Full-length transcriptome of Camellia perpetua reveals candidate SCPL1A gene family members involved in galloylated catechins biosynthesis
Yongbiao Deng, Bo Wang, Jingjian Li, Chao Xiong, Baojiao Huang, Lisheng Wang, Bo Zhao

TL;DR
This study identifies genes in Camellia flowers that may be responsible for producing antioxidant-rich galloylated catechins.
Contribution
The study provides new insights into the SCPL1A gene family's role in galloylated catechins biosynthesis in Camellia flowers.
Findings
Full-length transcriptome sequencing identified 17 CpSCPL1A genes in Camellia perpetua flowers.
11 candidate CpSCPL1A genes were linked to the biosynthesis of four types of galloylated catechins.
The CpSCPL1A proteins were grouped into five clusters with conserved motifs.
Abstract
Catechins includ galloylated catechins and non-galloylated catechins, among which galloylated catechins exhibit stronger antioxidant, anti-inflammatory and anti-cancer activities. Section Chrysantha Chang, the only group of yellow Camellia with rich catechins in their flowers, is a common health drink in southern China. To date, few studies have examined galloylated catechins biosynthesis in flowers of this group. To enrich the genetic information of the galloylated catechins biosynthesis, the ONT sequencing platform was used to perform full-length transcriptome sequencing of C. perpetua flowers and 7,972,574 transcripts was identified, including 42,883 simple sequence repeats (SSRs), 41,961 coding sequences (CDSs) and 2,602 long non-coding RNAs (lncRNAs). 36,516 transcripts were successfully annotated, and 147 critical enzyme-encoding genes were identified as involved in the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
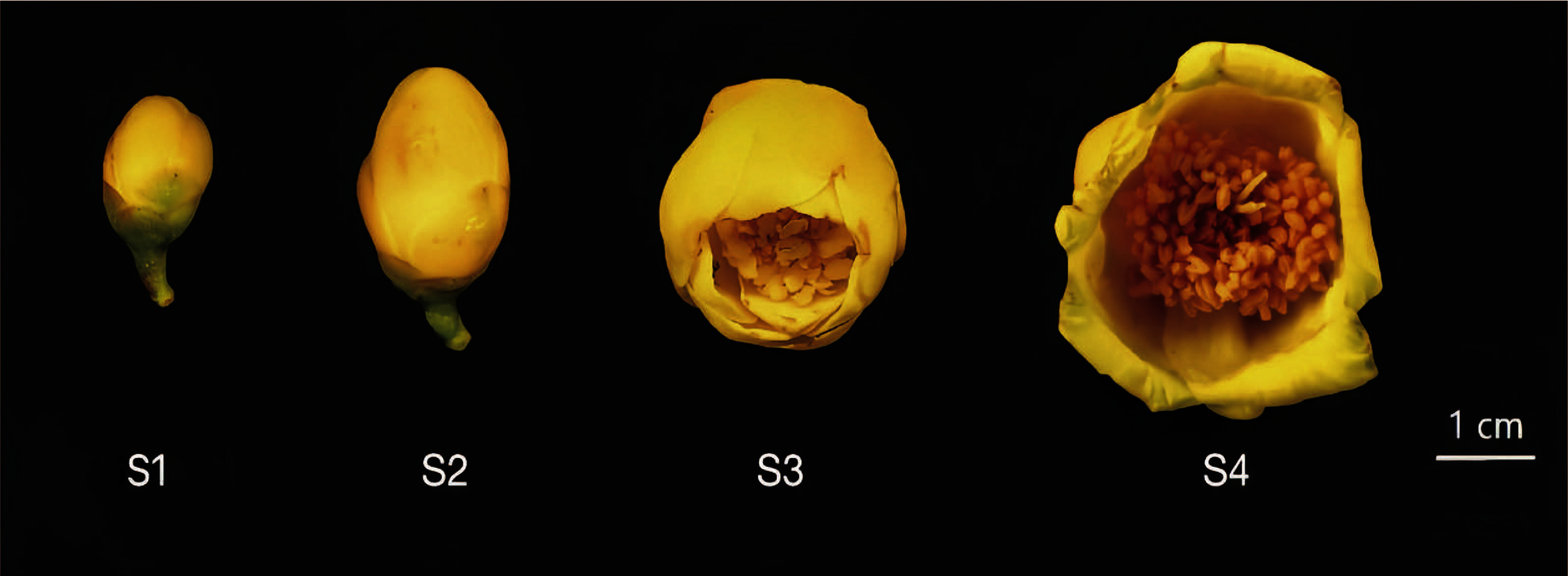 Figure 1
Figure 1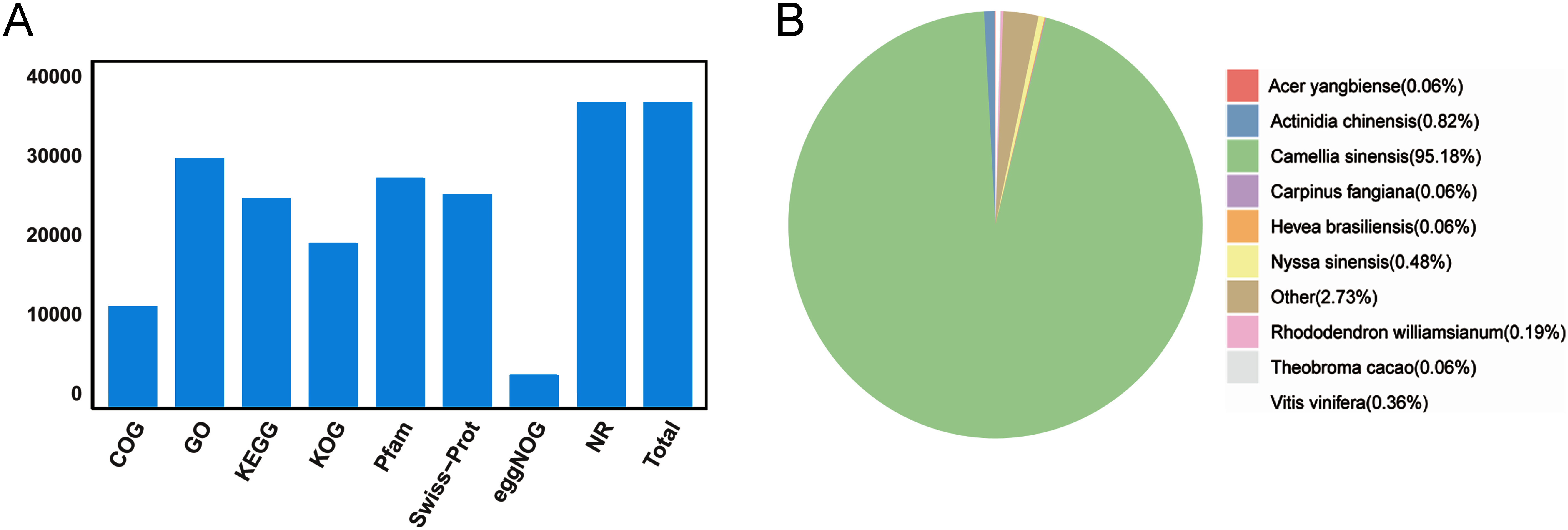 Figure 2
Figure 2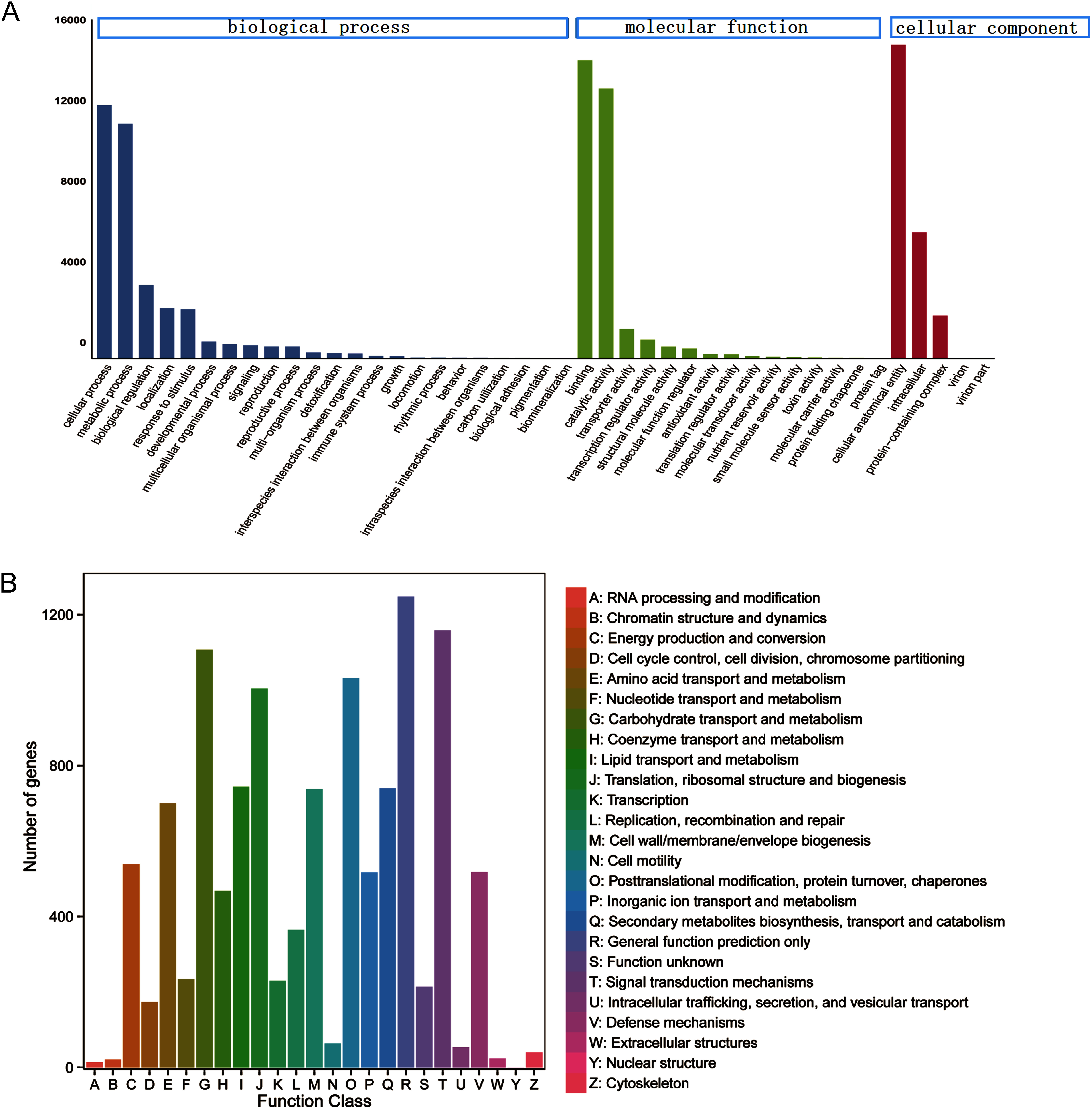 Figure 3
