cohort of 1592 NSCLC patients with METex14 skipping variants detected by comprehensive hybrid capture-based genomic profiling (69,219 NSCLC samples profiled) [25]. This study confirmed the comparable frequency of this alteration (2.3% versus 3.5% in our cohort), more frequent prevalence in older populations with smoking history, and co-existing mutations in tumor suppressor genes like TP53 and MDM2. Additionally, the feasibility of detecting METex14 in plasma (n = 134) was demonstrated to expand the applications of plasma as the sole source for testing.
To ensure the most reliable detection of METex14 variants, sufficient coverage on both splice sites should be granted, and this, as illustrated in Figure 1, varies between different DNA-NGS panels. It is important to use an NGS panel covering relevant sequences of both the SA and the SD sites, as well as the BS and PPT elements of METex14 [34]. Indeed, the CLv2 panel only covers the SS element of METex14 as well as parts of MET intron 13 and exon 14, which might explain why only approximately 10% of the possible METex14 splice site mutations can be detected using this panel [34]. In addition, not all METex14 skipping mutations can be reliably detected by DNA-based NGS alone, as larger genomic rearrangements or mutations within METex14 exonic SRE elements may be challenging to identify with certain DNA-NGS panels, particularly older panels such as the CLv2 panel. Therefore, to detect METex14- and other gene fusion-positive patients, the most optimal diagnostic approach is to use combined DNA- and RNA-NGS [35,36,37,38].
Therefore, optimal insight into this biomarker utilizing sequencing of DNA and RNA is needed to achieve a reliable detection supported by information from both materials. As shown in Figure 5, we present an optimized diagnostic workflow that has already been implemented at our institution. This diagnostic workflow reflects synergy of complementary DNA- and RNA-testing and allows optimal interpretation of both results, leading to a more sensitive approach, and provides direct access to an aberrant splicing product.
The five main diagnostic outcomes from combined DNA and RNA sequencing are outlined in Figure 5. The most common scenario (1) involves the detection of METex14 splice site mutations leading to exon skipping. When both mutations causing the exon 14 skipping and a shorter transcript are present, the concordant results support using MET-TKI. In the second instance (2), METex14 skipping is detected in the absence of identifiable splice site mutations within exon 14. Accurate quantification of METex14 transcripts is critical in these cases, as the presence of a shorter transcript is sufficient for producing abnormal protein, inaccessible for ubiquitination, and therefore can serve as a target for MET-TKI. Amplicon-based NGS analyses have occasionally demonstrated low-level expression of METex14 skipping. However, it is still uncertain whether this reflects physiological background expression or results from assay-related artifacts. Given that transcript detection is influenced by assay design and sequencing depth, it is essential for each laboratory to define the assay-specific thresholds to ensure reliable interpretation within the diagnostic context. In cases exhibiting high METex14 transcript expression, the most probable underlying mechanism is large-scale chromosomal rearrangements affecting MET exon 14 splicing, which are not detectable by the employed NGS panel due to its limited genomic coverage [34,39,40]. In the third scenario (3), where a potential METex14 splice site mutation is identified, but no corresponding exon 14 skipping transcript is observed, we recommend conducting a bioinformatic evaluation of the variant using, e.g., MaxEntScan. This tool aims to assess the splice site’s strength and to indicate whether the identified mutation has a functional impact on splicing. In this case, the usage of MET-TKI is unlikely to have an effect since MET protein stability should not be affected [41]. The fourth case (4) involves the identification of an exonic mutation within METex14 accompanied by exon 14 skipping, but without any detectable splice site mutation. In such instances, it is valuable to further investigate whether the exonic variant is located within an ESE motif, as disruption of ESE elements could potentially influence METex14 splicing. Notably, synonymous exonic mutations not leading to amino acid sequence change (i.e., silent mutations) are often filtered out during bioinformatic analyses, although they may affect ESE elements and consequently affect exon splicing. This rare scenario reflects the vulnerability of exon 14, which is its unique feature inducing aberrant splicing, while canonical splice sites are undisturbed. The utilization of MET-TKI is legitimized here due to the presence of METex14 skipping. The fifth scenario (5) involves the domain of METex14 where CBL binds, which can also result in sustained MET receptor activity. Importantly, mutations affecting amino acid residue Y1003—a critical site for CBL-mediated recruitment and subsequent ubiquitination of MET—can deregulate MET signaling. These mutations phenocopy the functional effects of METex14 skipping by impairing receptor downregulation, although they do not alter MET splicing [7,10]. Additionally, studies from cell lines have proved that altered CBL activity may also cause MET receptor activity, with CBL wildtype cells showing lower MET expression than CBL mutant cells. Since ubiquitination of MET was also decreased in CBL-mutant cells compared to CBL-wildtype cells, CBL status was proposed to be considered a potential positive indicator for MET-targeted therapy in NSCLC [7,42]. However, despite anecdotal case reports having described the clinical effect of the MET-TKI, Crizotinib, in NSCLC patients with MET Y1003S mutation [43,44], the efficacy of MET-TKI in this scenario remains uncertain [45].
