Identification and Analysis of Key lncRNAs for Adipose Differentiation
Xiujie Xie, Tianyu Li, Bohang Zhang, Junxiong Liao, Xing Zhang, Jing Gao, Xiaofang Cheng, Tiantian Meng, Yongjie Xu, Pengpeng Zhang, Cencen Li

TL;DR
This study identifies new long non-coding RNAs that may regulate fat cell differentiation and could help treat obesity by promoting fat browning.
Contribution
The study identifies novel lncRNAs associated with adipocyte browning and differentiation using transcriptomic data from mouse adipose tissues.
Findings
794 novel lncRNAs and 1499 differentially expressed genes were identified across white, brown, and beige adipose tissues.
Two lncRNAs, MSTRG.12661 and MSTRG.17758, are linked to extracellular matrix organization and fatty acid oxidation.
The identified lncRNAs may serve as new therapeutic targets for obesity treatment through white adipose tissue browning.
Abstract
Long non-coding RNAs (lncRNAs) are well-recognized regulators of adipocyte differentiation and metabolic processes. However, their specific roles in adipocyte browning remain poorly characterized. To address this knowledge gap, we performed transcriptomic analyses on publicly available RNA-seq datasets of mouse white (WAT), brown (BAT), and beige (BeAT) adipose tissues; these datasets were retrieved from the EMBL-EBI database under the accession number E-MTAB-2624. Through a sophisticated bioinformatics analysis pipeline, we ultimately identified 794 novel lncRNAs and 1499 differentially expressed genes (DEGs), among which 95 were shared across all three adipocyte types. Among these novel lncRNAs, two specific lncRNAs, MSTRG.12661 and MSTRG.17758, were found to be tightly associated with key biological processes, including extracellular matrix organization and fatty acid oxidation;…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
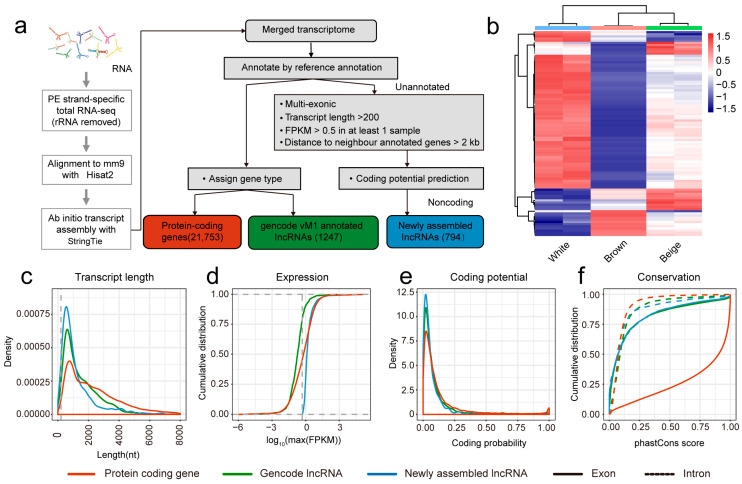 Figure 1
Figure 1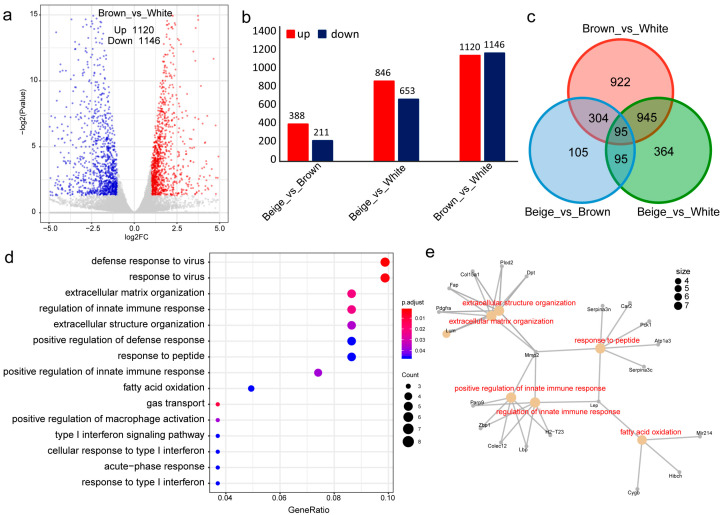 Figure 2
Figure 2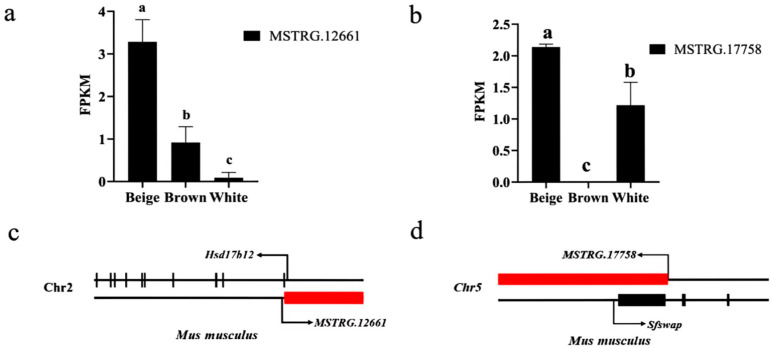 Figure 3
Figure 3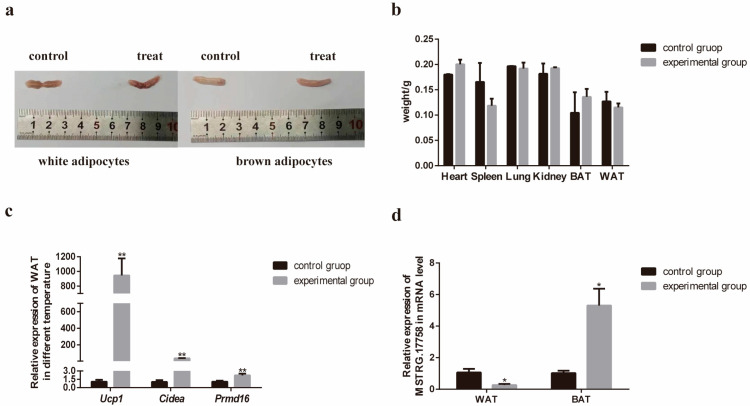 Figure 4
Figure 4- —National Natural Science Foundation of China
- —Department of Science and Technology in Henan Province
- —Natural Science Foundation of Henan Province
- —Postgraduate Education Reform and Quality Improvement Project of Henan Province
- —Key Research Projects of Higher Education Institutions in Henan Province