Figure 3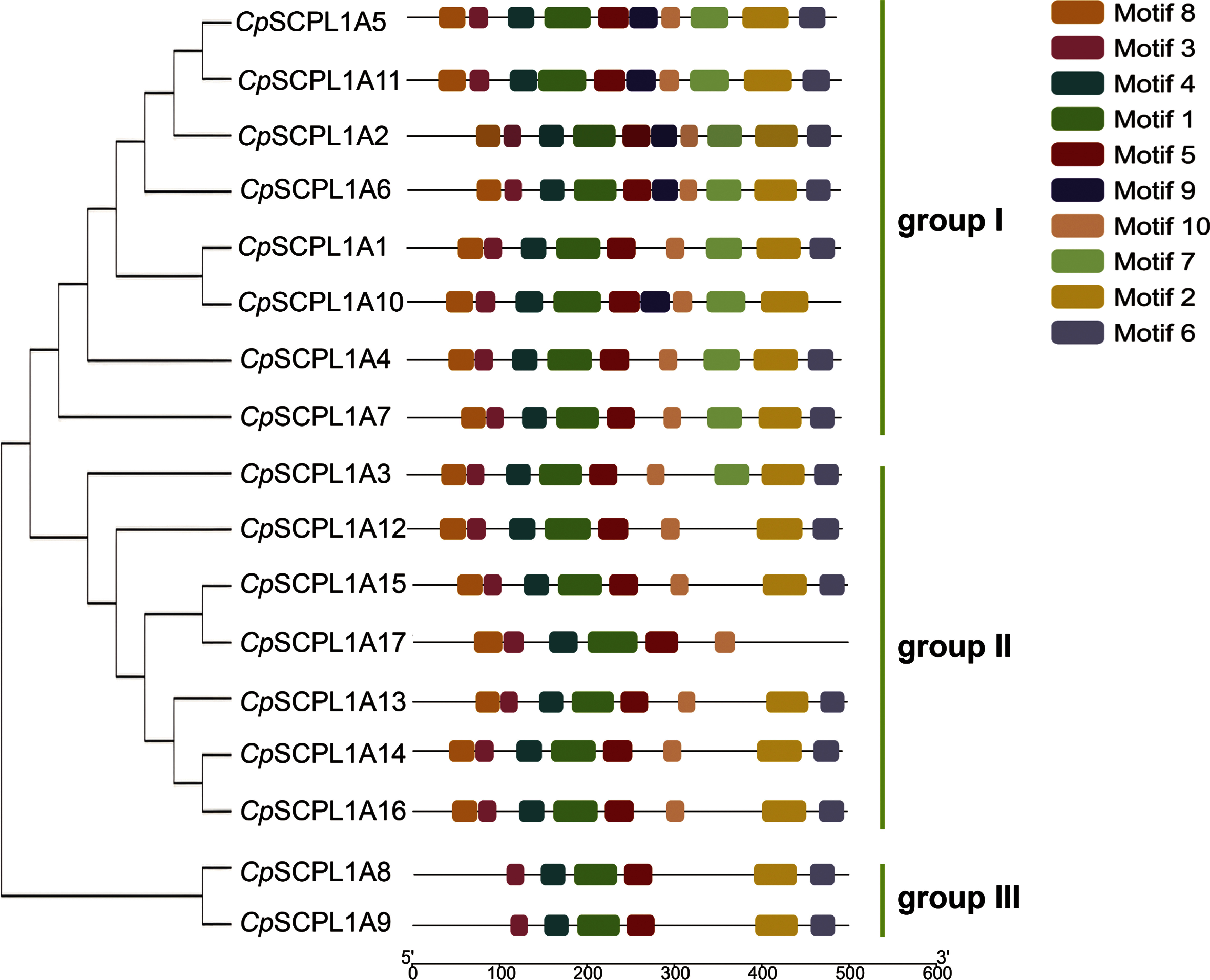 Figure 4
Figure 4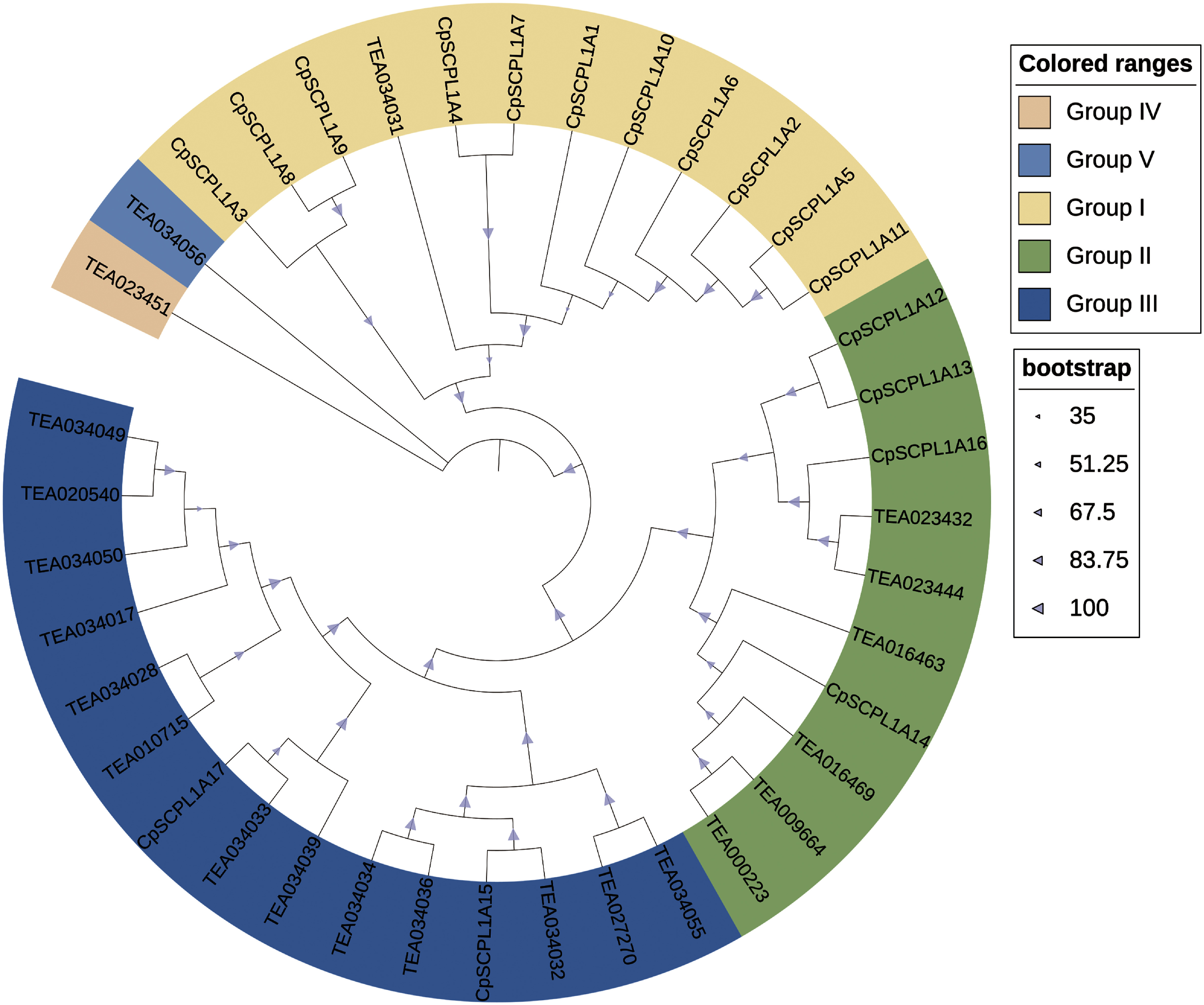 Figure 5
Figure 5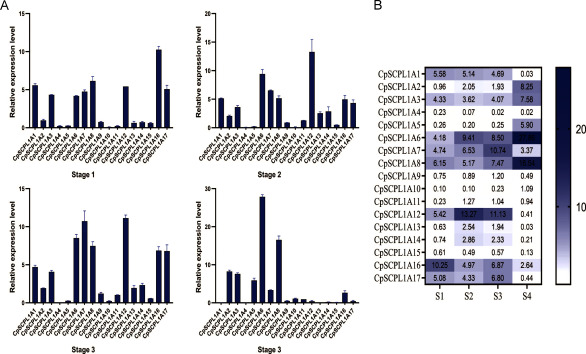 Figure 6
Figure 6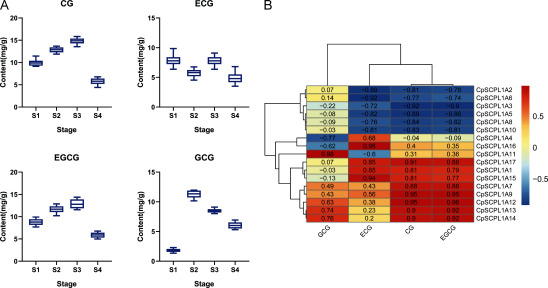 Figure 7
Figure 7| Data output | Number |
|---|---|
| Subread bases (Gb) | 38.30 |
| Subread number | 9,833,772 |
| Average subread length | 1,385 |
| Consensus N50 | 1,695 |
| FLNC | 7,972,574 |
| Consensus number | 90,245 |
| MaxLength | 9,574 |
| Transcripts | 57,487 |
| SSR | 42,883 |
| LncRNA | 2,602 |
| Gene name | Peptide lengths | Isoelectric point | Instability index | Subcellular localization | Alpha helix% | Extended strand% | Beta turn% | Random coil% |
|---|---|---|---|---|---|---|---|---|
| 480 | 6.83 | 39.18 | Vacuole | 31.04% | 14.17% | 6.46% | 48.33% | |
| 500 | 6.06 | 39.06 | Vacuole | 33.60% | 14.40% | 6.40% | 45.60% | |
| 496 | 5.34 | 33.76 | Peroxisome | 32.06% | 14.31% | 5.65% | 47.98% | |