Our workflow demonstrated in Figure 5 addresses the issue of discordance between DNA- and RNA-NGS and is primarily built on our experience with amplicon-based NGS. Hybrid-capture assay is an alternative technique that may also be considered in situations where DNA-NGS did not identify a METex14 mutation but a METex14 skipping transcript is present. In such cases, METex14 skipping can be caused by large structured chromosomal alterations, which cannot be detected by amplicon-based NGS. However, it requires more DNA and RNA input, which can be challenging with scarce diagnostic materials. Furthermore, it is a more costly and time-consuming approach [25,40].
The impact of each METex14 variant on splicing may play an important role in understanding this disease. The question is whether it is possible that each METex14 variant can be quantitatively defined in a way that determines its role as a driver alteration in a particular patient. To address the question of quantitative assessment of METex14 skipping variants, different approaches, like variant allele frequency (VAF) or number of RNA reads, may be taken into consideration. VAF means the percentage of reads of a given variant in relation to all reads in this position, and RNA reads give numbers of a given MET transcript. Despite these quantitative features that may inform about the measurable presence of a given METex14 variant, they all depend on spatial and temporal conditions determining the specific biopsy of the tumor at the given time. As NSCLC is an evolving disease, these conditions are dynamic and cannot be regarded as independent and robust factors, implying that the individual METex14 variant may or may not function as a strong driver in this disease.
Another approach may be in silico analysis providing a numerical assessment of a variant impact, which may be calculated by the MES tool. MES is a bioinformatics tool used to predict the effects of sequence variations on splicing signals by modeling the sequences of short sequence motifs involved in RNA splicing, and accounts for non-adjacent and adjacent dependencies between positions. We have calculated the impact of METex14 variants identified in our cohort by MES, and the results are presented in Figure 4. The varying levels of METex14 variant expression reflect differences in their functional impact—specifically, their capacity to drive a MET-dependent phenotype in NSCLC. The MES value determines the extent of MET-dependent signaling and contributes to disease heterogeneity. The impact of the individual MET variants and influence of co-existing alterations both create the individual molecular picture of each patient with the METex14 skipping variant. Then, these two factors: the MES score of MET variants and co-alterations may affect the response to targeted therapy with MET-TKIs. The observations from clinical studies show that the best response rates to the current available MET-TKIs reach about 70% in treatment-naïve patients [46]. It shows us that there might be several subgroups among NSCLC with METex14 skipping defined by different strengths of METex14 variants on aberrant splicing and MET signaling. Furthermore, co-existing molecular alterations may also have an impact on MET-TKI response [22,47,48]. Altogether, these factors may define individual subgroups of NSCLC patients with METex14 splice site variants.
Bioinformatics tools such as MES and SpTransformer may be used as complementary algorithms to prioritize variants [23,26]. When a variant is located within the splice site region, MES can be used to directly assess the effect on splice site strength (Figure 4), while SpTransformer allows for the estimation of the overall splicing impacts of variants both within and outside the splice site region (Figure 3). Variants flagged by SpTransformer can then be further analyzed with tools like MES to determine whether they alter splicing by creating a novel splice site, or by disturbing the balance between splicing enhancers and silencers.
Despite promising results of variant interpretation in silico by using DNA-NGS data, especially regarding MET variants not interrupting splice sites, the access to bioinformaticians may be a significant hurdle in the routine practice [49]. Furthermore, employing several recently developed machine learning-based algorithms, demonstrating a high potency to predict splicing and to classify transcript variants of METex14, may also necessitate collaboration with data scientists, who are still difficult to access in the real-world clinical diagnostics [50,51]. Finally, computational-based evidence predicting impact on skipping may not always be acceptable to categorize a new potentially (likely) pathogenic variant as pathogenic, and further functional studies may be required [52]. By using this approach, two METex14 splice site variants located outside the canonical GT/AG have recently been identified by integrating the combination of in silico prediction, RT-PCR, with Sanger sequencing and adopted to a laboratory standard procedure as routine practice [53].
4. Materials and Methods
4.1. Patient Cohort and Molecular Diagnostic Testing
We identified 34 NSCLC patient samples harboring METex14 variants as DNA splice site mutations and/or METex14 RNA transcript, from the targeted NGS data of the routine molecular testing of NSCLC-patients at the Department of Pathology, Rigshospitalet, Denmark, between January 2018 and August 2023. In this period, we used different DNA-NGS panels as a core diagnostic approach, and RNA-NGS as a supplementary and gradually implemented diagnostic tool. Molecular diagnostic NGS analyses of lung cancer patients were carried out initially (January 2018–August 2023) using the DNA-based AmpliSeq Colon and Lung Cancer Research Panel v2 (CLv2) (Thermo Fisher Scientific, Roskilde, Denmark) and in certain cases supplemented with broader NGS tests by using the DNA-based part of the Oncomine Comprehensive Panel v3 (Comp) (Thermo Fisher Scientific. Roskilde, Denmark), and/or the RNA-based FusionPlex Lung Panel v1.0 (FusionPlex) (Archer, Boulder, CO, USA). In August 2023, the NSCLC molecular diagnostic testing was substituted with combined DNA and RNA testing using the Oncomine Dx Express Test (ODxET) panel (Thermo Fisher Scientific, Roskilde, Denmark). Detailed gene lists of the NGS panels are provided in Supplementary Table S1. Samples without METex14 transcript and METex14 splice site mutation were retrospectively re-sequenced as part of this study using the ODxET panel. The Comp and FusionPlex panels were sequenced on an Ion GeneStudio™ S5 System (Thermo Fisher Scientific, Roskilde, Denmark) and the ODxET panel was sequenced on a Genexus™ Integrated Sequencer system (Thermo Fisher Scientific, Roskilde, Denmark). Both instruments were used for the CLv2 panel.
