DNA Damage Sensing and TP53 Function as Modulators of Sensitivity to Calicheamicin-Based Antibody–Drug Conjugates for Acute Leukemia
Camryn M. Pettenger-Willey, George S. Laszlo, Margery Gang, Frances M. Cole, Colin D. Godwin, Sarah Erraiss, Pritha Chanana, Allie R. Kehret, Junyang Li, Jacob W. Barton, Meghann M. Yochim, Eduardo Rodríguez-Arbolí, Roland B. Walter

TL;DR
This study explores how DNA damage pathways, especially TP53, influence the effectiveness of calicheamicin-based drugs in treating acute leukemia and identifies potential drug combinations to improve treatment outcomes.
Contribution
The study identifies TP53, ATM, and MDM2 as key modulators of calicheamicin sensitivity and proposes combination therapies with small-molecule inhibitors to enhance treatment efficacy.
Findings
TP53-mutant leukemia cells are significantly less sensitive to calicheamicin than TP53-wildtype cells.
MDM2 and ATM inhibitors enhance calicheamicin-induced cytotoxicity in acute leukemia cells.
Combination therapies with these inhibitors could improve the efficacy of calicheamicin-based ADCs.
Abstract
Acute leukemias are difficult-to-treat blood cancers which many patients will eventually die from despite intensive chemotherapies and bone marrow transplantation. Antibodies that recognize proteins on leukemia cells and deliver a cell toxin (so-called antibody–drug conjugates) have been developed to improve these outcomes. Two such drugs, gemtuzumab ozogamicin (GO, for acute myeloid leukemia) and inotuzumab ozogamicin (IO, for acute lymphoblastic leukemia), have been approved for use in patients but are not always effective. In our laboratory research, we undertook genome-wide screening to discover genes that are associated with response or resistance to the toxin (calicheamicin) that both GO and IO contain. Our studies revealed the importance of several DNA damage pathway regulation genes for the anti-leukemia activity of calicheamicin, including TP53, ATM, and MDM2. Building on these…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
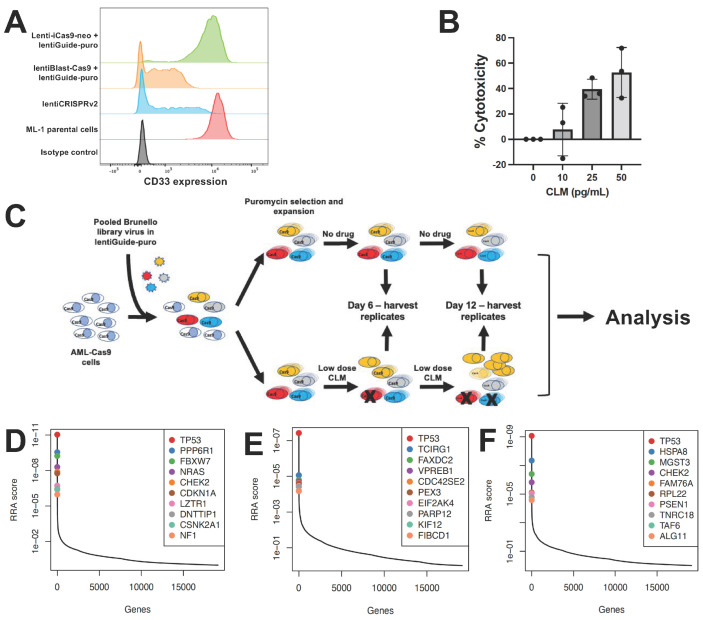 Figure 1
Figure 1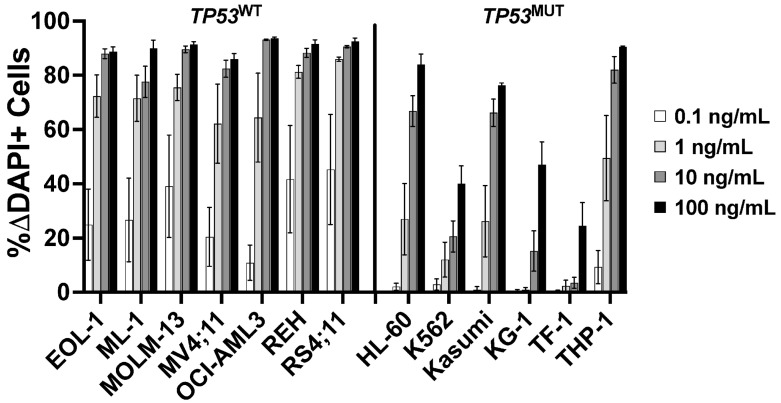 Figure 2
Figure 2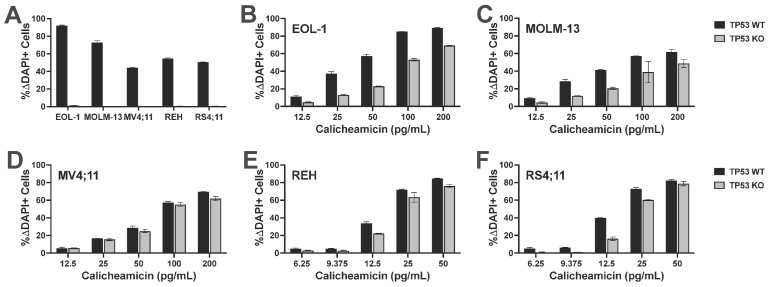 Figure 3
Figure 3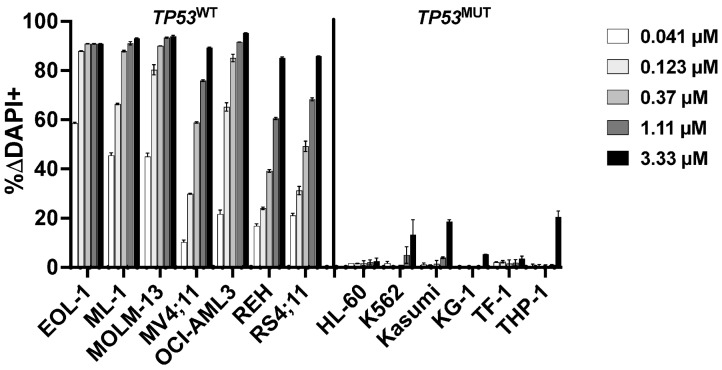 Figure 4
Figure 4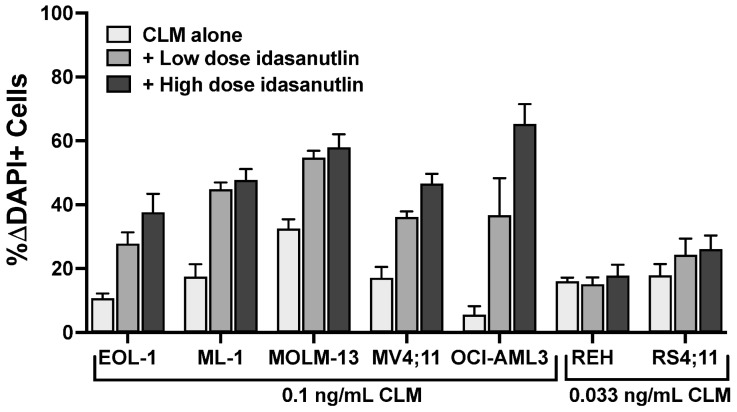 Figure 5
Figure 5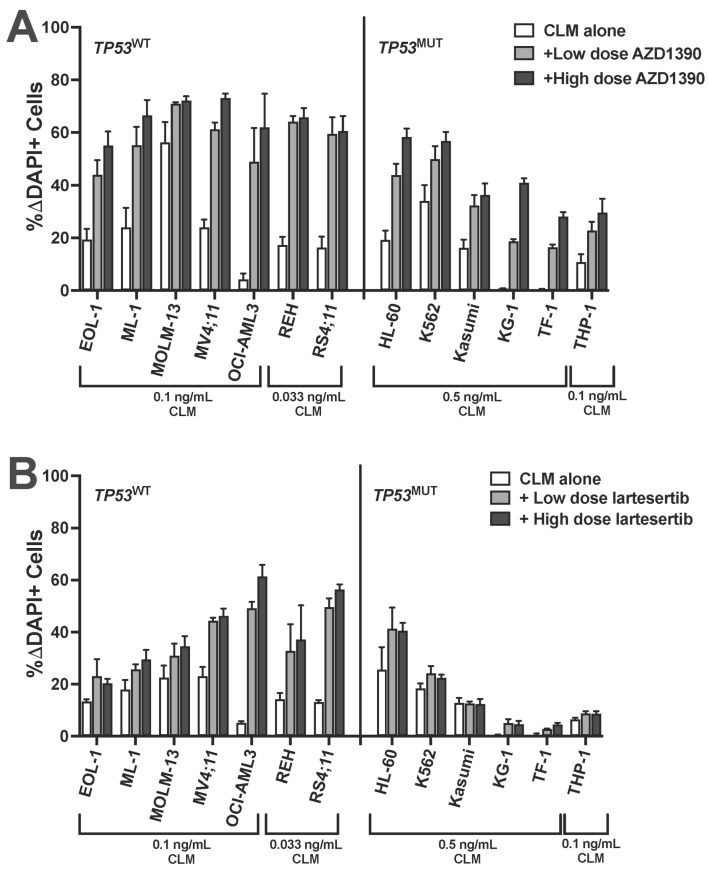 Figure 6
Figure 6- —Pfizer
- —José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research
- —NIH/NIDDK Cooperative Center of Excellence in Hematology
- —NIH/National Heart, Lung, and Blood Institute
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer therapeutics and mechanisms · HER2/EGFR in Cancer Research · Synthesis and pharmacology of benzodiazepine derivatives
1. Introduction
Acute leukemias remain difficult to treat despite multi-agent chemotherapy, allogeneic hematopoietic cell transplantation, and the increasing availability of immunotherapeutics and small-molecule inhibitors [1,2,3,4,5,6]. Antibody–drug conjugates (ADCs) have long been pursued to improve cure rates in these malignancies, with a major focus on CD33 (for acute myeloid leukemia [AML]) and CD22 (for B-acute lymphoblastic leukemia [B-ALL]). Improved outcomes with the CD33 ADC, gemtuzumab ozogamicin (GO) [7,8,9], and the CD22 ADC, inotuzumab ozogamicin (InO) [10,11,12], validate these efforts. Still, in many patients, GO and InO are insufficiently effective. Rational use of combinatorial therapies may be one strategy to widen the therapeutic reach of these ADCs in acute leukemia.