| 477 | 6.19 | 37.46 | Vacuole | 30.40% | 14.88% | 6.50% | 48.22% | |
| 454 | 5.78 | 37.99 | Vacuole | 29.96% | 15.20% | 7.49% | 47.36% | |
| 496 | 6.96 | 33.70 | Vacuole | 30.65% | 13.71% | 5.85% | 49.80% | |
| 502 | 5.72 | 36.45 | Peroxisome | 32.67% | 14.14% | 4.98% | 48.21% | |
| 446 | 6.31 | 40.74 | Vacuole | 30.94% | 15.02% | 6.95% | 47.09% | |
| 463 | 4.84 | 45.90 | Vacuole | 29.37% | 15.77% | 6.26% | 48.60% | |
| 506 | 5.43 | 42.89 | Vacuole | 34.78% | 14.23% | 5.53% | 45.45% | |
| 470 | 5.27 | 40.97 | Vacuole | 29.57% | 15.74% | 7.23% | 47.45% | |
| 483 | 5.26 | 40.38 | Vacuole | 28.78% | 15.73% | 6.21% | 49.28% | |
| 480 | 5.03 | 38.35 | Vacuole | 30.83% | 15.21% | 6.25% | 47.71% | |
| 427 | 5.32 | 39.70 | Vacuole | 33.72% | 15.46% | 4.68% | 46.14% |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTea Polyphenols and Effects · Microbial Metabolism and Applications · Tannin, Tannase and Anticancer Activities
Introduction
The Sect. Chrysantha Chang belongs to the Camellia and is distinct for its unique yellow flowers (Jiang et al. 2023). Predominantly found in Guangxi and Yunnan, China and parts of Vietnam, this rare and endangered golden-yellow Camellia has been termed “Giant Panda of the Plant Kingdom” and the “Queen of Camellia” (Wu et al. 2020). C. perpetua, a species of the Sect. Chrysantha Chang is only distributed in Chongzuo of Guangxi, China and adjacent northern Vietnam. It is the only yellow Camellia that blossoms year-round, thereby captivating the interest of the horticultural community (Yu et al. 2023). Flowers of C. perpetua with abundant flavonoids compounds, exhibit anti-aging, antihypertensive, hypolipidemic, and anti-allergic effects and are routinely utilized to treat hypertension, malignant tumors, nephritis, dysentery, sores and irregular menstruation (Li et al. 2020; Qi 2016).