- —Nanhu Scholars Program of Xinyang Normal University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdipose Tissue and Metabolism · Adipokines, Inflammation, and Metabolic Diseases · Cardiovascular Disease and Adiposity
1. Introduction
When dietary energy intake chronically exceeds the energy expended for growth and metabolism, surplus lipids, carbohydrates, and other nutrients are converted into triglycerides [1], which are stored as fat and contribute to the development of obesity [2]. Obesity is a major risk factor for numerous metabolic disorders, including type 2 diabetes and cardiovascular diseases, and has also been linked to an increased risk of several cancers [3,4,5,6,7]. It is estimated that over 4 million deaths annually are attributable to excess body weight [8]. Adipose tissue, composed primarily of lipid-storing adipocytes, serves as the body’s main energy reservoir and is classified into three major types: white, brown, and beige adipose tissue [9].
White adipose tissue is the most abundant form and is widely distributed in mammals. White adipocytes are characterized by a single large lipid droplet that displaces the nucleus to the periphery [10]. Beyond energy storage, WAT contributes to thermal insulation and endocrine regulation of systemic energy homeostasis [11]. In contrast, brown adipose tissue is thermogenically active, especially in neonates and under cold conditions [12,13]. Its brown coloration stems from high vascularity and mitochondrial density [14]. Brown adipocytes contain multilocular lipid droplets and express high levels of uncoupling protein 1 (UCP1), which dissipates the mitochondrial proton gradient to produce heat instead of ATP [15].
Beige adipose tissue arises within WAT depots in response to stimuli such as cold exposure, exercise, or pharmacological agents [16]. This process, termed the “browning”, leads to a cell type with thermogenic capacity similar to BAT [17]. Promoting WAT browning has emerged as a promising strategy to counter obesity and metabolic disease by enhancing energy expenditure.
LncRNAs are transcripts longer than 200 nucleotides with limited or no protein-coding potential, often exhibiting cell- or tissue-specific expression [18]. They participate in diverse biological processes-including epigenetic regulation, cell differentiation, and metabolic signaling-through various mechanisms of gene expression control [19].