Both GO and InO deliver a derivative of calicheamicin-γ_1_^I^ (N-acetyl gamma calicheamicin-γ_1_^I^ dimethyl hydrazide [CLM]) as a toxic payload. Once internalized and released from the antibody, CLM binds DNA in the minor groove and undergoes structural changes, leading to a diradical that abstracts hydrogens from the phosphodiester backbone of DNA, resulting in single- and double-strand breaks [8,13]. G_2_/M cell cycle arrest follows DNA damage sensing, and cell death—primarily via mitochondrial apoptosis pathways—ensues if damage is overwhelming. Modulation or loss of target antigen (for CD22), DNA damage response dysregulation, drug efflux pumps, and apoptotic dysregulation have been implicated in resistance to CLM-based ADCs [8,13,14,15]. However, the cellular mechanisms allowing acute leukemia cells to resist GO and InO remain incompletely understood. Consequently, to identify genetic vulnerabilities and new biomarkers that could inform selection of patients most likely to benefit from CLM-based ADCs and to develop combination therapies to augment the efficacy of these ADCs, we performed a genome-wide clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 screen to discover genes associated with CLM sensitivity.
2. Materials and Methods
2.1. Therapeutics and Chemicals
CLM was provided by Pfizer (New York, NY, USA). AZD1390, BML-277, ceralasertib, idasanutlin, lartesertib, and prexasertib were obtained from Selleck Chemicals (Houston, TX, USA). Veliparib was obtained from Apexbio Technology (Houston, TX, USA). For drug selection, blasticidin was purchased from Research Projects International (Mount Prospect, IL, USA), puromycin was obtained from Gold Biotechnology (Saint Louis, MO, USA), and G418 was obtained from ThermoFisher Scientific (Waltham, MA, USA).
