Targeting the MAPK Pathway in Brain Tumors: Mechanisms and Therapeutic Opportunities
Dimitrios Vrachas, Elisavet Kosma, Angeliki-Ioanna Giannopoulou, Angeliki Margoni, Antonios N. Gargalionis, Elias A. El-Habr, Christina Piperi, Christos Adamopoulos

TL;DR
This review explores how the MAPK pathway contributes to brain tumors and discusses current and future therapies targeting this pathway to improve treatment outcomes.
Contribution
The paper provides a comprehensive synthesis of MAPK pathway alterations and therapeutic strategies in CNS tumors, emphasizing precision treatment approaches.
Findings
MAPK pathway dysregulation is a common feature in many pediatric and adult CNS tumors.
Therapies targeting the MAPK pathway face challenges like drug resistance and limited BBB penetration.
Combination strategies and novel drug-delivery technologies are being explored to enhance treatment efficacy.
Abstract
Brain tumors remain among the most difficult cancers to treat, largely because of their biological complexity and the limited ability of many drugs to reach the brain. A major molecular pathway that drives the growth of many brain tumors is the MAPK signaling pathway. In this review, we explain how alterations in this pathway contribute to tumor development in both children and adults, and we summarize current and emerging therapies that specifically target this pathway. We also discuss the main challenges that limit treatment success, including drug resistance, tumor diversity, and the protective blood–brain barrier. By integrating recent advances in molecular biology with therapeutic strategies, this work aims to guide future research and improve precision treatment approaches for patients with brain tumors. Central nervous system (CNS) tumors consist of a diverse set of malignancies…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
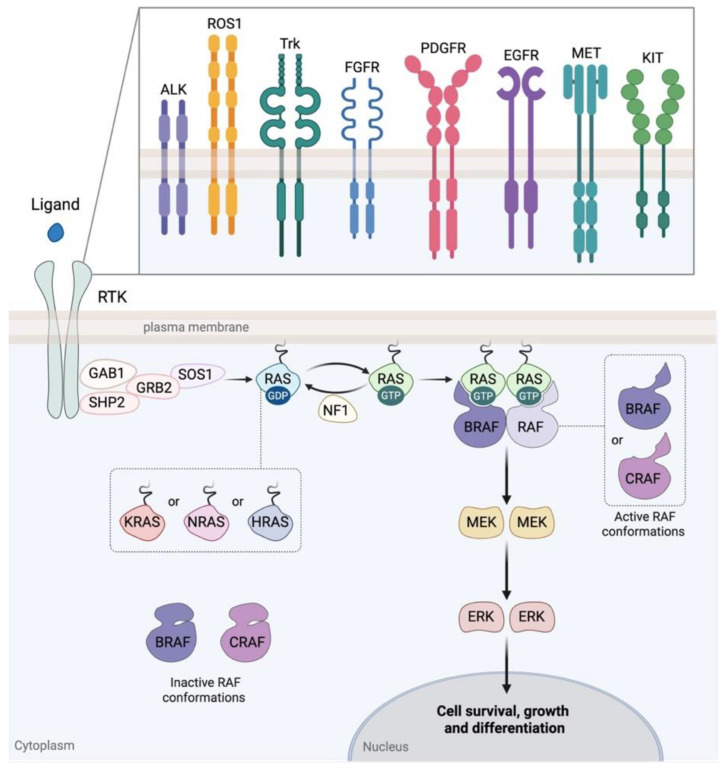 Figure 1
Figure 1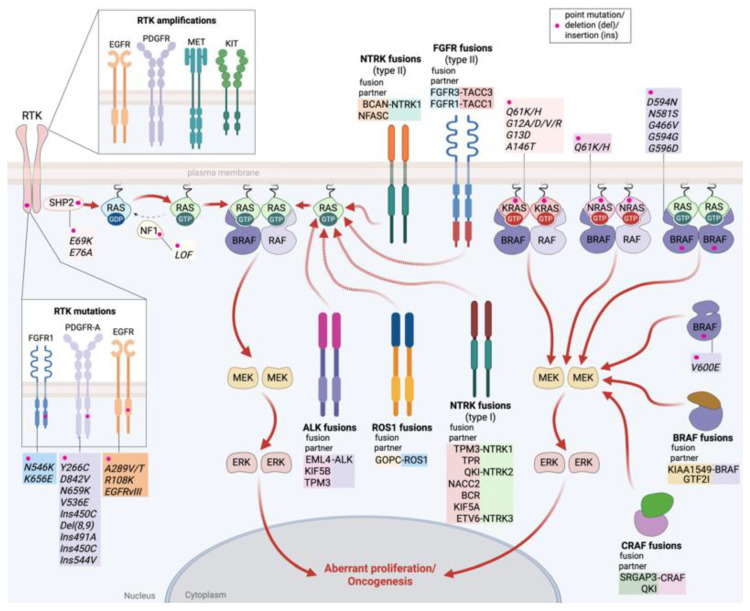 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMelanoma and MAPK Pathways · Glioma Diagnosis and Treatment · Brain Metastases and Treatment
1. Introduction
Central nervous system (CNS) tumors represent a heterogeneous group of both malignant and benign entities, characterized by varying clinical behavior, histological, and molecular traits. In 2020, CNS tumors accounted for 1.6% of all cancer cases globally, while in 2022, 321,624 new cases were estimated, corresponding to an age-standardized incidence rate (ASIR) of 3.5 per 100,000 people [1]. Despite their relatively low incidence rate, they impose a major disease burden due to their disproportionately high mortality rates, especially among children [2,3]. The average lifespan for adults with glioblastoma, the most aggressive type of brain tumor, is approximately 2 years [3]. A study utilizing the global burden of disease (GBD) database predicted that the total number of cases will gradually increase by 2040, even though the mortality rates in certain populations may decrease slightly [4]. To date, the current established treatment approaches include surgical resection, radiotherapy, and chemotherapy [5]. CNS tumors are characterized largely by intratumoral heterogeneity which can be defined as the coexistence of genetically, epigenetically, transcriptionally, and phenotypically distinct cell subpopulations within the same tumor mass. This molecular heterogeneity of these tumors, their anatomical location, in conjunction with the protective role of the blood–brain barrier (BBB) and intrinsic or acquired drug resistance, leads to limited treatment efficacy and poor clinical patient outcomes. Importantly, children and young adults who survive by receiving the standard of care experience long-term complications that largely affect their quality of life [3]. Moreover, recent molecular profiling studies have revealed that pediatric and adult gliomas constitute fundamentally distinct biological entities, driven by different oncogenic alterations and signaling dependencies [6,7]. Consequently, there is a pressing need for more targeted and personalized therapeutic strategies.