Flavonoids originate from the phenylpropanoid pathway, with the catechins biosynthesis pathway as a branch of this pathway (Supplementary Figure S1). Catechins serve as the terminal product of the flavonoid biosynthesis pathway in Camellia (Rani et al. 2012; Xiong et al. 2013). Clinical studies have demonstrated the myriad benefits of catechins for human health, including antioxidant effects on cardiovascular wellness, inhibiting the growth of cancer cells, preventing blood clot formation and reducing platelet aggregation and lipid regulation (Johnson et al. 2012; Sinija and Mishra 2008). Catechins, a subgroup of flavan-3-ols, are typically classified into seven categories: (+)-catechins (C), (−)-epicatechin (EC), (+)-gallocatechin (GC), (−)-epigallocatechin (EGC), (−)-epicatechin-3-gallate (ECG), (−)-epigallocatechin-3-gallate (EGCG) and (−)-gallocatechin gallate (GCG) (Tang et al. 2009; Wei et al. 2018). Among them, non-galloylated catechins, including C, EC, GC and EGC, are synthesized under the regulation of enzymes such as chalcone synthase (CHS), chalcone isomerase (CHI), dihydroflavonol reductase (DFR), leucoanthocyanidin reductase (LAR) and anthocyanidin reductase (ANR) (Guo et al. 2023; Punyasiri et al. 2004; Xiong et al. 2013). Finally, non-galloylated catechins (C, EC, GC and EGC) are enzymatically converted into galloylated catechins (CG, ECG, EGCG, GCG) by type 1A serine carboxypeptidase-like acyltransferases (SCPL1As), galloylated catechins exhibit enhanced biological activities compared to their non-galloylated counterparts, including antioxidant, anti-inflammatory and anti-cancer activities, and these attributes make them valuable for applications in the pharmaceutical, food and cosmetic industries (Baranwal et al. 2022; Huang F et al. 2022). However, the role of the SCPL1A gene family in galloylated catechins biosynthesis of yellow flowers of C. perpetua is still not well understood, although many flavonoids biosynthesis related genes have been reported in flowers of Sect. Chrysantha Chang (Peng et al. 2011).
The genome of C. perpetua remains unavailable, severely limiting the exploitation of gene families. Oxford Nanopore Technologies (ONT) sequencing is a versatile platform widely used for full-length transcriptome sequencing as well as whole-genome shotgun sequencing and other applications. Compared to PacBio-SMRT sequencing technology, ONT sequencing technology offers the advantage of generating exceptionally long read lengths (up to 2 M base pair), which enhances the detection of complex genomic features. Moreover, ONT sequencing eliminates the need for PCR amplification, minimizing the risk of amplification-induced errors. ONT sequencing has emerged as a powerful tool in the study of gene families, gene expression, metabolic pathways, and genetic diversity across a spectrum of medicinal plants (Jiang et al. 2023; Mestre-Tomás et al. 2023; Park et al. 2018; Petersen et al. 2019), including Iris lactea var. chinensis (Ni et al. 2021), Platycodon grandiflorus (Yu et al. 2021) and Pogostemon cablin (Chen et al. 2019).
In this study, we conducted ONT sequencing to obtain full-length transcriptome of C. perpetua flowers at four distinct developmental stages, subsequently screened and identified CpSCPL1A gene family Members to analyze their genetic information, conservative domain, evolutionary relationships, and correlation analysis between the gene expression and the content of galloylated-type catechins. Our research generated a valuable dataset of full-length transcriptome for C. perpetua flowers and explored the critical CpSCPL1A gene family involved in galloylated-type catechins biosynthesis, thereby contributing to a deeper comprehension of the complex mechanisms for the flavonoid biosynthesis pathways in the Sect. Chrysantha Chang.
Materials and methods
Plant materials and sequencing
The plants of C. perpetua were cultivated in a nursery at the Guangxi Institute of Botany, Chinese Academy of Sciences (Guangxi, China, E 110°18′54″, N 25°4′18″). Flowers at each of four developmental stages were collected with three biological replicates (from three similarly sized plants cultivated under identical conditions) on June 23, 2023 (Figure 1). The samples were rapidly frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
Figure 1. Flowers at four flowering stages: early buds (S1), yellowing flower buds (S2), semi-blooming flowers (S3) and full-bloomed flowers (S4).
Total RNA was extracted using the Total RNA Extractor Kit (Trizol) (B511311, Sangon Biotech Co., Ltd., Shanghai, China) following the manufacturer’s protocol after the collected flowers were ground and mixed in liquid nitrogen. The purity, concentration, and integrity of RNA were assessed using a Nanodrop (NanoDrop Technologies, Wilmington, USA) and an Agilent 2100 bioanalyzer (Agilent Technologies Inc, California, USA); only samples with an RNA Integrity Number (RIN)>8 were used in subsequent experiments. mRNA was enriched using oligo (dT) magnetic beads and reverse transcribed into cDNA with the SQK-PCS114 kit (Oxford Nanopore Technologies, Oxford, UK) for library construction and the MightyScript Plus First Strand cDNA Synthesis Master Mix (with gDNA diggalloylated) kit (B639252, Sangon Biotech Co., Ltd., Shanghai, China) for qRT-PCR. Sequencing was performed on the PromethION-48 system at Biomarker Technologies (Beijing, China) to obtain the raw sequencing data. The RNA-seq data were submitted to the NCBI Sequence Read Archive (SRA) database (Accession number: SRR30151566, SRR32367465 and SRR32367466).