2.2. Parental Human Acute Leukemia Cell Lines
Human myeloid and lymphoid EOL-1, HL-60, K562, Kasumi, KG-1, MOLM-13, REH, TF-1, and THP-1 cells were grown in RPMI-1640 medium with 10% fetal bovine serum (FBS; for EOL-1, K562, REH, and THP-1 cells), 20% FBS (for Kasumi, KG-1, and MOLM-13 cells), or 10% BCS (for HL-60 cells). TF-1 cells were supplemented with 4 ng/mL GM-CSF (Peprotech; Cranbury, NJ, USA). Human myeloid and lymphoid OCI-AML3 and RS4;11 cells were grown in alpha-MEM with 10% FBS. ML-1 and MV4;11 cells were grown in IMDM and 10% FBS, with MV4;11 cells supplemented with 5 ng/mL GM-CSF with 1x Insulin–Transferrin–Selenium supplement (ThermoFisher Scientific). All cell lines were grown with penicillin/streptomycin, tested for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit; Lonza, Basel, Switzerland), and authenticated using standard STR CODIS typing.
2.3. Lentiviral Expression Vectors
Lentiviral vectors containing Cas9 expression cassettes, LentiCas9-Blast (plasmid ID # 52962), Lenti-iCas9-neo (plasmid ID # 85400), lentiCRISPR v2 (plasmid ID # 52961), the lentiGuide-puro (plasmid ID # 52963) for expression of individual gRNAs, and the Brunello CRISPR knockout pooled library lentiviral particle prep with puromycin selection cassette, including 76,441 unique gRNAs covering over 19,000 genes, each gene targeted with 4 unique sgRNAs (ID # 73178-LV), were all purchased from Addgene (Watertown, MA, USA). The CD33 knockout (KO) sgRNA targeting exon 1 of CD33 (sequence 5′-CTGCTGCCCCTGCTGTGGGC-3′) was cloned into lentiGuide-puro with standard cloning techniques and confirmed by Sanger sequencing. Cas9 expression lentiviral vector particles and CD33 sgRNA lentiviral particles were prepared as described [16]. Lentivirally transduced sublines, when viral titering was possible, were generated at multiplicities of infection (MOI) of 0.25–25. EGFP-positive cells were isolated by fluorescence-activated cell sorting (FACS) and re-cultured for further analysis/use.
2.4. CRISPR/Cas9 Whole-Genome-Wide Screening
ML-1, K562 or OCI-AML3 cell lines were transduced with viral particles encoding LentiCas9-Blast [17] and selected with blasticidin to generate cells with constitutive Cas9 expression. To reduce the frequency of cells harboring > 1 sgRNA, cells were transduced with MOI of 0.3. Viral transduction of Brunello whole-genome targeting CRISPR/Cas9 library was performed at ~150–300x coverage. Two days later, transduced cells were selected with puromycin. Subsequently, cells were expanded to allow for treatment with either CLM or vehicle. CLM was administered every 3 days at a concentration aimed at inducing 15–30% cell death. Samples were collected on days 0, 6, and 12. Genomic DNA was purified and embedded sgRNA was amplified and barcoded by PCR, sequenced, and analyzed to determine sgRNAs under- or over-represented in CLM-treated populations compared to controls. Bowtie was used for alignment and guide quantification. Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) was used in conjunction with a modified robust ranking aggregation (RRA) algorithm to identify positive/negative selected genes across the two conditions, and hits with a false discovery rate (FDR) < 5% were considered for further study.
2.5. Quantification of CD33 Expression
CD33 cell surface expression was quantified with a PE-conjugated CD33 antibody (clone p67.6; BD Biosciences, Franklin Lakes, NJ, USA) and reported as median fluorescence intensity (MFI).
2.6. TP53 Genomic Analysis
We used 500 ng of purified genomic DNA from human acute leukemia cells to assess the TP53 allelic status as described [18].