The mitogen-activated protein kinase (MAPK) signaling pathway plays a fundamental role in cell physiology by regulating cell cycle, proliferation, survival, differentiation, apoptosis, and is implicated in various other developmental processes [8]. Core components of the MAPK pathway are serine/threonine-specific protein kinases, the mitogen-activated protein kinases (MAPKs), that transduce intracellular signals through sequential protein phosphorylation and activation events. Among them, the rapidly accelerated fibrosarcoma (RAF), the mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK), and the extracellular signal-regulated kinase (ERK) form the RAF-MEK-ERK signaling axis, the central and most thoroughly studied MAPK pathway [8,9] (Figure 1).
The MAPK pathway is usually activated upon binding of a ligand, such as a growth factor, to a receptor tyrosine kinase (RTK), leading to its activation and the following recruitment, intracellularly, of adaptor proteins/regulators, which in turn activate the membrane-anchored small guanosine triphosphatase (GTPase), rat sarcoma virus oncogene (RAS) [10] (Figure 1). Afterward, the active GTP-bound RAS recruits at the membrane and activates RAF through a complex process of dimerization and phosphorylation events [11]. RAF then phosphorylates and activates its substrate MEK, which consecutively phosphorylates and activates ERK [12,13] (Figure 1). Finally, activated ERK phosphorylates its targets, usually transcription factors or co-activators in the nucleus, thereby regulating the expression of several genes [8,14].
RAS-RAF-MEK-ERK signaling axis deregulation, primarily due to its aberrant activation, is a key driver of carcinogenesis [15,16,17,18]. Mutations in key effectors of the pathway, most frequently in RAS and RAF, have been identified in a wide variety of cancers, including melanomas, lung, colorectal, and ovarian cancers [18,19,20].
Consequently, therapeutic efforts that target MAPK pathway components have led to the approval by the Food and Drug Administration (FDA) of several small-molecule inhibitors, while other alternative targeting approaches are under pre-clinical investigation and development, including gene silencing, proteolysis-targeting chimeras (PROTACs), and bispecific antibodies [8,21,22,23].
Notably, MAPK pathway component alterations have also been detected in primary brain tumors. The most common alterations are the gene fusion between the BRAF and KIAA1549 genes and the BRAF V600E mutation, both of which are most prevalent in pediatric compared to adult tumors [24].
Beyond these, diverse genetic alterations involving RTKs, RAS, RAF kinases and pathway regulators lead to the aberrant activation of the MAPK pathway and highlight its central role in the pathogenesis of CNS tumors [8,25].
In this review, we provide a comprehensive overview of MAPK pathway alterations in CNS malignancies, with emphasis on pediatric and adult gliomas, glioneuronal tumors, and ependymomas. We summarize current strategies for MAPK pathway inhibition, including BRAF, MEK, and ERK inhibitors, and discuss how these approaches are being integrated into clinical management [26,27]. Furthermore, we address the major therapeutic challenges that limit efficacy, including restricted BBB penetration, tumor heterogeneity and resistance mechanisms, and the immunosuppressive tumor microenvironment [28,29]. Finally, we highlight emerging treatment concepts and combinatorial approaches that hold promise and shape future perspectives for MAPK-targeted therapy in brain tumors.
2. Central Nervous System (CNS) Tumors
Central nervous system (CNS) tumors can be classified as either primary, originating from cell types within the brain and spinal cord, or metastatic, arising from tumors that develop in distal organs, most commonly the lung and breast, and spread to the brain through the bloodstream or the lymphatic system [30,31,32]. Primary CNS neoplasms depict a heterogeneous group, consisting of gliomas, glioneuronal, neuronal, and ependymal tumors [33]. Although relatively rare, tumors of the brain and other parts of the central nervous system contribute substantially to morbidity and mortality across all age groups. The frequency of these neoplasms is higher in children aged up to 5 years old, with most being malignant gliomas, germ-cell, and embryonal tumors. In adults, malignant CNS tumors, especially gliomas, are among the leading causes of death [31,34].
The basic criteria for the characterization of these entities have traditionally depended on histological, immunohistochemical, and cytological observations, often linked to their likeness to an alleged cell type of origin. The recognition of CNS tumors solely based on morphological features began to hinder the diagnosis of several subgroups and, hence, proper treatment. Eventually, as described in the 5th edition of the World Health Organization (WHO) CNS tumor classification (CNS5, 2021), this obstacle was overcome by incorporating molecular and genetic alterations into the diagnostic criteria. According to the current classification, six families of both benign and malignant tumors have emerged, comprising adult-type diffuse gliomas, pediatric-type diffuse low-grade gliomas (DLGG), pediatric-type diffuse high-grade gliomas (DHGG), circumscribed astrocytic gliomas, glioneuronal/neuronal tumors, and ependymal tumors [33,35].
The implementation of molecular assays in their diagnosis, such as DNA/RNA sequencing, genome-wide methylation profiling, quantitative PCR (qPCR), and FISH, has revealed a broad spectrum of genetic alterations in CNS tumors, encompassing point mutations, insertions and deletions, copy number changes, and gene rearrangements [36]. The genes that are more frequently affected are vital to cellular homeostasis. For instance, alterations in genes encoding for the phosphoinositide 3-kinase (PI3K), epidermal growth factor receptor (EGFR), V-Raf murine sarcoma viral oncogene homolog B (BRAF), platelet-derived growth factor receptor α (PDGFRA), and Met tyrosine-protein kinase (MET) lead to defective receptor tyrosine kinase signaling. The regulation of the cell cycle is also affected by mutations in the p53, retinoblastoma susceptibility (RB1), cyclin-dependent kinase 4 (CDK4), cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A and CDKN2B), and v-myb avian myeloblastosis viral oncogene homolog (MYB) genes. Moreover, genetic changes in telomerase reverse transcriptase (TERT) and α-thalassemia intellectual disability X-linked (ATRX) genes affect the preservation of telomere integrity. Additionally, modifications in histone variants H3.1 and H3.3, predominantly the substitutions K27M and G34V/R, are implicated in abnormal chromatin arrangement and epigenetic regulation of gene expression [35,36,37,38]. Cell metabolism is also affected through the production of oncometabolites, such as 2-hydroxyglutarate (2-HG), which arises from mutations in the isocitrate dehydrogenase (IDH) gene [39]. The mutations in IDH1 and IDH2 often co-exist with concurrent deletion of chromosome arms 1p and 19q (1p/19q codeletion) and TERT alterations [40].
The treatment of CNS tumors continues to pose difficulties in both pediatric and adult age groups. More specifically, several parameters need to be considered in terms of tumor cell origin, location, genetic background, microenvironment and effective drug delivery. The standard clinical strategies, so far, involve surgery, radiation, and chemotherapy [30,41,42]. Over the last years, more targeted therapies, incorporating inhibitors, chimeric antigen receptor-T (CAR-T) cells, and vaccines, among others, have emerged. Regarding glioblastoma multiforme (GBM), the most frequent and aggressive form of glioma, immunotherapy may prove to be a promising treatment option [43]. Nonetheless, brain tumors still represent one of the main causes of cancer-related mortality, while survivors face a high risk of chronic health conditions, thus underscoring the pressing need for new treatment modalities [42,44].
