Vaccine therapy for pediatric high-grade glioma: current landscape, challenges, and future directions
Stuart D. Harper, Jacob A. Alderete, Shivani Baisiwala, Bianca H. Bergsneider, Linda M. Liau, Anthony C. Wang

TL;DR
This review explores vaccine-based immunotherapy as a potential treatment for aggressive childhood brain tumors called high-grade gliomas, highlighting current strategies, challenges, and future directions.
Contribution
The paper provides a comprehensive overview of vaccine therapy for pediatric high-grade gliomas, emphasizing novel antigen targets and clinical platforms.
Findings
Vaccine therapies targeting glioma-associated antigens and neoantigens show safety and immunogenicity in early-phase trials.
Challenges include the immunosuppressive tumor environment, low mutational burden, and logistical issues in vaccine production.
Future directions involve optimizing antigen selection, combining therapies, and improving delivery systems for pediatric use.
Abstract
Pediatric high-grade gliomas (pHGG) are among the most aggressive childhood brain tumors, with limited treatment options and poor prognosis. Vaccine-based immunotherapy offers a promising strategy by leveraging tumor-specific or associated antigens to stimulate durable anti-tumor immune responses with minimal toxicity. This review outlines the scientific rationale for vaccine therapies in pHGG, detailing key targets such as glioma-associated antigens (EphA2, IL-13Rα2, survivin), driver mutation–derived neoantigens (H3.3K27M, TP53, IDH1), and viral antigens (CMV pp65). We evaluate current vaccine platforms, including peptide vaccines, dendritic cell vaccines, mRNA-based vaccines, and neoantigen-personalized approaches, highlighting early-phase clinical trial results that demonstrate safety and immunogenicity. Despite encouraging preliminary data, several challenges hinder clinical…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
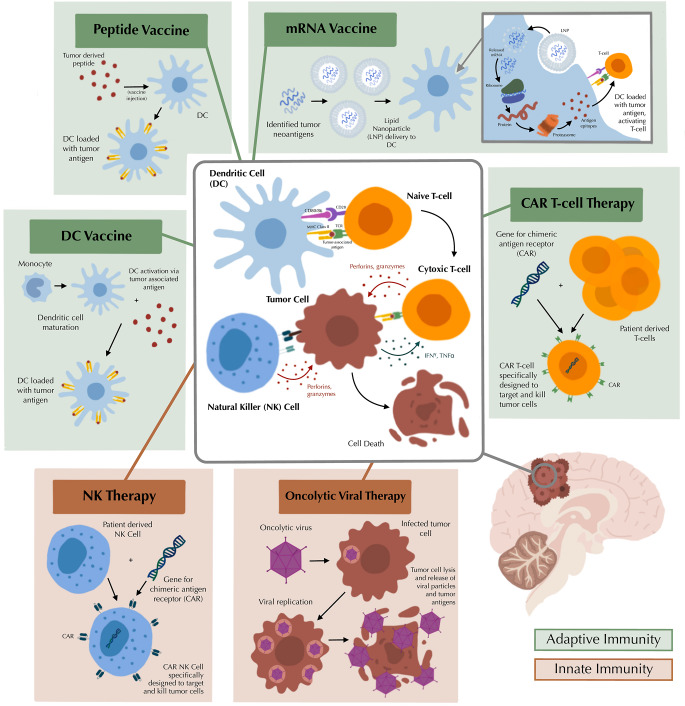 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlioma Diagnosis and Treatment · Immunotherapy and Immune Responses · Neuroinflammation and Neurodegeneration Mechanisms
Introduction
Pediatric high-grade gliomas (pHGG) represent the most aggressive and lethal of the childhood brain tumors. Prognosis remains dismal, with median survival less than 24 months in 70–90% of patients, and less than 1 year in most subtypes [1–5]. The current standard of care, which includes maximal safe surgical resection and radiotherapy, has limited efficacy due to the infiltrative nature of these tumors, their resistance to cytotoxic agents, and the frequent involvement of eloquent or surgically inaccessible brain regions such as the brainstem [1, 4–6].
Over the past decade, molecular profiling has revolutionized the classification of pHGG, distinguishing them from adult gliomas and uncovering key oncogenic drivers unique to the pediatric population. This shift from reliance upon pathologic appearance to molecular identity is reflected in the most recent 2021 WHO guidelines, which heavily depend upon direct tissue sequencing for tumor classification [7, 8]. A growing understanding of the key driver mutations in pHGG, such as H3 K27M in diffuse midline glioma, H3 K27-altered (DMG), and H3 G34R/V in diffuse hemispheric glioma, H3 G34-mutant (DHG), has not only improved diagnostic precision but also opened new avenues for targeted therapies [1, 7]. Nevertheless, translating these insights into effective treatments has remained challenging, particularly given the unique biology and immunology of pediatric brain tumors.
Immunotherapy has emerged as a promising strategy in oncology, with dramatic successes in hematologic malignancies [9, 10] and select adult solid tumors [11, 12]. However, its application in pediatric neuro-oncology has lagged, constrained by factors such as the distinct nature of the immune system within the central nervous system (CNS), the immune-suppressive tumor microenvironment (TME), and the low mutational burden characteristic of pHGG [6, 13–16]. Recent landmark trials utilizing chimeric antigen receptor (CAR) T-cell therapy have demonstrated meaningful biological activity and early clinical responses, sparking increasing interest in the potential of immunotherapy for pHGG. BrainChild-03 improved median survival for 21 DIPG patients to 19.8 months utilizing repeated intracerebroventricular (ICV) B7-H3 CAR T-cell dosing with only one dose-limiting toxicity [17]. Another phase-I clinical trial (NCT04196413) employed single-dose IV and subsequent ICV infusions of GD2-CAR T-cells in lymphodepleted DIPG or spinal DMG patients, and identified volumetric reductions in 7 of 11 patients receiving therapy [18]. Notably, three patients on the higher dosing regimen developed dose-limiting cytokine release syndrome, with additional instances of tumor inflammation-associated neurotoxicity (TIAN) or immune effector cell acute neurotoxicity syndrome (ICANS). These milestone studies highlight the promising therapeutic potential, as well as the logistical and safety hurdles still to be addressed, of CAR T-cell therapy for pHGG.