2.7. Quantification of Drug-Induced Cytotoxicity
Human acute leukemia cells were incubated in 96-well round-bottom plates at 5–10 × 10^3^ cells/well with various concentrations of CLM and/or AZD1390, BML-277, ceralasertib, idasanutlin, lartesertib, prexasertib, or veliparib. After 3 days, cell numbers and drug-induced cytotoxicity, using 4′,6-diamidino-2-phenylindole (DAPI) to detect non-viable cells, were determined flow cytometrically.
2.8. Generation of Human Leukemia Cell Lines with Deletion of TP53
Leukemia cell sublines with deletion of TP53 were generated as described [18]. Briefly, CRISPR/Cas9 editing was carried out by electroporating purified Cas9 protein (TrueCut Cas9 V2; ThermoFisher Scientific) complexed with a pool of synthetic guide RNAs (sgRNAs) targeting TP53 (sequences: 5′-CGCUAUCUGAGCAGCGCUCA-3′, 5′-GUGCUGUGACUGCUUGUAGA-3′, 5′-CAACAAGAUGUUUUGCCACC-3′; Synthego, Redwood City, CA, USA), using the ECM 380 Square Wave Electroporation system (Harvard Apparatus; Cambridge, MA, USA). TP53 knockout (TP53^KO^) cells were selected in culture with either 1 µM (myeloid cell lines) or 2.5 µM (lymphoid cell lines) idasanutlin, which was freshly added every 2–3 days. Cytotoxicity assays were performed 14–21 days after electroporation.
2.9. Statistical Considerations
Statistical analyses were performed with Prism (Graphpad; La Jolla, CA, USA).
3. Results
3.1. CRISPR/Cas9 Whole-Genome Screening Identifies TP53 as a CLM Sensitivity Gene
CRISPR whole-genome screening requires library recipient cells expressing Cas9. To identify an optimal screening platform, we compared conditional with constitutive Cas9 expression using a single guide RNA (sgRNA) to disrupt CD33 in ML-1 cells. In our studies, we contrasted an inducible Cas9 expression construct, zeocin-inducible Lenti-iCas9-neo, with constitutive Cas9 expression from the EF1a promoter-driven lentiBlast-Cas9 construct and an all-in-one single expression plasmid construct as control, lentiCRISPRv2, which combines constitutive expression of both Cas9 and sgRNA from one plasmid. While the inducible Cas9 system generated only a small subpopulation of CD33-negative cells, constitutive Cas9 expression yielded a robust CD33^KO^ phenotype (Figure 1A). Based on these results, ML-1 cells with lentiBlast-Cas9 were selected for transduction of the Brunello sgRNA library. Transduced cells were expanded under puromycin selection, and aliquots were simultaneously subjected to CLM titration to identify 25 ng/mL as the CLM dose inducing 15–30% cell death, the desired target cytotoxicity for the drug screen (Figure 1B). The screen was conducted by incubating human acute leukemia cells (ML-1, K562, OCI-AML3) in the presence or absence of CLM. Samples were collected at time 0 as well as after 6 and 12 days of culture. At these time points, cells aliquots were collected, sgRNAs sequenced, and bioinformatically analyzed (Figure 1C). As depicted in Figure 1D, this screen identified several DNA damage pathway regulation genes in ML-1 cells, including TP53 (as the highest ranked hit overall), PPP6R1, FBXW7, CHEK2, CDKN1A, DNTTIP1, and CSNK2A1 as conferring sensitivity to CLM. These findings were confirmed in K562 cells (for TP53; Figure 1E) and OCI-AML3 cells (for TP53 and CHEK2; Figure 1F).