2.1. MAPK Pathway Alterations in CNS Tumors
Aberrant activation of the MAPK pathway is a hallmark of several CNS tumors, frequently driven by genetic alterations such as point mutations, gene fusions, amplifications, or overexpression, most commonly involving RTKs, RAS, RAF, and regulators of the pathway such as SHP2 and NF1 [22,45,46] (Figure 2).
2.1.1. Receptor Tyrosine Kinase (RTK) Alterations
RTKs are critical oncogenic drivers in gliomas, affected by diverse mechanisms, which include point mutations, gene amplifications, and chromosomal rearrangements that create fusion oncoproteins (Figure 2). These aberrations commonly confer ligand-independent kinase activation, leading to persistent MAPK pathway signaling [47]. For instance, EGFR alterations, most notably gene amplification and the EGFRvIII deletion variant, are prevalent in GBM and drive aggressive proliferation and therapeutic resistance [48]. Similarly, PDGFR-A amplification is characteristic of the proneural GBM subtype, while MET amplification and PDGFR overexpression also contribute to glioma pathogenesis [49,50] (Figure 2). Importantly, in pediatric gliomas, gene fusions involving anaplastic lymphoma kinase (ALK), ROS proto-oncogene 1 (ROS1), neurotrophic tyrosine receptor kinase (NTRK2) and MET define a distinct, hemispheric high-grade subgroup with intermediate prognosis [51]. Fibroblast growth factor receptor (FGFR) gene family alterations, including FGFR3–transforming acidic coiled-coil containing protein 3 (TACC3) fusions, constitute actionable drivers in a subset (3–5%) of GBM, producing fusion proteins that promote oncogenesis [52].
In pilocytic astrocytomas (PAs), fusions of the NTRK2 gene, which encode for the tropomyosin receptor kinase B (TrkB) with either the transcriptional repressor nucleus Accumbens-associated protein 2 (NACC2), NACC2-NTRK2, or the pre-mRNA alternative splicing regulator Quaking homolog KH domain containing RNA binding (QKI), QKI-NTRK2, can induce MAPK pathway hyperactivity in a ligand-independent manner [53]. Additionally, in PA patients, the activating substitutions N546K and K656E in the FGFR1 gene have been linked to elevated phosphorylated/activated ERK levels [54]. In a small number of infant-type hemispheric gliomas (IHGs), gene fusions involving ALK, NTRK1, and ROS1 were detected without co-occurrence [54,55]. In the same study, a case of PA bore a fusion of breakpoint cluster region (BCR) and NTRK2 genes (BCR-NTRK2), while a patient with pleomorphic xanthoastrocytoma (PXA) harbored the tropomyosin 3 (TPM3)-NTRK1 fusion. Moreover, two cases of gangliogliomas exhibited the fusion FGFR3-TACC3 [54,55].
The localization of NTRK fusions varies depending on the fusion partner. Thus, when the 5′ fusion partner encodes a membrane or extracellular protein, such as the protein fusions of TrkA with the proteoglycan brevican (BCAN), BCAN–NTRK1, and the cell surface protein neurofascin (NFASC), NFASC–NTRK1, the fusion protein is membrane-associated (Type II) (Figure 2). Otherwise, when the fusion partners are cytosolic or nuclear, such as TPM3, QKI, Ets variant transcription factor 6 (ETV6), localization is cytoplasmic (Type I) (Figure 2). Yet all retain the NTRK kinase domain and drive constitutive MAPK signaling [56]. In GBM, Golgi-associated PDZ and coiled-coil motif-containing protein (GOPC)–ROS1 fusion proteins exhibit isoform-specific subcellular localization with the long form localizing to the Golgi, while the short form is cytoplasmic [57].
2.1.2. RAS Alterations
All three RAS small GTPases, Kirsten rat sarcoma viral oncogene homolog (KRAS), neuroblastoma rat sarcoma viral oncogene homolog (NRAS) and Harvey rat sarcoma viral oncogene homolog (HRAS), function as molecular switches that alternate from their inactive GDP-bound state to their active GTP-bound state (Figure 1). Oncogenic RAS mutations, typically missense substitutions at hotspot codons 12, 13, or 61, largely impair intrinsic GTP hydrolysis and lock RAS in its active form, resulting in constitutive downstream signaling [58] (Figure 2). While RAS mutations are among the most common oncogenic drivers in many cancers, they are relatively rare in gliomas. Nevertheless, accumulating evidence indicates that RAS alterations can contribute to gliomagenesis across different subtypes, either as point mutations or gene copy number gains, often cooperating with other oncogenic events to sustain tumor growth and progression [59].
In two distinct cases of PXA, a KRAS mutation at codon 61 was detected, which codes for glutamine in position 61 of KRAS protein [60]. More specifically, in one case, glutamine was substituted by lysine (Q61K), while in the other, it was replaced by histidine (Q61H). Interestingly, these two cases were the first in which a KRAS mutation was detected in PXA patients [60]. Despite its rarity, a case of ganglioma with an NRAS mutation was reported among a heterogeneous group of 30 patients with infantile CNS tumors [54,55]. Oncogenic missense mutations in KRAS and NRAS were present in 8 patients with IDH-mutant astrocytomas, including G12A/D/V, G13D, D33E, A146T, and K117N substitutions. In the same cohort, 3 tumor samples reported a high increase in KRAS gene copies [25,54,55].
2.1.3. MAPK Pathway Regulators Alterations
Beyond RTKs and RAS itself, several intracellular modulators of MAPK signaling are altered in CNS tumors. The most relevant include neurofibromin (NF1) and the associated sprouty-related EVH1 domain-containing protein 1 (SPRED1) and leucine zipper-like transcription regulator 1 (LZTR1), and Src homology region 2 domain-containing phosphatase-2 (SHP2). NF1 acts as a tumor suppressor by accelerating RAS GTP hydrolysis, a process facilitated by SPRED1, which recruits NF1 to the plasma membrane [24,61]. In PXAs, NF1 mutations were reported in 3 of 13 cases, including two missense and one truncating variant [58]. In IDH-mutant astrocytomas, NF1 alterations occurred in 17 of 27 cases, while LZTR1 mutations (nonsense, frameshift, splice site, or missense) were also observed, consistent with loss of its role in targeting RAS proteins, among others, for ubiquitin-mediated degradation [62]. Inactivating SPRED1 lesions, including biallelic deletion and frameshift changes, have also been described in this tumor type [24].
On the other hand, SHP2 is a positive effector that promotes MAPK pathway activation, as it functions as an adaptor protein and phosphatase downstream of multiple RTKs (Figure 1). Gain-of-function mutations, particularly E69K and E76A, enhance its activity, facilitating sustained RAS/MAPK signaling in PA [54,63] (Figure 2).