While adoptive cellular therapies including CAR T-cells have provided important data to validate that the immune system can indeed be therapeutically mobilized against pHGG, these ongoing challenges may underscore the need for complementary immunotherapeutic modalities. Anti-tumor vaccine strategies represent a particularly attractive approach due to their relatively non-invasive administration and more favorable toxicity profile compared to other immunotherapeutic modalities [19, 20]. Vaccines aim to stimulate the patient’s immune system to recognize tumor-associated antigens (TAA), thereby enabling a durable anti-tumor response with minimal off-target effects and without the need for direct cellular replacement. In early-phase trials, some vaccine platforms have shown encouraging safety and immunogenicity in children with high-grade gliomas [21–26]. Other pHGG vaccine modalities remain in the preclinical stage with encouraging preliminary data. However, numerous challenges remain, including the identification of optimal antigen targets, overcoming the barriers imposed by CNS relative immune privilege and glioma-mediated immunosuppression, and ensuring timely vaccine delivery given the rapid progression of pHGG. Additionally, ethical and logistical considerations in the pediatric population require careful navigation, particularly when pursuing highly individualized experimental therapies in patients with limited therapeutic alternatives.
This review summarizes the current landscape of vaccine-based immunotherapy for pHGG, including peptide, dendritic cell (DC), mRNA, shared mutant neoantigen, and personalized neoantigen vaccine therapies. We outline the scientific rationale for vaccine development in this population, review key tumor antigens and immunologic barriers, and evaluate the major vaccine platforms under clinical investigation.
Rationale for vaccine immunotherapy in pediatric HGG
Given the dismal prognosis and resistance to conventional therapies, novel treatment strategies for pHGG are critical. Advances in modern sequencing techniques and growing insight into how histone-based mutations drive tumorigenesis have enabled the development of targeted molecular approaches [27, 28]. Among these, vaccine-based therapies harness the body’s immune system to recognize and attack TAAs. Although still investigational, early-phase clinical trials of multiple vaccine platforms have demonstrated immunogenicity and favorable safety profiles. A schematic representation of the various vaccine modalities currently under investigation is presented in Fig. 1. Current efforts are focused on refining antigen selection, enhancing delivery platforms, and evaluating synergistic combinations with other immunotherapeutic agents. Continued clinical investigation is essential to confirm the therapeutic potential of pHGG vaccines in larger patient cohorts and to translate promising early findings into meaningful clinical benefit.
Tumor antigens and potential vaccine targets
Considerations for antigen selection
Antigen selection for vaccine therapies is predicated upon the identification of TAAs or neoantigens that are differentially expressed by tumor cells compared to healthy brain parenchyma. Comprehensive high-throughput genomic and proteomic sequencing enables the discovery of antigens, which must then be evaluated for major histocompatibility complex (MHC) binding affinity and potential to elicit immunogenic T-cell responses [29].
Fig. 1. Immunotherapeutic strategies under investigation for pediatric high-grade gliomas. Adaptive immune therapies include peptide vaccines, mRNA vaccines, dendritic cell (DC) vaccines and chimeric antigen receptor (CAR) T-cell therapies. Innate immune therapies include natural killer (NK) cell therapy and oncolytic viral therapy
Several critical considerations guide the selection of suitable antigens. First, candidate antigens must be abundantly and consistently overexpressed in tumor cells relative to normal tissue, ensuring sufficient antigen presence to elicit a robust immune response. Antigens that are minimally expressed in normal, healthy tissues are preferred to reduce the risk of off-target autoimmune effects and to enhance therapeutic specificity. Second, antigens should demonstrate strong immunogenicity, indicating their capacity to provoke an effective T-cell-mediated immune response. Third, selected TAAs should ideally exhibit high prevalence across patient populations to ensure broad applicability. Tumor-specific neoantigens derived from unique somatic mutations may offer heightened specificity and avoid off-target effects; however, their widespread applicability could be limited given tumor heterogeneity across patients and pHGG subtypes. Lastly, antigens implicated in poor prognosis or therapy resistance are particularly attractive as their immunologic targeting may not only induce tumor regression but also disrupt pathways involved in tumor progression. Given the limited number of eligible patients and the poor prognosis of pHGG, only the most promising and rigorously validated antigens must be advanced into clinical trials to maximize the potential for therapeutic success and minimize risk in this vulnerable population.
TAAs in Glioma
TAAs identified in gliomas represent a group of frequently overexpressed or uniquely mutated proteins that are conserved across many pHGG patients. This makes them appealing targets for vaccine-based therapies given their wide applicability across the population. Prominent TAAs currently under investigation include EphA2, IL-13Rα2, and survivin [22, 23]. These antigens are prioritized based on consistent overexpression in glioma cells relative to normal tissue, demonstrated capacity to induce antigen-specific T-cell responses, and established safety profiles in preliminary clinical studies [30, 31]. Beyond these well-established antigens, emerging mRNA-based vaccine strategies have expanded the repertoire of targetable antigens, encompassing proteins such as TP53, IDH1, C3, TCF12, ANXA5, FKBP10, MSN, and PYGL [32, 33]. These candidate antigens have been identified through integrative genomic and immunologic analyses as correlating strongly with poor prognosis and immune cell infiltration, underscoring their potential clinical significance in vaccine formulations for pHGG.