3.2. Validation of TP53 as a CLM Sensitivity Gene
Having identified TP53 as a possible CLM sensitivity gene in our screening, we evaluated the association between TP53 mutations and CLM-induced cytotoxicity in a set of 13 human leukemic cell lines. This set included seven TP53 wild-type (TP53^WT^: EOL-1, ML-1, MOLM-13, MV4;11, OCI-AML3, REH, and RS4;11) and six TP53 mutant (TP53^MUT^; HL-60, K562, Kasumi, KG-1, TF-1, and THP-1) cell lines we identified via DNA sequencing for all coding exons of TP53 [18]. Across these 13 acute leukemia cell lines, the 6 TP53^MUT^ cell lines were approximately 10- to 1000-fold less sensitive to CLM than the 7 TP53^WT^ cell lines. For example, at 1 ng/mL, the median (±SEM) CLM-induced cell death was 19 ± 8% in TP53^MUT^ cells and 72 ± 3% in TP53^WT^ cells (Figure 2). To test the relationship between TP53 and CLM sensitivity further, we then derived TP53^WT/KO^ syngeneic cell line pairs via deletion of TP53 from bulk cells via CRISPR/Cas9 and exposure to 1–2.5 µM of the mouse double minute 2 (MDM2) inhibitor, idasanutlin, to enrich for the population of TP53^KO^ cells [18]. As shown in Figure 3, TP53^KO^ sublines indeed showed reduced sensitivity to their TP53^WT^ counterparts in all five cell line models, although the degree of relative resistance varied significantly across cell lines.
3.3. Validation of Role of DNA Damage Pathways in CLM Sensitivity
Because TP53 and CHEK2 are in the same cellular DNA damage response pathway as MDM2 and ataxia–telangiectasia mutated (ATM) [19,20,21], we next tested which inhibitors of this pathway regulated response to CLM. We first tested the mouse double minute 2 (MDM2) inhibitor idasanutlin, a strong activator of p53. Nanomolar concentrations of idasanutlin effectively killed parental TP53^WT^ leukemia cell lines. By comparison, much higher doses (3–10 µM) were required to kill parental leukemia cell lines harboring TP53 alterations, in line with the notion that TP53 inactivation render cells insensitive to MDM2 inhibition [22] (Figure 4). We therefore assessed whether idasanutlin enhanced CLM-induced cytotoxicity in TP53^WT^ cells. Indeed, across the seven TP53^WT^ cell lines, drug-induced toxicity increased from a median of 7 ± 5% ΔDAPI+ cells (CLM alone at 0.05 ng/mL) to 31 ± 3% ΔDAPI+ cells when idasanutlin (10–20 nM) was added (Figure 5). In contrast, TP53^MUT^ cells were resistant (<5% ΔDAPI+ cells) to idasanutlin even at 1 µM, and no combinatory effect was seen with CLM (Supplementary Figure S1).
We then tested small-molecule inhibitors that could activate DNA damage pathways upstream of TP53. One such pharmacological target is ATM. Treatment of cells with the ATM inhibitor AZD1390 has been shown to cause cell death in TP53^MUT^ cells [23]. At relatively non-toxic doses (Supplementary Figure S2), AZD1390 enhanced CLM-induced cytotoxicity in all 13 tested human leukemia cell lines, including all 6 TP53^MUT^ cell lines (Figure 6A). Specifically, in TP53^WT^ cell lines, median drug-induced cell death increased from 7 ± 6% with 0.05 ng/mL CLM alone to 41 ± 9% with the addition of 0.25–2.5 µM of AZD1390; across TP53^MUT^ cell lines, the median drug-induced cell death increased from 19 ± 13% with 0.5 ng/mL of CLM to 50 ± 10% with the addition of 1–2.5 µM of AZD1390. Testing of a second ATM inhibitor, lartesertib, yielded qualitatively similar results, except for one cell line (Kasumi) which failed to show any evidence of sensitivity to lartesertib (Figure 6B and Supplementary Figure S3). In contrast, no significant or consistent effects on CLM-induced cytotoxicity were found in combination treatments with an ATR inhibitor (ceralasertib; Supplementary Figure S4), a Chk1/Chk2 inhibitor (prexasertib; Supplementary Figure S5), a Chk2 inhibitor (BML-277; Supplementary Figure S6), or a PARP inhibitor, veliparib. With the latter, for example, in TP53^WT^ cell lines, median drug-induced cell death changed from 3 ± 3% with 0.05 ng/mL CLM alone to 7 ± 6% with the addition of 1–10 µM of veliparib across TP53^WT^ cell lines. Across the leukemia cell lines with TP53 alteration, the median drug-induced cell death changed from 5 ± 5% with 0.5 ng/mL of CLM to 7 ± 7% with the addition of 2.5–10 µM of veliparib (Supplementary Figure S7).