2.1.4. RAF Alterations
Within the RAF family of serine/threonine kinases, BRAF is the predominant oncogenic driver in cancers, including gliomas, followed less frequently by CRAF and only rarely by ARAF. RAF proteins under physiological conditions signal as BRAF homodimers or BRAF-CRAF heterodimers, constituting the most common and biologically relevant signaling forms [64,65] (Figure 1). Oncogenic BRAF alterations, through gene fusions or hotspot mutations, result in sustained constitutive downstream MAPK activation.
The most prevalent mutation in pilocytic astrocytomas is a genomic rearrangement that leads to the fusion of the KIAA1549 and BRAF genes. The structural rearrangement involves the duplication of a DNA segment between the KIAA1549 5′-end and BRAF 3′-end genes in the 7q34 chromosomal region, spanning approximately 2 Mb. From this event, five different in-frame variants have been identified: KIAA1549^ex16^-BRAF^ex9^, KIAA1549^ex15^-BRAF^ex9^, KIAA1549^ex19^-BRAF^ex9^, KIAA1549^ex16^-BRAF^ex11^, and KIAA1549^ex18^-BRAF^ex10^. All the resulting chimeric proteins exhibit constitutive activation, as they all lack the N-terminal domain responsible for the autoregulation of BRAF, due to substitution from KIAA1549. At the same time, they maintain the kinase domain of BRAF [66,67,68,69]. Interestingly, in a small cohort of PAs several other fusion partners have been identified for BRAF, including family with sequence similarity 131 member B (FAM131B), ring finger protein 130 (RNF130), chloride voltage-gated channel 6 (CLCN6), makorin ring finger protein 1 (MKRN1), guanine nucleotide-binding protein subunit alpha-11 (GNA11), quaking homolog KH domain RNA binding protein (QKI), fizzy and cell division cycle 20 related 1 (FZR1), microtubule actin crosslinking factor 1 (MACF1), and general transcription factor II-I (GTF2I). Although biologically and functionally disparate, these fusion partners render domains that converge on the same outcome: hyperactivation of BRAF and its downstream signaling [24,54,70,71].
Regarding BRAF mutations, the second most common alteration detected in PAs is a substitution of valine in position 600 by glutamic acid, which results in BRAF V600E the most frequent BRAF mutation in human cancers [69,72,73]. This point mutation disrupts the regulatory conformation of BRAF, resulting in loss of its N-terminal autoinhibitory domain and conferring monomeric kinase activity with hyperactivation of the MAPK pathway (Figure 2) [72,73]. BRAF V600E is observed across several glioma subtypes. In a study by Zou et al., who evaluated mutations in a cohort of 13 PXA patients using next-generation sequencing, the BRAF V600E mutation was present in 38% of the cases [60]. In a comprehensive analysis of 30 infantile (<12 months old) CNS tumors, 7/10 cases of desmoplastic infantile ganglioglioma (DIG) harbored alterations in BRAF (5 mutations, 1 duplication and 1 fusion), 1/2 cases of PXA carried the CAP-Gly domain containing linker protein 2 (CLIP2)-BRAF fusion, 1/2 cases of PA had the KIAA1549-BRAF fusion, and a single case of DLGG was BRAF V600E-mutant [55]. Tumors from 3 young-adult patients with IDG-mutant astrocytomas possessed a rare in-frame protein tyrosine phosphatase receptor type Z1 (PTPRZ1)-BRAF gene fusion and two class III BRAF mutations, the substitutions G464E and D594G [25]. G464E affects the kinase domain of BRAF, producing a kinase-impaired protein that requires RAS activation, whereas D594G affects the activation segment of BRAF, resulting in a kinase-dead protein, both relying on upstream RAS/RTK activity [74,75].
In the context of chimeric proteins, the gene encoding CRAF protein (CRAF or RAF1) has been observed to fuse either with the nuclear transcription factor 1A (NF1A) or SLIT-ROBO Rho GTPase activating protein 3 (SRGAP3) gene in some rare PA case reports at chromosomal regions 1q31.3 and 3p25, respectively. The end-product of both translocations is an oncoprotein that augments constitutive MAPK pathway activation [68,76].
Alterations are not limited to BRAF. In the study of Tauziède-Espariat et al., 2/30 infantile patients with tumors characterized as DIGs carried CRAF fusions, particularly one of these cases presented with the protein kinase cAMP-dependent type II regulatory subunit a (PRKAR2A)-RAF1 fusion [55]. CRAF fusions have been described in rare PAs, involving the nuclear transcription factor 1A (NF1A) or SLIT-ROBO Rho GTPase activating protein 3 (SRGAP3) as fusion partners [68,76]. In infantile DIGs, CRAF fusions have also been identified, including a protein kinase cAMP-dependent type II regulatory subunit α (PRKAR2A)-CRAF chimera [55]. Additional RAF1 fusions, contributing to constitutive MAPK activity, have been documented across gliomas and other tumor types [77].
3. RAS/MAPK Pathway Inhibitors in CNS Tumors
The high frequency of activating mutations and other genetic alterations in the RAS/RAF/MEK/ERK signaling axis and its subsequent hyperactivation has highlighted their association with cancer development and progression [78]. Consequently, components of this pathway have become promising therapeutic targets through their selective inactivation by small-molecule inhibitors. In addition, alternative medicinal chemistry strategies with the development of PROTACs or molecular glues have emerged. All these targeting efforts have been directed towards CNS tumors as well [79].
3.1. RAF Inhibitors
The high frequency of the BRAF V600E mutation, accounting for 95% of all BRAF mutations, and the increased kinase activity of the BRAF V600E oncoprotein made it an ideal pharmacological target for small-molecule inhibitors. This led to the development of the first- and eventually the more selective second-generation RAF inhibitors targeting the mutant-BRAF kinase [72,73]. The increased selectivity for the monomeric mutated BRAF versus the dimeric wild-type BRAF is the basis of the high therapeutic index of the second-generation RAF inhibitors [74]. These discoveries resulted in the FDA’s approval of vemurafenib in 2011 and dabrafenib in 2013, as single agents, for the treatment of metastatic BRAF V600E-mutant melanoma [72,73]. Since then, combination therapies using the RAF inhibitors vemurafenib, dabrafenib and encorafenib, along with the MEK inhibitors trametinib, cobimetinib and binimetinib or the EGFR inhibitor cetuximab, have gained FDA approvals in subsequent years for other types of cancer, harboring the BRAF V600E mutation [72,73,80]. However, the effectiveness of these monomer-selective RAF inhibitors is often hindered by the development of adaptive resistance, primarily mediated by the formation of RAF dimers, through MAPK-pathway reactivation because of the relief of negative feedback or via secondary genetic alterations [72,73,74]. To overcome the dimer-forming resistance mechanisms, next-generation RAF inhibitors that target the dimeric form of RAF have been developed [72,73,74]. Recently, one such inhibitor, tovorafenib, was clinically approved for treating pediatric patients with low-grade glioma (LGG) carrying genetic alterations in the BRAF gene [26].