Growing evidence implicates lncRNAs in the regulation of adipocyte differentiation. For instance, Sun identified 175 lncRNAs dynamically regulated during adipogenesis [20], while Wang revealed the involvement of MEK6-AS1 in adipogenic processes [21]. Mostafa proposed LINC00312, LINC00607, and TYMSOS as key regulators in white adipogenesis [22]. Moreover, Tran reported that the primate-specific LINC00473 is highly expressed in human thermogenic adipocytes, correlates with UCP1, and is reduced in obesity and type-2 diabetes [23].
To further elucidate the role of lncRNAs in adipose biology, we analyzed RNA-seq data from mouse WAT, BAT, and BeAT. Our objectives were to identify novel adipose-associated lncRNAs and refine existing annotations. Through differential expression analysis, we aimed to pinpoint key lncRNAs involved in adipocyte differentiation and browning. This study enhances the understanding of lncRNA-mediated regulatory mechanisms in adipose tissue and may contribute to identifying novel therapeutic targets for obesity and related metabolic disorders.
2. Materials and Methods
2.1. Animals and Tissue Preparation
All animal experiments in this study were approved by the Institutional Review Board of Xinyang Normal University. Mice were housed under conditions of 40–70% humidity and an ambient temperature of 22 ± 2 °C, with a standard 12 h light/12 h dark cycle (8:00 a.m.–8:00 p.m.), and had ad libitum access to food and water. The basal diet used in this study was purchased from Xietong (Nanjing, China). In the study, 6 healthy 8-week-old male C57BL/6 mice (body weight: ~20.0 g) were randomly allocated into two experimental groups, namely the control group and the cold stimulation group, with 3 mice per group serving as biological replicates (n = 3). Specifically, mice in the control group were reared at room temperature, while those in the cold stimulation group were maintained in a constant-temperature environment of 4 °C. Except for the temperature condition, all other rearing conditions were kept consistent between the two groups. The experimental treatment lasted for 3 days. After the treatment, all mice were euthanized by cervical dislocation. The mice were rapidly dissected to collect major organs, including the heart, spleen, lung, and kidney, as well as BAT and WAT. All collected samples were immediately transferred to a −80 °C ultra-low temperature refrigerator for storage until subsequent experimental analysis.
2.2. Quality Control Procedures of RNA-Seq Data
RNA-seq datasets corresponding to mouse white, beige, and brown adipocytes were retrieved from the EMBL-EBI database under accession E-MTAB-2624 [24]. The dataset included two biological replicates for each adipocyte type (Table 1). Raw sequence files in SRA format were converted to FASTQ format using the fastq-dump utility with default parameters. Quality assessment of the raw reads was performed using FastQC (v0.11.5) [25,26] to verify that all samples met standard quality criteria prior to downstream analysis.
2.3. RNA-Seq Data Analysis
Raw sequencing reads were initially processed with Trimmomatic (v0.33) [27] to remove adapter sequences and low-quality bases. The trimming steps included: (1) adapter removal; (2) Sliding window trimming (4-base window, minimum Phred quality score of 15); and (3) exclusion of reads shorter than 63 nucleotides after trimming.
The high-quality reads were then aligned to the mouse mm9 reference genome (genome.NCBIM37.clean.fa) using HISAT2 (v2.1.0) [28]. The resulting SAM files were sorted and converted to BAM format using SAMtools (v1.9) [29]. Transcript assembly was performed with StringTie (v2.0) [30], and individual transcriptomes from all samples were merged into a unified annotation file using the StringTie --merge function.
2.4. Identification of Novel lncRNAs
Novel lncRNAs were identified from the merged transcriptome annotation through a multi-step filtering pipeline implemented with custom Python (V2.7) scripts (File S1). Transcripts were retained if they met the following criteria: (1) length > 200 nucleotides and an FPKM > 0.5 in at least one sample; (2) no overlap with known exons and located >2 kb away from any single-exon transcript; and (3) coding potential score < 0 as predicted by both CPAT (V1.2.4) [31] and COME (V1.4.0) [32] tools.