4. Discussion
With GO and InO, CLM-based ADCs are routinely employed in patients with acute leukemia nowadays. However, while they are effective in subsets of patients with CD33+ AML or CD22+ B-ALL, respectively [7,8,9,10,11,12], others do not meaningfully benefit from these drugs as they are currently used. To identify genetic vulnerabilities and new biomarkers that could inform the selection of patients most likely to benefit from CLM-based ADCs and to develop rational combination therapies that augment the efficacy of these ADCs, we performed genome-wide CRISPR/Cas9 screening to discover genes associated with CLM sensitivity. In this screen, human acute leukemia cell lines with genetic lesions introduced by CRISPR/Cas9 were exposed to CLM and surviving cells were analyzed for over- or under-representation of genetic lesions relative to the starting cell population as a read-out for resistance or sensitivity to CLM, respectively.
As a main finding, our screen identified TP53 as a key sensitivity gene for CLM, with genetic TP53 deletion being the top hit and leading to relative CLM resistance in all three acute leukemia cell backgrounds tested, including K562 cells which harbor a gain-of-function TP53 point mutation (Q136P) [24] that was possibly selected against in our screening. Consistent with these results, we found human leukemia cell lines with TP53 alterations to be 10- to 1000-fold less sensitive to CLM than TP53^WT^ cell lines across the panel of 13 cell lines we studied. To study the impact of TP53 on CLM sensitivity more directly, we also derived TP53^KO^ sublines from five TP53^WT^ cell lines. In these studies, TP53^KO^ cells were not completely resistant to CLM but were significantly less sensitive to CLM than their TP53^WT^ counterparts, further demonstrating the impact TP53 has on CLM-induced cytotoxicity. Together, these data point to the acute leukemia cells’ TP53 status—a key marker for the sensitivity of both AML and B-ALL to conventional chemotherapy [25,26,27,28]—as a biomarker for patient selection or, in clinical drug testing, patient stratification when CLM-based ADCs are used. Whether TP53-mutated leukemia cells’ sensitivity to CLM-based ADCs is affected by co-mutations that may be present is an open question in need of further study.
Having identified TP53 as a key sensitivity gene for CLM, we then focused our efforts on genes in DNA damage pathways that are affected by TP53, in particular MDM2 and ATM [19,20,21]. In TP53^WT^ but not TP53^MUT^ cells, the MDM2 inhibitor and p53 activator, idasanutlin, enhanced CLM cytotoxicity, demonstrating that decoupling of cells from MDM2-p53 regulation sensitizes leukemia cells to CLM. Moreover, both ATM inhibitors we tested, AZD1390 and lartesertib, significantly enhanced CLM efficacy. Consistent with our data, previous investigations with SV40-transformed fibroblasts derived from ataxia telangiectasia (AT) patients showed hypersensitivity to a calicheamicin derivative [29]. Of potential clinical interest, the effect of ATM inhibitors was independent of the TP53 status, unlike the effect of the MDM2 inhibitor. In contrast to reports by others [30,31], a PARP inhibitor (veliparib) only minimally affected CLM-induced cytotoxicity in our panel of human acute leukemia cell lines. Likewise, CLM-induced cytotoxicity was not significantly modulated by an ATR inhibitor, a Chk1/Chk2 inhibitor, or a Chk2 inhibitor. The latter was unexpected considering that the Chk2-p53 pathway is well established and both TP53 and CHEK2 were identified in our screen and may suggest a role of residual non-catalytic functions of Chk2 in the leukemia cells’ response to CLM-induced cytotoxicity.