Neoantigens Derived from Somatic Mutations
Neoantigens, derived from tumor-specific somatic mutations, represent highly specific targets for vaccine therapies and can be broadly categorized into shared mutant antigens (arising from recurrent cancer-specific mutations) or personalized neoantigens (patient-specific genetic alterations). Because neoantigens are not present in normal tissues, they bypass central immune tolerance and reduce the risk of autoimmunity. Among pHGG, the most extensively studied shared mutant neoantigen is the H3.3K27M mutation, a hallmark alteration in DMG [34, 35]. This mutation generates a unique and immunologically distinct peptide sequence which is absent in healthy tissues, making it an attractive target for antigen-specific vaccine development. Additional shared mutant neoantigens, including recurrent mutations in IDH1 and TP53, have also emerged as potential vaccine targets [32, 35]. In contrast, personalized neoantigens arise from unique, patient-specific somatic mutations and give rise to de novo peptide sequences that vary between patients. These personalized neoantigens form the basis for individualized neoantigen vaccine strategies. Together, neoantigen targets highlight the potential of personalized vaccine strategies to elicit robust, tumor-specific immune responses in pHGG while avoiding off-target autoimmune effects.
Immunologic challenges in pediatric brain tumors
The CNS exhibits an immune landscape fundamentally distinct from the rest of the body, characterized by tightly regulated immune responses to protect neural tissue from inflammatory damage. While the brain has historically been described as immune privileged, this privilege is now understood to be relative rather than absolute, as activated T-cells are capable of trafficking across the BBB and mounting immune responses under physiological and therapeutic conditions such as systemic immunotherapy [36, 37]. Nevertheless, CNS immune responses remain tightly regulated through the BBB, specialized lymphatic drainage, and immunoregulatory mechanisms at CNS interfaces such as the meninges and perivascular spaces. While immune surveillance does occur, robust immune response within the parenchyma is limited to prevent collateral damage to neural tissue with limited regenerative capacity [36, 38]. As a result, immune-based therapies in the CNS (particularly in critical midline structures such as the brainstem) carry a risk of immune-mediated pseudo-progression. In these regions with limited tolerance for treatment-induced inflammation and edema, this phenomenon can mimic tumor progression, lead to worsening neurologic compromise and necessitate urgent administration of corticosteroids [22]. Therefore, therapeutic strategies aiming to stimulate antitumor immunity in the CNS must overcome these barriers to enable effective immune cell infiltration and activation within the tumor microenvironment.
In addition, as part of their resistance mechanism, pHGGs actively create an immunosuppressive microenvironment that inhibits effective antitumor immunity. This environment is marked by low cytotoxic T-cell infiltration, abundant immunosuppressive myeloid populations, and secretion of inhibitory cytokines like TGF-β and IL-10 [13, 36]. Single-cell transcriptomics have demonstrated that myeloid cells represent the predominant immune population in pHGG, yet their composition and functional states are distinct from those observed in adult glioblastoma and vary according to tumor location and histone mutations [6, 39, 40]. Pediatric gliomas, particularly H3.3K27M DMG, are enriched for heterogeneous disease-associated myeloid populations that exhibit impaired interferon signaling, limited antigen presenting capacity, and express chemokines to recruit additional suppressive myeloid cells and restrict lymphocyte infiltration [39]. Within this myeloid compartment, resident microglia and infiltrating bone-marrow derived macrophages (BMDMs) play complementary roles. Microglia preferentially localize to tumor margins and perivascular regions, whereas BMDMs infiltrate the tumor core and actively suppress T-cell proliferation [39, 41, 42]. Together, this myeloid landscape restricts T-cell access to the tumor core and suppresses their function, providing a possible mechanistic explanation for limited intratumoral immune activity despite detectable peripheral vaccine responses.
Another challenge is the low tumor mutational burden seen in pHGGs. Compared to many tumor types seen in adults, which are generally thought to develop over years as numerous somatic mutations accumulate, pHGGs exhibit a characteristically low mutational burden and instead arise from developmental and epigenetic alterations. This scarcity of somatic mutations limits the number of neoantigen targets for vaccine therapies as fewer TAAs are available for immune recognition [6, 14, 43].
Aside from the tumor-specific challenges, there are also challenges inherent to the pediatric population. For example, the pediatric immune system is characterized by a higher proportion of naïve T-cells and fewer memory T-cells and thus presents unique challenges for vaccine therapies. These less-experienced immune systems have reduced capacity for robust and durable adaptive responses to novel antigens [44]. When compounded by the relative immune privilege of the CNS and an immunosuppressive tumor microenvironment, the efficacy of vaccine-based interventions may be severely limited. In addition, emerging evidence suggests that T-cell dysfunction in pHGG differs from the classical T-cell exhaustion observed in adult glioblastoma. Whereas glioblastoma is characterized by profound T-cell exhaustion with upregulation of immune checkpoint molecules and clonal T-cell expansion [45], pHGG instead exhibit sparsely infiltrating T-cells in immature states, limited clonal expansion, and reduced effector functionality [40, 42]. These differences suggest that immune failure in pHGG may reflect impaired T-cell priming and recruitment rather than exhaustion alone.