2.5. Characterization of Novel lncRNAs
A systematic comparison was conducted among newly identified lncRNAs, annotated lncRNAs, and protein-coding genes based on their biophysical and expression features, including transcript length, expression level (FPKM), coding potential, and conservation. The results of these comparative analyses are presented using the ggplot2 (V 3.4.4) package in R (V 4.3.3).
2.6. Methods for Differential Expression Analysis and Functional Annotation
Differential expression analysis of both novel and annotated lncRNAs across the three adipose tissue types was conducted using the DESeq2 (V1.40.2) package in R. Significantly differentially expressed lncRNAs were defined as those with an adjusted p-value (padj) < 0.05 and an absolute log2 fold change >1. Putative target genes of the differentially expressed lncRNAs were predicted, and their potential biological functions were investigated through GO and KEGG pathway enrichment analyses to identify key lncRNAs implicated in adipocyte differentiation.
2.7. Total RNA Extraction and RT-qPCR
Total RNA of tissues was extracted and purified using RNAiso Plus (TaKaRa, Dalian, China) according to manufacturer’s instructions. Total RNA was dissolved in RNase-free H_2_O, and the concentration and purity were measured using a NanoDrop 2000 spectrophotometer (Thermo Scientific, Waltham, MA, USA). All samples had absorbance ratios of 1.8–2.0 at 260 and 280 nm. RNA integrity and contamination were verified by 1% agarose gel electrophoresis. 1 μg of total RNA from different samples was prepared using a reverse transcription kit (TaKaRa, Dalian, China) to be reverse-transcribed into cDNA. RT-qPCR reactions were performed using TB Green^®^ Fast qPCR Mix (TaKaRa, Dalian, China) and the CFX-96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Relative expression levels were calculated using 2^−ΔΔt^ method, and 18S rRNA was used as the internal reference gene for normalization. All primer sequences (Table 2) were synthesized by General Biol (Anhui) Co., Ltd. (Chuzhou, China).
3. Results
3.1. Quality Control Results of RNA-Seq Data
Initial quality assessment of the raw RNA-seq data confirmed its high quality and suitability for downstream analysis. Key metrics included an N-content of 0 across all sequencing positions, an average base quality score peaking at 38, and per-base quality scores consistently above 28. The observed GC content distribution across sequences aligned well with the theoretical distribution, indicating no significant bias or contamination (Figure S1).
3.2. Identification and Characterization of lncRNAs
Transcriptomic alignment and assembly identified 21,753 annotated protein-coding genes and 1247 known lncRNAs. A stringent filtering pipeline was applied to the remaining unannotated transcripts to identify novel lncRNAs (Figure 1a). This multi-step process, based on exon count, transcript length (>200 bp), expression level (FPKM > 0.5 in at least one sample), and coding potential, yielded 794 novel lncRNAs (Files S2–S5). A global view of gene expression in WAT versus BAT showed that the majority of genes were expressed at higher levels in WAT (Figure 1b).
Comparative analysis revealed distinct features of the novel lncRNAs. Both novel and annotated lncRNAs were typically shorter than protein-coding genes, which showed a greater proportion of transcripts exceeding 2 kb (Figure 1c). As expected, lncRNAs exhibited lower expression levels than protein-coding genes. Notably, the novel lncRNAs were expressed at a higher level than annotated lncRNAs (Figure 1d). Coding potential analysis confirmed that the novel lncRNAs had minimal protein-coding capacity, with scores significantly lower than those of annotated lncRNAs and protein-coding genes (Figure 1e). Conservation analysis indicated that exon regions of both novel and annotated lncRNAs were less conserved than those of protein-coding genes, while all three categories showed low conservation in intronic regions (Figure 1f).
3.3. Differential Expression Analysis and Functional Annotation
Differential expression analysis between BAT and WAT identified 2266 differentially expressed genes, with 1120 upregulated and 1146 downregulated in WAT relative to BAT. Among these, 181 lncRNAs exhibited differential expression between BAT and WAT (Figure 2a,b). Comparison of BeAT and WAT revealed 1499 DEGs, with 846 upregulated and 653 downregulated in WAT. Specifically, 74 and 31 lncRNAs were upregulated and downregulated in WAT, respectively (Figure 2b). The contrast between BeAT and BAT yielded 599 DEGs, including 45 differentially expressed lncRNAs (Figure 2b). An intersection analysis identified 95 common DEGs consistently differentially expressed across all three adipose cell types (Figure 2c).