4.2. Genomic Profiling by Next-Generation Sequencing
Genomic DNA was isolated from tissue sections of formalin-fixed paraffin-embedded (FPPE) tumor resections or core needle biopsies (n = 18) using the One-tube FFPE extraction method [54] or from cytological tumor fine-needle aspirates (n = 16), (Supplementary Table S3) using Maxwell RSC DNA Blood kit (Promega, Madison, WI, USA). RNA was isolated from both materials using the Maxwell RSC RNA FFPE kit.
4.3. Bioinformatic Analysis
Splice site mutations were in silico analyzed using MES (https://github.com/Congenica/maxentscan (accessed on 24 November 2025)) [23]. Prediction of exonic splicing enhancers (ESEs) was performed by using ESE finder 3.0 (https://esefinder.ahc.umn.edu/cgi-bin/tools/ESE3/esefinder.cgi (accessed on 24 November 2025)) [55] and SpTransformer (http://tools.shenlab-genomics.org/tools/SpTransformer (accessed on 24 November 2025)) [26]. All bioinformatic analyses were performed using MET reference transcript sequence NM_001127500.1 (https://www.ncbi.nlm.nih.gov/nuccore/NM_001127500.3/ (accessed on 24 November 2025)).
5. Conclusions
This study has demonstrated:
- When using DNA-NGS technology to detect METex14 skipping variants, it is important to note that different panels, such as CLv2, ODxET, and Comp, are designed to capture splice sites mutations in specific regions of exon 14.
- Complementary DNA- and RNG-NGS are needed for optimal detection of METex14 skipping in real-world NSCLC patients.
- The presence of the aberrant MET transcript is the most predictive biomarker for using MET-TKIs.
- Bioinformatics tools such as MES and SpTransformer provide additional information regarding impact of each METex14 mutation on aberrant splicing and the altered binding site, respectively.
- Two novel exonic mutations are also capable of causing abnormal splicing of METex14, in addition to variants localized in canonical splice sites.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Cooper C.S. Park M. Blair D.G. Tainsky M.A. Huebner K. Croce C.M. Vande Woude G.F. Molecular cloning of a new transforming gene from a chemically transformed human cell line Nature 1984311293310.1038/311029 a 06590967 · doi ↗ · pubmed ↗
- 2Frampton G.M. Ali S.M. Rosenzweig M. Chmielecki J. Lu X. Bauer T.M. Akimov M. Bufill J.A. Lee C. Jentz D. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors Cancer Discov.2015585085910.1158/2159-8290.CD-15-028525971938 · doi ↗ · pubmed ↗
- 3Serna-Blasco R. Mediavilla-Medel P. Medina K. Sala M.Á. Aguiar D. Díaz-Serrano A. Antoñanzas M. Ocaña J. Mielgo X. Fernández I. Comprehensive molecular profiling of advanced NSCLC using NGS: Prevalence of druggable mutations and clinical trial opportunities in the ATLAS study Lung Cancer 202520410855010.1016/j.lungcan.2025.10855040300279 · doi ↗ · pubmed ↗
- 4National Comprehensive Cancer Network Non-Small Lung Cancer (Version 1.2026). Guidelines Detail (nccn.org)Available online: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf(accessed on 24 November 2025)
- 5Hendriks L.E. Kerr K.M. Menis J. Mok T.S. Nestle U. Passaro A. Peters S. Planchard D. Smit E.F. Solomon B.J. ESMO Guidelines Committee. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment, and follow-up Ann. Oncol.20233433935710.1016/j.annonc.2022.12.00936872130 · doi ↗ · pubmed ↗
- 6Subramanian J. Tawfik O. Detection of MET exon 14 skipping mutations in non-small cell lung cancer: Overview and community perspective Expert Rev. Anticancer Ther.20212187788610.1080/14737140.2021.192468333957836 · doi ↗ · pubmed ↗
- 7Peschard P. Fournier T.M. Lamorte L. Naujokas M.A. Band H. Langdon W.Y. Park M. Mutation of the c-Cbl TKB-domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein Mol. Cell 20018995100410.1016/S 1097-2765(01)00378-111741535 · doi ↗ · pubmed ↗
- 8Recondo G. Che J. Jänne P.A. Awad M.M. Targeting MET Dysregulation in Cancer Cancer Discov.20201092293410.1158/2159-8290.CD-19-144632532746 PMC 7781009 · doi ↗ · pubmed ↗