Transcriptomic analysis
Raw sequencing data were converted from fast5 to fastq format using GUPPY v4.2.2 (https://github.com/nanoporetech/minknow_api). Quality assessment and control were conducted with NanoComp and pychopper (https://github.com/nanoporetech/pychopper), Full-length reads were identified based on the presence of 5′ primers, 3′ primers, and poly-A tails, Chimeric reads were filtered out using NanoPack2, Error correction was performed, and the resulting high-quality reads were clustered to generate FLNC transcripts (De Coster and Rademakers 2023). These sequences are further processed and corrected using isONcorrect (Sahlin and Medvedev 2021). The processed data were then reconstructed using Lyric (https://github.com/guigolab/LyRic) to generate non-redundant transcripts. Protein-coding sequences were predicted using TransDecoder v5.7.1 (https://github.com/TransDecoder/TransDecoder) with default settings, where full-length transcripts longer than 100 amino acids were scored, and the highest-scoring ORF was selected to represent the coding sequence (CDS). Using BLAST, functional annotation of the non-redundant transcripts was performed by aligning them to several databases, including GO, KEGG, KOG, Pfam, SwissProt, COG, eggNOG and NR.
Simple sequence repeats (SSRs) were identified in all transcripts longer than 500 base pairs using MISA v1.0 with default parameters (Beier et al. 2017). Transcripts without protein-coding potential were identified as candidate lncRNAs using Pfam, CPAT v1.2 (https://rna-cpat.sourceforge.net/), CNCI v2.0 (https://github.com/www-bioinfo-org/CNCI) and CPC v1.0 (https://cpc.gao-lab.org/) (Kong et al. 2007). Finally, open reading frames (ORFs) in all candidate lncRNAs were predicted using EMBOSS getorf (Rice et al. 2000), and sequences over 100 base pairs were excluded from further analysis.
Identification of CpSCPL1A gene family in C. perpetua
SCPL1A protein sequences of C. sinensis and Arabidopsis thaliana were downloaded from the Tea Plant Information Archive 2.0 (TPIA, http://tpia.teaplants.cn/index.html) and The Arabidopsis Information Resource (TAIR, https://www.arabidopsis.org/) (Supplementaty Table S1) (Berardini et al. 2015; Buchfink et al. 2015; Gao et al. 2024), respectively. A database was constructed using DIAMOND to search for potential CpSCPL1A protein sequences in C. perpetua transcripts (Buchfink et al. 2015). HMMER was utilized to download and build a hidden Markov model (HMM) with SCPL1A protein sequences from C. sinensis and A. thaliana as background files (Tang 2024); candidate SCPL1A protein sequences were identified by aligning these with C. perpetua transcripts (Potter et al. 2018). By integrating results from DIAMOND (Buchfink et al. 2015) and HMMER, CpSCPL1A proteins were identified. Additionally, GO annotations for CpSCPL1A proteins were retrieved using STRING (https://string-db.org/) (Szklarczyk et al. 2023). The gfanoo software was used to identify other key genes involved in the flavonoid synthesis pathway.
Physicochemical properties and motif analysis of CpSCPL1A proteins
The Expasy tool (https://web.expasy.org/protparam/) was utilized to analyze the number of amino acids (aa), theoretical pI, instability index (II), and other physicochemical properties of the CpSCPL1A proteins (Huang et al. 2023). The NPSA server (https://npsa.lyon.inserm.fr/cgibin/npsa_automat.pl?page=/NPSA/npsa_sopma.html) was employed to determine the proportions of alpha helices, beta turns, extended strands and random coils in the CpSCPL1A proteins (Deléage 2017). The Plant-mPLoc tool (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/) was employed to predict the subcellular localization of CpSCPL1A proteins (Chou and Shen 2010). Additionally, the MEME suite (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/) identified motifs within the CpSCPL1A proteins, with parameters set to default except for the number of motifs, which was set to 10, Tbtools was used for motif visualization (Chen et al. 2020).