GO enrichment analysis of these 95 common DEGs highlighted significant involvement in biological processes critical to adipose biology, including extracellular matrix organization and fatty acid oxidation (Figure 2d). This suggested their potential regulatory roles in adipocyte differentiation. Notably, two lncRNAs, MSTRG.12661 and MSTRG.17758, were prioritized as key candidates. A gene network analysis further implicated two genes (Cygb and Hibch) and one microRNA (mir214) as potential players in fatty acid oxidation pathways (Figure 2e).
3.4. Functional Prediction of Candidate lncRNA
Expression profiling showed that MSTRG.12661 was highly expressed in beige adipocytes but expressed at low levels in white adipocytes (Figure 3a). Genomic localization analysis placed MSTRG.12661 on the antisense strand downstream of the Hsd17b12 gene (Figure 3c), a known regulator of adipocyte differentiation. This positional relationship suggests a potential role for MSTRG.12661 in modulating adipocyte differentiation.
The second candidate, MSTRG.17758, was also highly expressed in beige fat and absent in brown adipocytes (Figure 3b). This lncRNA was located upstream of Sfswap (Figure 3d), a protein-coding gene involved in mRNA splicing. We hypothesize that MSTRG.17758 may influence adipocyte differentiation by interacting with Sfswap to regulate the splicing of key transcripts.
3.5. Validation of Gene Expression in RNA-Seq
To investigate the regulatory effects of cold stimulation on mouse tissues, we compared tissue morphological differences between the cold-stimulated group and the room temperature-cultured group, with the cold stimulation treatment lasting for 3 consecutive days. There was no significant difference in the morphology of the adipose tissue between the two groups (Figure 4a). Subsequently, we collected the heart, spleen, lung, kidney, brown adipose tissue (BAT), and white adipose tissue (WAT) from both the control and experimental groups for weighing. The results indicated no significant differences in body weight between the two groups (Figure 4b). To further evaluate the reliability of the RNA sequencing data, we first examined the relative expression levels of three adipose tissue-specific marker genes (Ucp1, Cidea, and Prdm16) in WAT following cold stimulation via RT-qPCR. Our data revealed a statistically significant reduction in the expression of these genes in the experimental group versus the control group. (Figure 4c). Meanwhile, we also detected the expression levels of these three marker genes in BAT, and their relative expression levels were significantly increased compared with the control group; this result is presented in Figure S2. Additionally, a candidate gene (MSTRG.17758) was randomly selected for RT-qPCR validation. The results demonstrated that the expression of this gene was significantly decreased in white adipose tissue but significantly increased in brown adipose tissue, a trend fully consistent with the RNA-seq data (Figure 4d).
Collectively, these findings indicate that the RNA-seq data obtained in this study are highly reliable and accurate, providing a solid foundation for further investigation into the mechanisms by which cold stimulation regulates adipose metabolism in mice.
4. Discussion
In this study, we employed a comprehensive transcriptomic analysis of mouse white, beige, and brown adipose tissues to elucidate the landscape of long non-coding RNAs. Our investigation led to the identification of 794 novel lncRNAs, significantly expanding the catalog of non-coding transcripts associated with adipocyte biology. The molecular characterization of these transcripts reinforced the established paradigm that lncRNAs generally exhibit shorter lengths, lower expression, lower coding potential, and reduced evolutionary conservation compared to protein-coding genes [33]. This consistency with canonical lncRNA features underscores the robustness of our identification pipeline and provides a reliable foundation for further functional exploration.
Beyond cataloging, our comparative analysis across the three adipocyte types revealed distinct lncRNA expression signatures, suggesting their specific roles in defining adipose tissue identity and function. Notably, we pinpointed two lncRNAs, MSTRG.12661 and MSTRG.17758, as high-priority candidates involved in the browning of white adipose tissue. The genomic context of MSTRG.12661, located downstream of the Hsd17b12 gene, is particularly intriguing. Hsd17b12 encodes 17β-hydroxysteroid dehydrogenase type 12, a key enzyme in fatty acid elongation and lipid metabolism [34]. The antisense orientation and proximity of MSTRG.12661 to this metabolic regulator suggest a potential cis-regulatory mechanism, possibly influencing Hsd17b12 expression to modulate lipid utilization and energy dissipation, thereby promoting a beige adipocyte phenotype.