3.1.1. Vemurafenib
Vemurafenib is a selective BRAF V600E inhibitor that competes with ATP binding, thus preventing downstream MEK activation. It exhibits limited penetration across the BBB, which restricts its efficacy in primary brain tumors (Table 1) [81]. Initially approved for metastatic melanoma, vemurafenib has shown partial efficacy in BRAF-mutant gliomas in case series and small trials [82,83,84]. Responses tend to be short-lived due to the development of adaptive resistance and insufficient CNS concentrations. Common adverse effects include rash, joint pain, fatigue and paradoxical activation of wild-type BRAF leading to secondary malignancies like squamous cell carcinoma [82,83,84].
3.1.2. Dabrafenib
Dabrafenib is another selective BRAF V600E inhibitor with superior BBB penetration and a more favorable safety profile compared to vemurafenib [85]. Clinical trials have demonstrated that dabrafenib is effective in pediatric patients with BRAF-mutant LGGs, leading to tumor regression and improved progression-free survival [86]. On 16 March 2023, dabrafenib combined with the MEK inhibitor trametinib gained FDA approval for pediatric BRAF V600E-mutant LGGs (Table 1) [86]. This synergistic regimen shows improved tolerability, with fewer secondary skin malignancies when used in combination therapy. Patients may exhibit pyrexia, fatigue, skin rash and arthralgia [87].
3.1.3. Encorafenib
Encorafenib is a newer first-generation BRAF V600E inhibitor developed to reduce paradoxical activation and enhance the duration of response [88]. While it is primarily used in melanoma and colorectal cancer, preclinical studies are investigating its use in brain tumors [89]. Although encorafenib features a longer dissociation half-life from BRAF V600E and potentially better pharmacodynamic suppression of MAPK signaling, its efficacy in CNS tumors may be limited by its lower BBB penetration (Table 1) [90].
3.1.4. NST-628
NST-628 is a non-degrading molecular glue that binds to both RAF and MEK proteins, stabilizing their complex in a way that prevents MEK phosphorylation by RAF, effectively blocking downstream signaling [91]. This mode of action avoids resistance mechanisms common in traditional kinase inhibitors. NST-628 inhibits all RAF isoforms (ARAF, BRAF, CRAF) and works across multiple RAS- and RAF-mutant cancers, including those resistant to existing therapies. Unlike many inhibitors, NST-628 is brain-penetrant, making it potentially effective against CNS tumors (Table 1). The compound induces long-lasting suppression of the MAPK pathway in both in vitro and, also, in vivo models, including mouse xenografts and organoids derived from human tumors. As a result, due to its broad activity, resistance-evasion capacity, and brain penetration, NST-628 shows promise for treating a wide range of RAS- and RAF-driven CNS cancers, including those with KRAS, NRAS, or BRAF mutations [91].
3.2. MEK Inhibitors
Selective MEK inhibitors have been developed to effectively block the MAPK pathway activation, especially after its reactivation due to the relieved negative feedback mechanisms following BRAF inhibitor therapy [72,73,74]. Thus, combinatorial targeting of MEK inhibitors (trametinib, cobimetinib, binimetinib) with RAF inhibitors (vemurafenib, dabrafenib, encorafenib) has been FDA-approved, from 2014 to 2018, for patients with metastatic melanoma, non-small cell lung cancer (NSCLC), and anaplastic thyroid cancer carrying the BRAF V600E mutation [72,73,92,93,94]. In 2021, the FDA approved the MEK inhibitor selumetinib for pediatric patients with neurofibromatosis type 1, a genetic disorder in which NF1 loss predisposes to peripheral nerve sheath tumors and other cancers (Table 1) [95,96]. Most MEK inhibitors disrupt the formation of the RAF-MEK complex, inhibiting MEK phosphorylation and activation [97].
3.2.1. Selumetinib
Selumetinib is an allosteric MEK inhibitor that prevents ERK activation and has demonstrated significant efficacy in NF1-associated pLGGs [98], as well as in non-NF1-associated pLGGs [99], including disease stabilization and, in some cases, prolonged disease control (Table 1). Furthermore, it is an orphan drug designation for NF1-altered gliomas [100]. Generally, it is well tolerated in children, but adverse effects, like gastrointestinal symptoms, skin rash, rare cardiomyopathy and ocular toxicity may arise [98,99,100]. Ongoing clinical trials comparing selumetinib with conventional chemotherapy in both NF1-associated and non-NF1 pLGG will further clarify its therapeutic value and long-term safety. Notably, emerging evidence indicates that a subset of patients can maintain durable progression-free survival (PFS) even after treatment cessation, underscoring the potential of MEK inhibition as a promising disease control strategy [99,100].
3.2.2. Trametinib
Trametinib is a potent selective allosteric MEK inhibitor, often used in combination with dabrafenib [92,93,94,97,101]. Approved in combination for BRAF V600E-mutant tumors, trametinib enhances efficacy and reduces adverse effects such as secondary skin cancers [92,93,94,101,102]. Current results from an ongoing clinical trial demonstrate significant clinical benefit to the majority of both pLGG and plexiform neurofibromas (PNs) patients, including measurable responses and prolonged PFS (Table 1) [102]. Common adverse effects are diarrhea, skin rash, fatigue, and hypertension [102].
3.2.3. Binimetinib and Cobimetinib
The MEK inhibitors binimetinib and cobimetinib have been tested primarily in non-CNS malignancies but are currently under investigation in gliomas [89]. Ongoing trials are evaluating their BBB permeability and potential in combination with BRAF and mTOR inhibitors, supported by favorable pharmacokinetics and CNS bioavailability [103]. Binimetinib is currently under clinical investigation in brain tumors, including high-grade glioma (HGG) [89]. Cobimetinib provided efficacy when tested in combination with vemurafenib in a refractory case of BRAF V600E-mutated ganglioglioma [104]. Its role in neuro-oncology, however, remains to be fully defined. Given their pharmacologic profiles, both agents represent promising candidates for rational combination strategies targeting multiple signaling pathways in gliomas.
3.2.4. Mirdametinib
Mirdametinib is an orally bioavailable MEK inhibitor that has recently achieved its first regulatory approval in the United States for the treatment of symptomatic, inoperable NF1-associated PNs in both adult and pediatric patients (Table 1) [105]. Beyond neurofibromatosis type 1, ongoing trials are evaluating its efficacy in pLGGs and other RAS/MAPK-driven tumors, providing a rationale for its potential application in primary brain tumors [105,106]. With established clinical activity in NF1-associated CNS tumors and a favorable oral dosing profile, mirdametinib represents a promising candidate for expanding MEK-directed strategies in neuro-oncology [105,106].