This challenge is further compounded by steroid therapy, which is frequently used to manage cerebral edema in pHGG patients. Unfortunately, although beneficial for edema, steroids suppress immune function, resulting in reduced lymphocyte counts and impaired antigen presentation. Coupled with lymphopenia resulting from tumor progression and conventional therapies including temozolomide and radiation therapy, these immunosuppressive effects greatly impair vaccine-induced T-cell responses [21, 24]. The decision to offer steroids must be carefully considered in patients eligible for vaccine-based therapies, weighing the dangers of worsening cerebral edema against a possible reduction in vaccine efficacy. Future clinical investigations should rigorously monitor if patients are concurrently receiving steroid treatment to better evaluate if steroid use is contraindicated in pHGG vaccines.
Vaccine strategies, trial outcomes, and biomarkers of response
Peptide-based vaccines
Peptide-based vaccines utilize short amino acid sequences corresponding to epitopes derived from TAA to elicit an immune response against cancer cells expressing the chosen antigen. Upon administration, these peptides are internalized by antigen-presenting cells (APCs) and presented on the cell surface by MHC molecules. This activates antigen-specific CD8⁺ cytotoxic T lymphocytes or CD4⁺ helper T-cells, thereby initiating a targeted immune response [21–24]. Peptide-based vaccines currently under investigation for pHGG target both shared TAA and tumor-specific neoantigens.
H3.3K27M peptide vaccine
The H3.3K27M_26 − 35_ peptide vaccine is an investigational therapy targeting the H3.3K27M mutation characteristic of DMG. A phase I trial (NCT02960230) evaluating this vaccine enrolled 29 children with H3.3K27M-positive DMG and demonstrated that the vaccine was well tolerated without severe immune-related adverse events (Table 1) [21]. Patients who mounted an H3.3K27M-specific CD8⁺ T-cell response exhibited significantly improved median overall survival (OS) compared to non-responders. However, concurrent corticosteroid use was associated with diminished CD8⁺ T-cell responses, potentially blunting therapeutic efficacy. While this vaccine was initially investigated in patients with the human leukocyte antigen (HLA) subtype A02:01, emerging analyses suggest that immune responses to this vaccine can involve multiple HLA class II alleles and B-cell activation, indicating broader immunogenic potential.
Table 1. Summary of clinical trials evaluating active immunotherapies in pediatric high-grade gliomaVaccine TypeTrial #Paper TitlePopulationTarget AntigenDelivery MethodAdjuvantsNumber of PatientsImmune ResponsePFSOSSafety ProfilePeptide Vaccine (H3.3K27M)NCT02960230Mass cytometry detects H3.3K27M-specific vaccine responses in diffuse midline glioma [21]Age 3–21 years, newly diagnosed DIPG or DMG. HLA-A*02:01 + and H3.3K27M+H3.3K27M_26–35_Subcutaneous every 3 weeks for first 8 doses, then every 6 weeks. Maximum treatment of 96 weeks totalTetanus Toxoid (TT) peptide, poly-ICLC and Montanide-ISA 51 VG29 (19 DIPG, 10 DMG)39% had H3.3K27M-reactive CD8⁺ T cell expansion4.9 mo (DIPG), 3.5 mo (DMG). Immune responders had greater PFS than non-responders (p = 0.05)Median 16.1 months (immune responders) vs. 9.8 months (non-responders), p = 0.05; overall 12-month OS ~ 40%Grade 1–2 AEs, injection site reactions most common. One case of suspected aseptic meningitis (resolved)Peptide Vaccine (H3K27M long peptide)Compassionate use basis; ongoing INTERCEPT-H3 trial: NCT04808245A H3K27M-targeted vaccine in adults with diffuse midline glioma [46]Adults > 18 years with progression of histologically-confirmed H3K27M^+^ DMG after standard radiotherapy and chemotherapy with TMZH3K27M_14–40_Subcutaneous injection bi-weekly for 6 weeks, then monthly for 4 months, followed by quarterly thereafterMontanide ISA-50; topical imiquimod8 (all H3K27M^+^ DMG)H3K27M-specific CD4^+^ T cell-dominated immune response in 5/8 patients (62.5%) by IFN-γ ELIspot. One patient developed pseudoprogression followed by sustained complete remission.Median: 6.2 months from first vaccinationMedian: 12.8 months from first vaccinationOne patient with sustained complete remission for > 31 monthsNo regimen-limiting toxicity. Grade 1 injection site reactions in 25%. One patient developed pseudoprogression. No grade 3 or higher vaccine-related AEsPeptide Vaccine (GAA)NCT01130077Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas [22]Age 1–21 with high grade glioma and HLA-A2+EphA2, IL-13Rα2, and survivinSubcutaneous injection every 3 weeks for 8 doses, then doses every 6 weeks for up to 2 yearsTetanus Toxoid (TT) peptide, poly-ICLC and Montanide ISA-5126 (20 brainstem glioma, 6 other high grade glioma)62% of patients had immune response to at least one GAA epitopeUnspecified, 5 patients with pseudoprogression (19%)Median 13.3 months for total cohort, 25.1 months among HGG patientsGrade 1 and 2 AEs, including injection-site reactions (100%), flu-like symptoms (92%), grade 1 GI symptoms (31%), grade 1 leukopenia (15%)Peptide Vaccine (GAA)NCT01130077Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: Results of a pilot study [23]Age 1 to < 22 with high grade glioma and HLA-A2+EphA2, IL-13Rα2, and survivinSubcutaneous injection every 3 weeks for 8 doses, then doses every 6 weeks for up to 2 yearsTetanus Toxoid (TT) peptide, poly-ICLC and Montanide ISA-5112 (6 glioblastoma, 5 AA, 1 malignant gliomatosis cerebri)9 of 10 evaluable patients (90%) had immune reactivity to at least one GAAMedian 4.1 months. 