Similarly, MSTRG.17758, which is upregulated in beige fat and located upstream of the Sfswap gene, presents another compelling case. Sfswap encodes a splicing factor involved in alternative mRNA splicing, a critical layer of post-transcriptional regulation [35]. It is plausible that MSTRG.17758 interacts with Sfswap to influence the splicing patterns of a network of genes governing adipocyte differentiation and thermogenic programming. Such a mechanism would represent a novel pathway through which lncRNAs can orchestrate complex phenotypic changes, like the white-to-beige transition, by rewiring the transcriptome via splicing regulation.
While our bioinformatic predictions provide strong mechanistic hypotheses, they await experimental validation. A critical next step involves functional studies, such as gain-of-function and loss-of-function experiments in adipocyte cell models, to confirm the roles of MSTRG.12661 and MSTRG.17758 in browning. Furthermore, techniques like RNA antisense purification (RAP) or chromatin isolation by RNA purification (ChIRP) could be employed to verify the physical interactions between these lncRNAs and their putative genomic targets (Hsd17b12) or protein partners (Sfswap) [36,37]. Understanding the upstream regulators, such as specific transcription factors induced by cold exposure or β-adrenergic signaling [38], that control the expression of these lncRNAs would add another crucial layer to the regulatory network.
5. Conclusions
In conclusion, our study not only provides a valuable resource of novel adipocyte-associated lncRNAs but also delineates specific candidates with high potential for regulating adipose tissue browning. The proposed mechanisms—linking one lncRNA to lipid metabolism and another to mRNA splicing—highlight the diverse ways lncRNAs can fine-tune cellular identity. Future work focused on validating these pathways will not only advance our fundamental understanding of adipocyte plasticity but may also uncover novel, RNA-based therapeutic targets for combating obesity and its associated metabolic disorders.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Santos-Baez L.S. Ginsberg H.N. Hypertriglyceridemia-Causes, Significance, and Approaches to Therapy Front. Endocrinol.20201161610.3389/fendo.2020.0061632982991 PMC 7492386 · doi ↗ · pubmed ↗
- 2González-Muniesa P. Mártinez-González M.A. Hu F.B. Després J.P. Matsuzawa Y. Loos R.J.F. Moreno L.A. Bray G.A. Martinez J.A. Obesity Nat. Rev. Dis. Primers 201731703410.1038/nrdp.2017.3428617414 · doi ↗ · pubmed ↗
- 3Lu D. He A. Tan M. Mrad M. El Daibani A. Hu D. Liu X. Kleiboeker B. Che T. Hsu F.F. Liver ACOX 1 regulates levels of circulating lipids that promote metabolic health through adipose remodeling Nat. Commun.202415421410.1038/s 41467-024-48471-238760332 PMC 11101658 · doi ↗ · pubmed ↗
- 4Sakers A. De Siqueira M.K. Seale P. Villanueva C.J. Adipose-tissue plasticity in health and disease Cell 202218541944610.1016/j.cell.2021.12.01635120662 PMC 11152570 · doi ↗ · pubmed ↗
- 5Donohoe C.L. Lysaght J. O’Sullivan J. Reynolds J.V. Emerging Concepts Linking Obesity with the Hallmarks of Cancer Trends Endocrinol. Metab.201728466210.1016/j.tem.2016.08.00427633129 · doi ↗ · pubmed ↗
- 6Sarma S. Sockalingam S. Dash S. Obesity as a multisystem disease: Trends in obesity rates and obesity-related complications Diabetes Obes. Metab.20212331610.1111/dom.1429033621415 · doi ↗ · pubmed ↗
- 7Chew N.W.S. Ng C.H. Tan D.J.H. Kong G. Lin C. Chin Y.H. Lim W.H. Huang D.Q. Quek J. Fu C.E. The global burden of metabolic disease: Data from 2000 to 2019 Cell Metab.202335414428.e 310.1016/j.cmet.2023.02.00336889281 · doi ↗ · pubmed ↗
- 8Alsaqaaby M.S. Cooney S. le Roux C.W. Pournaras D.J. Sex, race, and BMI in clinical trials of medications for obesity over the past three decades: A systematic review Lancet Diabetes Endocrinol.20241241442110.1016/S 2213-8587(24)00098-638723646 · doi ↗ · pubmed ↗