3.3. ERK Inhibitors
Although mutations in ERK proteins are rare, selective ERK inhibitors are under preclinical development, given that ERK is the terminal kinase of the RAS/RAF/MEK/ERK signaling cascade, seeking a more durable inhibitory response [107]. These agents are particularly promising in tumors that develop resistance to BRAF and/or MEK inhibitors.
Ulixertinib (BVD-523)
Ulixertinib is an oral, ATP-competitive ERK inhibitor that has demonstrated preclinical efficacy in various tumor models, including those resistant to BRAF and MEK inhibitors. Phase I clinical trials have shown acceptable tolerability and preliminary antitumor activity in patients with advanced solid tumors harboring MAPK pathway alterations. In gliomas, its ability to cross the BBB and suppress ERK-driven transcription makes it a promising candidate, although its application is under exploration (Table 1) [108]. Elevated liver enzymes, diarrhea and fatigue are the main observed side effects of this drug [108].
3.4. SHP2 Protein Inhibitors
Alongside direct targeting of the RAS/RAF/MEK/ERK axis components, selective inhibitors have been developed against the SHP2 phosphatase (e.g., TNO155 and RMC-4630), which block upstream activation of RAS by inhibiting the GRB2-SOS1 interaction [109] (Figure 1). Specifically, a study has shown that SHP2 inhibition, using the SHP2 inhibitor SHP099, could efficiently reduce RAS-GTP loading, block RAS-mediated RAF/MEK/ERK signaling and abrogate tumor growth in NF1-malignant peripheral nerve sheath tumors (MPNSTs) (Table 1) [110]. Furthermore, combining SHP2 inhibition treatment with hydroxychloroquine (HQ), a pharmacological inhibitor of autophagy, showed enhanced effectiveness in mouse and human NF1-MPNST models [110]. Additionally, Sang and colleagues examined the efficacy of SHP099 in GBM with activated PDGFR-A. SHP099 exhibited antitumor activity either as a single agent or in combination with temozolomide (TMZ) and provided significant survival benefits for GBM tumor xenograft-bearing animals [111].
3.5. Combinatorial Therapies
Combined inhibition of multiple MAPK pathway components enhances treatment efficacy and reduces the risk of resistance or overcomes the already developed adaptive resistance [72,74,97,112,113]. BRAF plus MEK inhibitor is the most validated combination, especially in GBM and in pediatric LGG [114,115]. It delays resistance, lowers toxicity, and provides better PFS compared to monotherapy. Furthermore, ongoing trials are exploring a multi-combinatorial strategy of BRAF, MEK, and AKT inhibitors [116,117]. Lastly, MAPK inhibitors could combine with immunotherapy, given that MAPK inhibition may increase immune recognition, making combination with immune checkpoint inhibitors (ICIs) a promising therapeutic avenue [118,119].
3.6. Clinical Application and Efficacy
3.6.1. Pediatric Low-Grade Glioma (LGG)
Pediatric LGGs are the most frequent pediatric brain tumors and are characterized by indolent growth but can cause significant morbidity. Molecular profiling has revealed that most of these tumors harbor MAPK pathway alterations. The combination of dabrafenib and trametinib has demonstrated remarkable efficacy in pediatric LGGs with BRAF V600E mutations [101,114,115]. In clinical trials, response rates exceeded 70%, and the combination was associated with PFS and tolerable side effects [114,115]. Selumetinib has also shown clinical benefit in NF1-associated pediatric LGGs [98]. Results from Phase II trials indicated that selumetinib led to tumor shrinkage and visual improvement in children with optic pathway gliomas [99,100].
3.6.2. High-Grade Glioma (HGG)
In HGG, the MAPK pathway is often only one of many dysregulated networks, and monotherapy with BRAF or MEK inhibitors has generally been less effective [120]. However, in select cases, such as BRAF V600E-positive GBM, targeted therapies have resulted in durable responses [101,121]. Combination therapy is currently under active investigation in clinical trials, including regimens that pair MAPK inhibitors with other targeted or immunotherapeutic agents [122,123]. Specifically, Arbour and colleagues reported an 18-year-old female with a grade III PXA treated upfront with dabrafenib and trametinib [122]. Also, Fusco et al. describe a similar case of a 19-year-old male patient with grade III PXA, who achieved durable PFS with BRAF and MEK inhibitors combination [123].
3.6.3. Ganglioglioma
Gangliogliomas are usually low-grade brain tumors containing both neuronal and glial elements, most often occurring in children and young adults. A large proportion of these tumors harbor activating mutations in the MAPK signaling pathway, particularly BRAF V600E, which renders them responsive to MEK inhibitors [124]. Nonetheless, some gangliogliomas lack identifiable MAPK pathway alterations and therefore have not traditionally been considered candidates for MEK-targeted therapy. Interestingly, a recent report described a young adult patient with ganglioglioma who did not carry MAPK pathway mutations but achieved a marked and durable response to the MEK inhibitor trametinib [125].
3.6.4. Medulloblastoma
Medulloblastoma is a common malignant pediatric brain tumor. While existing treatments can be effective, they often cause significant long-term side effects [126]. A major clinical challenge is resistance to therapy and recurrence, often driven by tumor stem-like cells [127]. The protein BMI1, a known regulator of stem cell renewal and tumorigenesis, is overexpressed in medulloblastoma and supports tumor growth [128]. A study investigates whether targeting BMI1, alone or in combination with MAPK/ERK pathway inhibitors, could be an effective treatment strategy against medulloblastoma [129]. The study used the PD325901, a MEK inhibitor that blocks ERK phosphorylation, as the MAPK/ERK pathway inhibitor in combination with BMI1 inhibition to evaluate synergistic effects on medulloblastoma cells [129].
3.7. Current and Ongoing Clinical Trials
Several ongoing clinical trials assess the MAPK inhibition in different CNS tumor types (Table 2). Selectively, some of them include the evaluation of the dabrafenib plus trametinib combination in pLGGs [114], which paved the way for the FDA approval of this combination for treatment, the investigation of the effectiveness of selumetinib in NF1-associated gliomas [96], and the study of the role of tovorafenib in relapsed pLGG with BRAF alterations (FIREFLY-1/NCT04775485) [130]. These studies are refining indications, dosing and combinations, and will help define future standard-of-care approaches.