33% reached 6 months PFS.Median OS: 12.9 months. 73% reached 6 months OSGrade 1 and 2 AEs, including injection-site reactions (100%) and flu-like symptoms (100%), grade 1 GI symptoms (42%), grade 1 anemia (8%) and grade 1 leukopenia (8%)Peptide vaccine (PEP-CMV)NCT03299309A peptide vaccine targeting the CMV antigen pp65 in children and young adults with recurrent high-grade glioma and medulloblastoma: a phase 1 trial [24]Age > 18, recurrent medulloblastoma or malignant gliomaCMV pp65 (PEP-CMV). Synthetic long peptide (SLP) of 26 amino acids encoding epitopes of pp65Intradermal injection of PEP-CMV every 2 weeks for first 3 doses, then every month. Maximum treatment of 10 yearsPEP-CMV injection given in Montanide ISA-51 following Td toxoid preconditioning and 5-day temozolomide course to induce lymphopenia36 (HGG patients who received ≥ 1 vaccine)ELISpot in 21 patients with known CMV status: 76.2% (16/21) showed increased IFN-γ pp65-directed T cell activity2.5 months (95% CI: 2.2–3.2)6.5 months (95% CI: 4.6–8.4)17 grade 1 AEs, 15 grade 2, 1 grade 3 (pyramidal tract syndrome), 1 grade 4 (cerebral edema). Most common AE was grade 1 injection site reaction (81%)Dendritic cell (DC) VaccineNoneAdjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours [25]Children with malignant brain tumorAutologous tumor lysateIntradermal injections of DCs pulsed with tumor lysate via 4 distinct schedulesTopical imiquimod one night prior and 2 nights following each vaccination45 (33 HGG, 5 MB/PNET, 4 ependymoma, 3 ATRT)Of 23 tested patients, 19 (83%) showed vaccine-specific T-cell proliferation; 6 patients had > 24 months survivalMedian 4.4 months. 42% reached 6 months PFSMedian OS: 13.5 months; 6 patients > 24 months survivalOnly mild vaccine-related adverse events: fever, local skin reactions, fatigue, vomiting, headacheDendritic cell (DC) VaccineNCT00107185Autologous tumor lysate-pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high-grade gliomas [26]Age 1–18 with confirmed WHO grade III or IV gliomaAutologous tumor lysateIntradermal injection biweekly. Patients received between 2–4 dosesNone3 (2 glioblastoma, 1 anaplastic oligoastrocytoma)Mild response of Th1/Th2 cytokines including IFN-γ, IL-2, IL-4, IL-6, TNFαUnspecified.Two patients survived at least 51 and 40 months following surgery. Last patient had 9 month OSMild adverse events (headache, injection-site erythema). One Grade 4 reaction with elevation of alkaline phosphatase (resolved)Personalized Neoantigen Peptide VaccineNoneA real-world observation of patients with glioblastoma treated with a personalized peptide vaccine [47]Age 9–87 years; histologically-confirmed IDH-wildtype glioblastomaPatient-specific tumor neoantigens (median 19 peptides per patient)Intracutaneous injection 4 times in first 2 weeks, with subsequent booster vaccinations every 4–6 weeksSargramostim (GM-CSF) injection; topical imiquimod on injection site173 (70 primary IDH-wt GBM, 103 recurrent IDH-wt GBM)Neoantigen specific T cell responses were detected in 87/97 evaluable patients (90%).Immunological responders Median: 28.7 monthsNon-responders median: 17.4 months.HR 0.47 (0.24–0.94), p = 0.03Median 31.9 months from diagnosisAE predominantly grade 1–2. Four grade 3 reactions (allergic reaction, anaphylaxis, skin reaction). No grade 4 AE observed.CAR T cell (B7-H3)NCT04185038Intracerebroventricular B7-H3-targeting CAR T cells for diffuse intrinsic pontine glioma: a phase I trial [17]Age 1–26 years with high grade glioma or diffuse midline glioma H3K27M-altered following standard radiation therapyB7-H3 (CD276)Repetitive intracerebroventricular (ICV) infusion every 2 weeks for 8 weeks with escalating doses, then continued dosing every 2–4 weeksNone; no lymphodepletion21 (17 DMG via molecular diagnosis, 1 pontine anaplastic astrocytoma, 3 suspected DMG via radiographic diagnosis)CAR T detected in CSF in 13/18 tested patients (72%). CSF elevation of CXCL10, IP-10 and GM-CSFUnspecified. 9 patients were enrolled before progression, 12 patients enrolled after progression.Median survival from first CAR T cell infusion: 10.7 monthsMedian survival from diagnosis: 19.8 months3 long term survivors > 44 monthsMostly grade 1–2 adverse events (most common: headache, nausea/vomiting, fatigue, fever). One grade 4 intratumoral hemorrhageCAR T cell (GD2)NCT04196413Intravenous and intracranial GD2-CAR T cells for H3K27M^+^ diffuse midline gliomas [18]Age 2–30 years with biopsy-confirmed H3K27M-mutant DMG, including DIPG or spinal DMG, who had completed standard radiotherapyGD2Single IV infusion of GD2-CAR T cells, followed by sequential IV or ICV infusions for patients with radiographic or clinical benefitLymphodepleting chemotherapy prior to IV infusion; no lymphodepletion for ICV infusions13 enrolled;11 treated (9 DIPG, 2 spinal DMG)Peripheral expansion and persistence of GD-2 CR T cells in blood and CSF with repeat ICV infusions. 8/11 patients classified as “responders”.Tumor volume reduction was observed in 7/11 patients, with > 50% reduction in 4 patients.Median survival from diagnosis: 20.6 months2 long term DIPG survivors (30, 33 months)All patients (11/11) experienced CRS following initial IV GD2-CAR T infusion, DLT at higher dose level (3 patients with grade 4 CRS). No DLT with ICV infusion observed. TIAN observed in 71% of ICV infusions.