Our observation that inhibition of ATM or MDM2 sensitizes acute leukemia cells to CLM may provide the rationale for the clinical exploration of a combination therapy with either GO or InO. While efficacy has been limited thus far, several MDM2 inhibitors have been tested in patients with acute leukemia [32,33,34,35]. Testing of ATM inhibitors in acute leukemia is so far largely confined to preclinical investigations but early-phase clinical trials in other cancers have been completed [36] or initiated (e.g., NCT03423628, NCT05116254, NCT05182905, NCT05396833, NCT06433219, NCT06894979). Our data would support the testing of an MDM2 inhibitor with either GO or InO in patients with TP53^WT^ acute leukemia, whereas the combination of GO or InO with an ATM inhibitor could be explored in patients independent of the TP53 mutational status. It is interesting to speculate—and a goal of future studies—whether p53 reactivators like eprenetapopt (APR-246) [37] might be of value to sensitize acute leukemias with TP53 mutations to CLM-based ADCs.
In our studies, we exclusively focused on CLM-containing ADCs. While our findings do not immediately apply to ADCs delivering other small-molecule toxins, as resistance mechanisms may differ, our experimental approach may offer a blueprint for the identification of genetic vulnerabilities that are critical for the anti-tumor activity of other ADCs.
5. Conclusions
Our study identified ATM, MDM2, and TP53—which exhibit the same cellular response to DNA damage pathways—as key modulators of CLM-induced cytotoxicity in acute leukemia cells. While these findings may not be surprising per se, considering the mechanism of action of CLM [8,13], the observation that our genomic screening identified these modulators as key hits may suggest their potential value as targets for combination therapies. Thus, our results support further evaluation of combination therapies with corresponding small-molecule inhibitors targeting MDM2 or ATM toward clinical testing as novel strategies to increase the efficacy of CLM-based ADCs such as GO and InO.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Döhner H. Wei A.H. Löwenberg B. Towards precision medicine for AML Nat. Rev. Clin. Oncol.20211857759010.1038/s 41571-021-00509-w 34006997 · doi ↗ · pubmed ↗
- 2Döhner H. Wei A.H. Appelbaum F.R. Craddock C. Di Nardo C.D. Dombret H. Ebert B.L. Fenaux P. Godley L.A. Hasserjian R.P. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN Blood 20221401345137710.1182/blood.202201686735797463 · doi ↗ · pubmed ↗
- 3Jabbour E. Short N.J. Jain N. Haddad F.G. Welch M.A. Ravandi F. Kantarjian H. The evolution of acute lymphoblastic leukemia research and therapy at MD Anderson over four decades J. Hematol. Oncol.2023162210.1186/s 13045-023-01409-536927623 PMC 10018889 · doi ↗ · pubmed ↗
- 4Aldoss I. Roboz G.J. Bassan R. Boissel N. De Angelo D.J. Fleming S. Gökbuget N. Logan A.C. Luger S.M. Menne T. Frontline treatment of adults with newly diagnosed B-cell acute lymphoblastic leukaemia Lancet Haematol.202411 e 959e 97010.1016/S 2352-3026(24)00285-039638543 · doi ↗ · pubmed ↗
- 5Kantarjian H.M. Di Nardo C.D. Kadia T.M. Daver N.G. Altman J.K. Stein E.M. Jabbour E. Schiffer C.A. Lang A. Ravandi F. Acute myeloid leukemia management and research in 2025 CA Cancer J. Clin.202575466710.3322/caac.2187339656142 PMC 11745214 · doi ↗ · pubmed ↗
- 6Kantarjian H. Aldoss I. Jabbour E. Management of adult acute lymphoblastic leukemia: A review JAMA Oncol.20251177177810.1001/jamaoncol.2025.061340310617 PMC 12529734 · doi ↗ · pubmed ↗
- 7Hills R.K. Castaigne S. Appelbaum F.R. Delaunay J. Petersdorf S. Othus M. Estey E.H. Dombret H. Chevret S. Ifrah N. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials Lancet Oncol.20141598699610.1016/S 1470-2045(14)70281-525008258 PMC 4137593 · doi ↗ · pubmed ↗
- 8Godwin C.D. Gale R.P. Walter R.B. Gemtuzumab ozogamicin in acute myeloid leukemia Leukemia 2017311855186810.1038/leu.2017.18728607471 · doi ↗ · pubmed ↗