4. Therapeutic Challenges of Targeting the MAPK Pathway in Brain Tumors
4.1. Blood–Brain Barrier (BBB) and Drug Delivery Limitations
The therapeutic management of intracranial tumors such as gliomas, meningiomas, pituitary adenomas and craniopharyngiomas is limited by the presence of both the BBB and the blood–tumor barrier (BTB). BBB’s fundamental role through its high selectivity is to maintain cerebral homeostasis, but at the same time, it restricts the entry of many pharmacological agents, especially large or hydrophilic molecules [131,132,133]. In contrast, the BTB, which arises from abnormal tumor-induced angiogenesis, displays heterogeneous permeability [134]. This results in uneven intratumoral drug distribution, particularly in aggressive tumors such as GBM and craniopharyngiomas. These anatomical and physiological characteristics affect the uniform delivery and eventually the efficacy of systemically administered drugs [28,135]. Thus, effective brain tumor treatment requires the development of compounds that both target oncogenic signaling pathways and, at the same time, achieve adequate penetration through the BBB. However, even small, lipophilic molecules can fail to accumulate sufficiently in the CNS due to active efflux mechanisms mediated by ATP-binding cassette (ABC) transporters, which include the P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) [136,137]. Such transporters are located at the BBB/BTB interface and within tumor cells and contribute to chemoresistance by actively extruding therapeutic agents from the brain parenchyma [138]. Importantly, ABC transporter expression is heterogeneous across tumor subtypes and can be upregulated in response to treatment. For instance, exposure to doxorubicin has been demonstrated to induce the expression of multiple transporters, such as P-gp, BCRP, MRP-1, -2, -3, and -6, in gliomas, further compounding resistance [139].
Many RTK inhibitors, including erlotinib, gefitinib, and afatinib, are known substrates of P-gp and BCRP, restricting their CNS bioavailability [140,141,142]. Certain compounds, such as sunitinib and sorafenib [143], and third-generation EGFR inhibitors, such as osimertinib, rociletinib, and HM61713, demonstrate improved BBB penetration and activity against resistance mutations like EGFR T790M challenges persist [142]. Similar pharmacokinetic barriers are encountered with RAF inhibitors, like vemurafenib and dabrafenib, and MEK inhibitors like trametinib, cobimetinib, binimetinib, selumetinib and pimasertib, many of which are subject to efflux via P-gp and BCRP. Other MEK inhibitors such as PD0325901 and E6201 have shown favorable BBB permeability in preclinical studies [144,145]. Furthermore, newer-generation RAF inhibitors, including dabrafenib, encorafenib, and the molecular glue, dual RAF-MEK inhibitor NST-628, have demonstrated enhanced CNS distribution, thus improving their therapeutic usage in intracranial malignancies [87,89,91]. Compounds that target KRAS G12C, like sotorasib and adagrasib, and ERK, like ulixertinib, are in active clinical evaluation. However, their pharmacokinetic properties and association with efflux transporters are not yet defined [108,146,147]. Several innovative drug delivery strategies are currently being explored, including nanoparticles, focused ultrasound-mediated BBB disruption and convection-enhanced delivery [28].
4.2. Tumor Heterogeneity and Resistance Mechanisms in MAPK Pathway-Targeted Therapies
Genetic alterations in the MAPK signaling pathway vary across different brain tumor types, affecting disease progression and therapeutic response. Hyperactivating mutations such as BRAF V600E and BRAF–KIAA1549 fusions are frequently observed in pediatric and adult low-grade gliomas, such as PAs, gangliogliomas, and PXAs [54,148]. Mutations in genes including ROS1, ALK, NF1, KRAS, MEK, and CRAF have been identified across glioma subtypes, highlighting the necessity for molecular classification beyond traditional histopathology [35,149,150]. Additionally, fusions involving FGFR and NTRK family genes and fusions and amplifications in ALK, ROS1, and MET, have been detected predominantly in pediatric brain tumors [51,151,152]. Mutations in PIK3CA and AKT1 are frequent in meningiomas, whereas activating mutations affecting the RAS/RAF/MEK/ERK, PI3K/AKT, and Wnt pathways have been described in pituitary adenomas [153,154]. Apart from primary CNS tumors, genetic alterations within the MAPK pathway, such as RTK and BRAF mutations, are often observed in brain metastases originating from lung, breast, and melanoma primary cancers [155]. Despite the considerable therapeutic advances of the MAPK pathway-directed targeted therapies, their clinical success is frequently limited by the development of drug resistance [22,156]. These resistance mechanisms often include compensatory activation of parallel signaling cascades like PI3K/AKT/mTOR pathway [72,157,158]. Furthermore, reactivation and/or hyperactivation of the MAPK pathway through the relief of negative feedback loops upon treatment with MAPK inhibitors, fosters epigenetic reprogramming by inducing expression of key transcription factors associated with cellular stemness and mesenchymal transition. This process involves chromatin remodeling, enhancer reconfiguration, and rewiring of the transcription factor network, including the transcription factors SOX2, OLIG2, STAT3, KLF4, and NOTCH, promoting therapeutic evasion and adaptive resistance [72,73,74,157,158,159,160].
4.3. Tumor Microenvironment (TME)
The TME functions as a dynamic ecological system that actively shapes tumor evolution, therapeutic response, and resistance through complex and reciprocal interactions between tumor cells and their surrounding stromal and immune compartments [161]. Genetic and epigenetic alterations influence the transcriptional and secretory programs of cancer cells, thereby reprogramming the TME, which in turn contributes to the emergence of resistance to MAPK pathway inhibitors. Certain oncogenic mutations in both the RAS/MAPK and PI3K/AKT signaling pathways can support an immunosuppressive TME [162,163]. Moreover, in GBM, elevated levels of phosphorylated ERK have been associated with an increased TME infiltration by M2-type tumor-associated macrophages (TAMs) [164]. This altered microenvironment corresponds to “cold tumors,” characterized by minimal infiltration of immune cells and poor response to immunotherapies [165]. Moreover, sustained MAPK pathway activation can induce a senescence-associated secretory phenotype (SASP) that further modifies the TME, promoting the secretion of cytokines, chemokines and growth factors that enhance cancer cell viability and foster therapeutic resistance [166]. In BRAF V600E-mutant HGG, dual BRAF and MEK inhibition affects glioma plasticity, promoting an immunomodulatory phenotype with elevated PD-L1 expression and improving the synergy with ICIs [167]. Furthermore, TME can drive resistance upon combinatorial BRAF inhibitor and ICI treatment in brain metastatic acral melanoma [168]. A comprehensive understanding of the effects of the MAPK pathway inhibition in TME, along with tumor cells, will allow the rational design of novel therapeutic strategies suitable for brain tumors [29].