In parallel, important insights have emerged from adult studies utilizing the long 27-mer H3K27M_14 − 40_ peptide vaccine in the INTERCEPT H3 trial (NCT04808245). In a compassionate-use cohort of eight adult DMG patients, vaccination was well-tolerated with only grade 1 adverse events, and the vaccine induced mutation-specific immune responses detectable in both peripheral blood and cerebrospinal fluid [48]. Subsequent in-depth immune profiling of a single patient demonstrated that the 27-mer H3K27M peptide resulted in a robust CD4^+^ T-cell response with polyclonal T-cell receptor expansion across multiple HLA alleles [46]. While these findings are derived from adult DMG patients and cannot be directly extrapolated to pediatric populations, they provide evidence that H3K27M peptide vaccines can induce coordinated adaptive immune responses.
However, recent work by Wang et al. challenges the feasibility of this approach [49]. Using patient-derived H3.3K27M DMG models, they demonstrated that the H3.3K27M_26–35_ peptide is not endogenously presented at detectable levels on HLA-A02:01⁺ tumor cells. Mass spectrometry failed to identify the peptide-HLA complex on multiple diffuse intrinsic pontine glioma (DIPG) cell lines and engineered T-cells specific for H3.3K27M-HLA-A*02:01 failed to kill patient-derived tumor cells. These findings suggest that, despite in vitro binding capacity, the absence of natural peptide presentation may render vaccines or T-cell therapies targeting this epitope ineffective. Further investigation is ongoing.
TAA peptide vaccine
TAA peptide vaccines targeting EphA2, IL-13Rα2, and survivin have demonstrated safety and modest immunogenicity in pHGG (NCT01130077, Table 1) [22]. There were no observed dose-limiting toxicities, and results of enzyme-linked immunosorbent spot analysis (ELISA) demonstrated the TAA peptide vaccine was able to induce antigen-specific T-cell responses in 62% of patients to at least one of the three TAA epitopes. Immunologic pseudo-progression was reported in five patients (19%), who required corticosteroid management and cessation of subsequent TAA vaccine, although some patients were able to resume at a lower poly-ICLC dose after symptomatic resolution [22]. Another cohort study of twelve children with recurrent pHGG from the same trial (NCT01130077) showed median progression-free survival (PFS) and OS were 4.1 and 12.9 months, respectively. This study demonstrated only grade 1 and 2 adverse events with a favorable safety profile, and 90% of evaluable patients had TAA immune reactivity (Table 1) [23].
CMV pp65 (PEP-CMV) peptide vaccine
Human cytomegalovirus (CMV) epitopes have been found in 67% of pediatric high-grade gliomas, with prior work utilizing a dendritic cell vaccine showing modest efficacy [24]. The PEP-CMV pp65 peptide vaccine is a 26-amino-acid chain that targets the CMV pp65 antigen on pHGG cells. Notably, this antigen is absent in healthy brain tissue. A phase I trial (NCT03299309) showed the safety of the CMV pp65 vaccine with mostly grade 1–2 adverse events, although rare severe CNS toxicities such as cerebral edema were reported. The vaccine elicits pp65-specific T-cell responses in 76% of patients with a median PFS of 2.5 months and OS of 6.5 months (Table 1) [24]. A multi-institutional phase II trial (NCT05096481) is ongoing to evaluate efficacy and immunological response in a larger cohort.
Notably, the presence of CMV within gliomas remains an area of ongoing controversy. While multiple groups have reported detection of CMV proteins or nucleic acids in glioma specimens using immunohistochemistry or PCR-based approaches [50, 51], others have failed to detect CMV [52, 53]. These discrepancies have raised concerns regarding assay sensitivity, specificity and reproducibility. Importantly, even in the absence of productive viral replication, low-level CMV antigen expression has been proposed as a potential immunotherapeutic target [53], providing a rationale for continued investigation of CMV peptide vaccines despite this ongoing controversy.
In summary, peptide vaccines targeting H3.3K27M, shared TAA (EphA2, IL-13Rα2, survivin), and CMV pp65 are actively under clinical investigation for pHGG. Early-phase trials consistently demonstrate safety and modest immune response generation in over half of patients, though efficacy data remain to be validated in larger, controlled studies.
Dendritic cell vaccines
Dendritic cell (DC) vaccines leverage the antigen-presenting function of dendritic cells to stimulate immune responses against high-grade gliomas. Patient-derived monocytes are differentiated, loaded with tumor antigens, and administered subcutaneously or intradermally. These DCs migrate to draining lymph nodes, activating CD8⁺ cytotoxic and CD4⁺ helper T-cells [25, 26]. This approach aims to overcome glioma-induced immune suppression by generating robust, tumor-specific immunity.
While DC vaccines have undergone extensive evaluation in the adult glioblastoma population, few trials evaluate their efficacy in the pHGG population. Ardon et al. demonstrated the feasibility of autologous tumor-lysate loaded DC vaccines in 45 children with relapsed malignant brain tumors, including 33 with pHGG [25]. Their study reported a median OS of 13.5 months for the HGG cohort, with six long-term survivors exceeding 24 months. DC vaccination was well tolerated, with only mild adverse events (Table 1). The ADDICT-pedGLIO (NCT04911621) trial is an ongoing investigation into a promising evolution of DC vaccinations utilizing Wilms’ tumor 1 (WT1) mRNA, an antigen overexpressed in pHGG including DIPG. This Phase I trial will establish safety and feasibility of DC vaccine leukapheresis protocols targeting the WT1 antigen in 10 children.
Lasky et al. conducted tumor lysate-pulsed DC vaccination in three children with high grade glioma (NCT00107185) [26]. They showed a favorable safety profile and only mild adverse events, including one patient who had a transient, asymptomatic elevation of alkaline phosphatase. Two patients had extended PFS of at least 51 and 40 months (Table 1). This study also underscored the significant feasibility challenges in DC vaccine administration, as rapid disease progression precluded vaccine administration in four patients. Trials investigating alternative splice variant-targeted peptide-pulsed DCs (NCT06342908) and the CMV pp65-LAMP mRNA-pulsed DCs (NCT03688178) are ongoing.
Despite encouraging safety profiles and signals of potential efficacy in select patients, DC vaccines face significant challenges in pHGG, including lengthy manufacturing times that can delay treatment in rapidly progressing disease, variability in immune responsiveness, and the profound immunosuppressive tumor microenvironment that may limit durable antitumor activity. Additionally, limited patient numbers in early-phase trials limit definitive conclusions regarding survival benefit. Future studies are critical to refine antigen selection, optimize timing of vaccine delivery, and explore synergistic combinations with other immunomodulatory or standard therapies, including radiation.
mRNA-based vaccines
Messenger RNA (mRNA) vaccines represent another therapeutic approach utilizing the patient’s adaptive immune system to target malignant cells. Unlike peptide vaccines, delivery of mRNA sequences encoding TAAs allows APCs to translate the full-length antigen and present multiple epitopes on MHC molecules without restriction to only certain HLA types [54]. This allows for a more robust immune response by stimulating cytotoxic CD8⁺ T lymphocytes and helper CD4⁺ T-cells through antigen presentation from both MHC-I and MHC-II molecules. Ex vivo loading of patient-derived DCs with mRNA encoding glioma antigens allows APCs to prime naïve T-cells upon reinfusion. Alternatively, direct injection of lipid nanoparticle (LNP)-encapsulated mRNA enables in situ translation and immune activation without ex vivo processing [55]. These strategies enable the immune system to mount a multi-epitope response with low rates of immune rejection and no risk of genome integration, offering unique advantages compared to other vaccine modalities.
Several TAA have been identified as possible targets for mRNA-based vaccines for gliomas including TP53, C3, TCF12, and IDH-1 [29, 32]. A comprehensive analysis of glioma datasets from The Cancer Genome Atlas (TCGA) and the Chinese Glioma Genome Atlas (CGGA) identified these antigens as overexpressed and correlated with increased infiltration of APCs, suggesting their potential utility as targets in mRNA-based vaccines. Studies have also noted the expression of immunosuppressive molecules such as PD-L1 and TIM-3 in pHGGs, supporting the rationale for combining vaccines with immune checkpoint blockade to enhance antitumor response [56, 57]. However, checkpoint inhibition alone has demonstrated only limited clinical success to date in pHGG, underscoring that any potential synergistic approach with vaccine therapies will require careful clinical validation.
Clinical translation of mRNA vaccines specifically for pediatric gliomas remains in its infancy, with no completed trials reported in children to date. However, adult trials targeting similar antigens, including early-phase studies using mRNA-loaded DC vaccines and direct LNP-encapsulated mRNA delivery, have demonstrated safety and immunogenicity, laying the groundwork for currently ongoing pediatric trials [58]. Ongoing identification of glioma antigens and evolving understanding of the unique pediatric tumor immune microenvironment are critical for the success of this vaccine modality. Trials investigating the CVGBM mRNA vaccine (NCT05938387) and RNA-lipid Particle (RNA-LP) vaccines (NCT04573140) are ongoing.
Neoantigen-personalized vaccines
Neoantigen-personalized vaccines represent a unique, highly targeted approach for immunotherapy in pHGG, driven by the unique genomic landscape of individual tumors. Unlike shared TAAs central to other glioma vaccine therapies, neoantigens arise from tumor-specific mutations, presenting entirely novel peptide sequences not found in healthy tissues that are specific to each individual patient [34]. In pediatric medulloblastoma, for instance, Rivero-Hinojosa et al. show aberrant splice junctions as significant neoantigen sources through proteogenomic analysis [59].
While pediatric trials utilizing neoantigen-personalized vaccines have not been conducted, early-phase clinical trials in adult glioblastoma have shown promising results. The personalized neoantigen vaccine “NeoVax”, which combines patient-specific neoantigens identified from tumor sampling, has successfully generated immune responses characterized by an increase in interferon-gamma (IFN-γ) producing T-cells and intratumoral T-cell infiltration [60]. In a cohort of 173 adult patients with IDH-wildtype glioblastoma, personalized multi-peptide neoantigen vaccines were feasible to manufacture (median time from tissue acquisition to vaccine administration was 16 weeks), well tolerated with only grade 1 or 2 adverse events, and induced durable T-cell responses in 77 of 97 patients tested [47]. Importantly, patients who were found to mount an effective immune response against multiple vaccinated neoantigens were associated with prolonged survival compared with low- or non-responders. These data provide important early evidence for the potential of neoantigen vaccines, albeit exclusively in an adult population. Although data specific to pHGG are limited at this time, the biological rationale and encouraging early-phase adult studies highlight the translational promise of neoantigen vaccines for pediatric populations, especially given the significant heterogeneity in pHGG mutations.