4.4. Toxicities Associated with MAPK Pathway Inhibitors
Targeted inhibition of the MAPK signaling cascade has significantly improved clinical outcomes in subsets of brain tumor patients. However, it is often associated with adverse effects and toxicities that affect treatment tolerability and limit long-term utilization [169,170,171,172,173,174,175,176,177]. BRAF inhibitors such as dabrafenib and vemurafenib are most commonly associated with cutaneous toxicities, including follicular or acneiform eruptions, xerosis, fatigue, and photosensitivity. While most dermatologic side effects are mild and manageable, treatment discontinuation may be required in cases of severe toxicity [169,170,171,172]. MEK inhibitors, including trametinib and selumetinib, also frequently cause dermatologic adverse events (rash, xerosis, paronychia). In addition, systemic toxicities such as fatigue and cardiovascular complications, including hypertension and bradycardia, have been reported. Given the risk of cardiotoxicity, especially in pediatric patients with CNS tumors, routine cardiac monitoring is strongly recommended [173,174]. Trk inhibitors, such as larotrectinib and entrectinib, target aberrant activation of Trk receptors resulting from NTRK gene fusions. Although generally well tolerated, these compounds have been linked to a spectrum of adverse effects, including gastrointestinal symptoms such as nausea, vomiting, diarrhea, hepatotoxicity, peripheral edema, cutaneous rashes, cardiac dysfunction, and neurological effects including dizziness, headache, and peripheral neuropathy [152,175,176,177].
5. Conclusions
The therapeutic targeting of the MAPK signaling pathway has clinical benefits in various CNS tumors, particularly in pLGGs that express the BRAF V600E mutation [113]. Monotherapy with BRAF or MEK inhibitors is often associated with drug resistance and substantial toxicities, while combination strategies, especially dual inhibition of RAF and MEK, have demonstrated superior efficacy [97,170]. For instance, the dabrafenib–trametinib combination has received FDA approval for BRAF V600E-mutant low-grade gliomas [154,178]. Drug resistance is frequently caused by reactivation of the RAF/MEK/ERK axis and alternative escape mechanisms from MAPK inhibition such as enhanced PI3K/AKT/mTOR signaling [79]. As a result, current efforts in preclinical models and early-phase clinical trials (e.g., NCT02023905, NCT02133183) focus on multi-targeted strategies, combining MAPK pathway inhibitors with inhibitors targeting parallel signaling pathways such as PI3K/AKT/mTOR. Resistance to MAPK inhibition may also arise through mechanisms that bypass the main signaling pathway, including activating mutations in PI3KC, AKT-mediated feedback loops, PTEN loss, mTOR upregulation, and autophagy-associated survival responses [22]. Therapeutic approaches focus on modulating apoptosis, disrupting tumor-associated metabolic reprogramming, regulating autophagy, and inhibiting phenotypic plasticity to enhance treatment efficacy [22,179,180,181]. Novel strategies aiming to reverse the immunosuppressive TME [182]. In this context, current approaches include the assessment of CAR T cells targeting tumor-associated antigens such as IL-13Rα2 and EGFRvIII, as well as inhibitors of PD-1/PD-L1 axis [183,184]. Consistently, PD-L1 overexpression in GBM has been associated with poor clinical outcomes [184,185,186]. Blockade of PD-1/PD-L1 interaction aims to suppress the PD-1-mediated inhibitory signaling, restore cytotoxic T-cell function and enhance anti-tumor immunity [187]. Current clinical trials assessing combinatorial immunotherapies involving ICIs, CAR T cells, and anti-angiogenic drugs (e.g., bevacizumab), showing promising preliminary results [28,188,189]. Combination approaches involving immunotherapies with MAPK pathway inhibition are being evaluated mostly to overcome resistance mechanisms [189,190]. Cytokine therapies are being explored for their potential to reactivate immune function within the TME. Such therapies have shown promise in augmenting immune responses without significant toxicities [188,191,192]. Moreover, inhibitors focused on metabolic reprogramming at the IDH1/2-mutant gliomas are under investigation [193]. Possible synergies of all these strategies with MAPK pathway inhibition in certain contexts could be proven highly beneficial, providing both sustained tumor suppression and enhanced TME immunomodulation.
Despite meaningful progress, the clinical management of high-grade brain tumors, such as GBM, continues to be hindered by tumor heterogeneity, adaptive resistance mechanisms, and the restrictive nature of the BBB. Moving forward, multimodal therapeutic strategies that address both the tumor and its surrounding microenvironment, along with personalized molecular profiling, will be critical for improving survival outcomes. The future of MAPK-targeted therapy in CNS tumors lies in precision medicine, with treatment paradigms tailored to each patient’s unique molecular and immunological landscape.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Kim S. Son Y. Oh J. Kim S. Jang W. Lee S. Smith L. Pizzol D. Lee J. Lee H. Global burden of brain and central nervous system cancer in 185 countries, and projections up to 2050: A population-based systematic analysis of GLOBOCAN 2022 J. Neurooncol.202517567368510.1007/s 11060-025-05164-040720078 · doi ↗ · pubmed ↗
- 2Huang J. Chan S.C. Lok V. Zhang L. Lin X. Lucero-Prisno D.E. Xu W. Zheng Z.-J. Elcarte E. Withers M. Disease burden, risk factors, and trends of primary central nervous system (CNS) cancer: A global study of registries data Neuro Oncol.202325995100510.1093/neuonc/noac 21336048182 PMC 10158137 · doi ↗ · pubmed ↗
- 3Aldape K. Brindle K.M. Chesler L. Chopra R. Gajjar A. Gilbert M.R. Gottardo N. Gutmann D.H. Hargrave D. Holland E.C. Challenges to curing primary brain tumours Nat. Rev. Clin. Oncol.20191650952010.1038/s 41571-019-0177-530733593 PMC 6650350 · doi ↗ · pubmed ↗
- 4Zhao X. He M. Yang R. Geng N. Zhu X. Tang N. The global, regional, and national brain and central nervous system cancer burden and trends from 1990 to 2021: An analysis based on the Global Burden of Disease Study 2021 Front. Neurol.202516157461410.3389/fneur.2025.157461440606128 PMC 12213423 · doi ↗ · pubmed ↗
- 5Angom R.S. Nakka N.M.R. Bhattacharya S. Advances in glioblastoma therapy: An update on current approaches Brain Sci.202313153610.3390/brainsci 1311153638002496 PMC 10669378 · doi ↗ · pubmed ↗
- 6Wood M.D. Beadling C. Neff T. Moore S. Harrington C.A. Baird L. Corless C. Molecular profiling of pre- and post-treatment pediatric high-grade astrocytomas reveals acquired increased tumor mutation burden in a subset of recurrences Acta Neuropathol. Commun.20231114310.1186/s 40478-023-01644-437670377 PMC 10481558 · doi ↗ · pubmed ↗
- 7He C. Xu K. Zhu X. Dunphy P.S. Gudenas B. Lin W. Twarog N. Hover L.D. Kwon C.H. Kasper L.H. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma Nat. Commun.202112408910.1038/s 41467-021-24168-834215733 PMC 8253809 · doi ↗ · pubmed ↗
- 8Ullah R. Yin Q. Snell A.H. Wan L. 8 RAF-MEK-ERK pathway in cancer evolution and treatment Semin. Cancer Biol.20228512315410.1016/j.semcancer.2021.05.01033992782 · doi ↗ · pubmed ↗