Despite the considerable potential, several challenges remain for neoantigen vaccine implementation. pHGGs typically possess fewer somatic mutations compared to adult tumors, which may complicate neoantigen identification and limit target availability [6, 13–15]. Furthermore, the tumor microenvironment in pediatric gliomas often demonstrates pronounced immunosuppression, reducing T-cell infiltration and activity, potentially undermining vaccine efficacy [13, 36]. Additionally, access to tissue samples for sequencing and neoantigen identification can be especially challenging in certain pHGG, depending on anatomical location and surgical accessibility. The requirement for patient-specific manufacturing also introduces logistical complexity, substantial costs, and potential delays that may be incompatible with the rapid progression of HGG. Given the rapid expansion of these tumor populations, the potential for intratumoral heterogeneity may also further complicate vaccine design [56]. Despite these physiologic and logistical challenges, the incorporation of combination approaches such as neoantigen vaccines alongside immune checkpoint inhibitors may enhance antitumor efficacy.
Ethical, regulatory, manufacturing, and translational challenges
Developing vaccines for pHGG presents a host of challenges. In this vulnerable population, characterized by an aggressive disease course and limited treatment options, maintaining a favorable risk-benefit balance is essential. Protecting patient welfare requires thoughtful, transparent communication between providers and families. Additional services to support the patient and family should be involved early, including social work, child life specialists, psychiatry, spiritual or cultural support, and palliative care. In the adolescent population, assessing decision-making capacity and navigating informed consent add further complexity, requiring a sensitive and individualized approach.
Regulatory oversight plays a critical role, mandating rigorous safety evaluations and continuous monitoring of vaccine trials by institutional review boards and ethics committees. Manufacturing challenges are substantial, particularly for personalized neoantigen vaccines, which require tumor sequencing, epitope selection, strict quality control, sterile production protocols, and efficient logistical coordination. In practice, this process ranges from 10 to 16 weeks from tissue acquisition to vaccine availability, a timeline that may be incompatible with the rapid clinical progression in many pHGG patients. Given this narrow therapeutic window, delays in manufacturing or trial enrollment may render patients ineligible for treatment due to worsening neurologic decline or tumor progression [26].
Logistical and financial barriers also further limit clinical translation. Vaccine platforms remain primarily accessible only within clinical trials, and reimbursement for investigational or personalized vaccines outside of a trial setting is often unclear and logistically challenging to navigate. These constraints limit equitable access and disproportionately restrict availability to patients treated at large academic centers with specialized infrastructure to host these clinical trials. Translational barriers also include the limited predictive value of preclinical models, a lack of validated biomarkers to monitor immune response and tumor progression, and the inherent difficulties in designing and executing clinical trials for a rare disease with a small and often rapidly deteriorating patient population. Overcoming these multifaceted challenges is essential to ensure the safe, timely, and effective advancement of vaccine-based therapies for children with high-grade gliomas.
Conclusions & future directions
Despite encouraging early-phase data, significant knowledge gaps remain in understanding how vaccine therapies can be optimized for pHGG. Key uncertainties include the most effective antigen targets for broad or personalized application, the optimal timing and sequencing of vaccine administration relative to standard therapies, and the impact of immune-modulating factors such as steroid use on vaccine efficacy. Moreover, the pediatric-specific immune response to tumor vaccines remains poorly characterized, particularly in the setting of CNS relative immune privilege and glioma-mediated suppression. The lack of validated biomarkers to predict or monitor treatment response further complicates trial design and clinical implementation.
An additional priority for future investigation is defining the optimal clinical windows for vaccine administration as pHGG treatment continues to evolve. Vaccine-based therapies may be the most effective during periods of minimal disease burden, such as following maximal safe surgical resection or in the post-radiation setting, when tumor antigen release and blood-brain barrier disruption may enhance immune priming. In contrast, concurrent administration during upfront chemoradiation may be limited by treatment-induced lymphopenia and the frequent requirement for corticosteroids. The recent approval of ONC201 for H3.3K27M-mutant DMG may also introduce a new therapeutic axis and expand the window of opportunity to maximize response to vaccine therapies by enabling disease stabilization with a favorable toxicity profile and reduced steroid dependence, thereby creating more favorable conditions for vaccine integration.
Future research must prioritize the development of integrated pipelines to rapidly identify high-quality tumor antigens in individual patients given the rapid progression of pHGG. Studies exploring combination therapies, such as vaccines with radiation or immune checkpoint inhibitors, are essential to enhance immunogenicity and overcome tumor-induced suppression or intratumoral heterogeneity. Pediatric-focused clinical trials with biomarker-correlated endpoints and immune profiling will be critical to refine dosing, timing, and patient inclusion criteria. Advancements in delivery platforms, such as focused ultrasound-assisted delivery across the BBB, may further increase therapeutic penetration and efficacy.
Vaccine-based immunotherapy represents a promising yet still nascent strategy in the treatment of pediatric high-grade gliomas. While initial trials have demonstrated safety and immunogenic potential, clinical efficacy has yet to be conclusively proven. Addressing the biological, logistical, and ethical challenges unique to this population will require sustained multidisciplinary collaboration across institutions. With continued innovation and rigorous clinical validation, vaccine therapies may one day complement or even transform the therapeutic landscape for children facing these devastating tumors.