Phylogenetic analysis of CpSCPL1A proteins
The CpSCPL1A protein sequences of C. sinensis were downloaded from the Tea Plant Information Archive 2.0 (TPIA, http://tpia.teaplants.cn/index.html) (Supplementary Table S2). The local pair algorithm in the MAFFT was employed to align these sequences (Rozewicki et al. 2019). The resulting alignments were trimmed using TrimAI software (Capella-Gutiérrez et al. 2009). A phylogenetic tree of CpSCPL1A proteins was constructed using the neighbor-joining (NJ) method in MEGA-X, with 1,000 bootstrap replicates (Kumar et al. 2018). Finally, iTOL (https://itol.embl.de/itol.cgi) was employed to visualize and annotate the phylogenetic tree (Letunic and Bork 2021).
qRT-PCR analysis
The expression level of CpSCPL1A genes was detected via qRT-PCR using pre-synthesized cDNA as a template. qRT-PCR analysis was conducted using RotorGene Q (Qiagen, Beijing, China). Specific primers for CpSCPL1A genes were designed using Primer Premier 5.0 software (Supplementary Table S3), with the 18S gene as the housekeeping gene for normalization. Each 20 µl reaction consisted of 10 µl of 2X SGExcel FastSYBR Mixture, 0.4 µl each of 0.4 µM forward and reverse primers, 2 µl of 50 ng cDNA and 7.2 µl of RNase-Free ddH2O. Amplification conditions were: initial denaturation at 95°C for 3 min, followed by 40 cycles of 95°C for 5 s and 60°C for 20 s. Fluorescence signals were collected in the FAM channel following the annealing/extension step, and melting curve analysis was performed at the end. Gene expression levels were quantified using the 2^−ΔΔCt^ method (Livak and Schmittgen 2001). Each experiment was conducted using three independent biological replicates and three technical replicates.
Extraction and HPLC analysis of galloylated catechins
Galloylated catechins were quantified using high-performance liquid chromatography (HPLC). Flowers at various developmental stages were frozen and uniformly ground in liquid nitrogen. A 0.1 g sample (accurate to 0.0001 g) was precisely weighed and transferred to a 10 ml centrifuge tube. Subsequently, 10 ml of preheated (70°C) 70% methanol solution was added. The mixture was homogenized and incubated in a metal bath at 70°C for 10 min. After cooling to room temperature, the mixture was centrifuged at 3,500 rpm for 10 min, and the supernatant was collected. The residue was re-extracted using the same procedure, and the supernatants were combined and diluted to 10 ml with 70% methanol. The final solution was filtered through a 0.22 µm organic membrane and analyzed via HPLC. Galloylated catechins content was quantified using an Elite P1201 High-Performance Liquid Chromatograph (Elite Analytical Instrument Co., Ltd., Dalian, China) equipped with a reversed-phase C18 column (4.6 mm inner diameter, 250 mm length, 5 µm particle size) (Agilent Technologies, USA). UV detection was carried out at 278 nm, with the column temperature at 35°C. The injection volume was 10 µl, gradient elution at a 0.5 ml/min flow rate. Mobile phase A comprised 8% acetonitrile, 2% acetic acid and 90% pure water, while mobile phase B contained 90% acetonitrile, 8% pure water and 2% acetic acid. Standard solutions for EC, GCG, EGCG and ECG were prepared at 2.00 mg/ml concentrations (Shanghai Shifeng Biological Technology Co., Ltd, China). The HPLC chromatograms of the standard compounds and samples are shown in Supplementary Figure S2.
Results
Full-length transcriptome sequencing data output
After filtering out low-quality reads, Oxford Nanopore sequencing yielded 38.30 Gb of clean reads (Table 1). We obtained 7,972,574 full-length non-chimeric (FLNC) transcripts from these clean reads, representing 88.82% of the total. The average read length of transcripts, including 5′ primers, 3′ primers and poly (A) tails of FLNCs, was 1,163 base pairs (bp). Additionally, we acquired 90,245 consensus isoforms with an average length of 1,327 bp and an average quality score of 0.99, and the N50 length was 1,695 bp. Further sequence-structure analysis of the transcripts resulted in 57,487 unique transcripts with an average length of 1,385 bp. We also identified 41,961 coding sequences (CDSs), 42,883 simple sequence repeats (SSRs) and 2,602 long non-coding RNAs (lncRNAs) (Table 1).
**:
Functional annotation of transcripts and gene families related to galloylated catechins biosynthesis
To obtain transcript annotation, BLAST was used to align transcripts against the NR, SwissProt, GO, COG, KOG, Pfam and KEGG databases. A total of 36,516 transcripts were successfully annotated, with 29,508, 24,593, 18,948, 2,717, 25,092, 10,753, 2,361 and 36,444 aligned to the GO, KEGG, KOG, Pfam, SwissProt, COG, eggNOG and NR databases, respectively (Figure 2, Supplementary Table S4). Notably, 97.74% of unigenes were annotated, demonstrating extensive functional coverage. Further analysis of the NR database revealed the closest phylogenetic relationship with C. sinensis (95.18%), indicating high accuracy in the transcriptome assembly and annotation of C. perpetua. Analysis of the GO and COG annotations (Figure 3) revealed that 11,624 transcripts were classified under the “metabolic process” category in GO. In comparison, 740 transcripts were assigned to the “Secondary metabolites biosynthesis, transport and catabolism” category in COG. A hidden Markov model (HMM) search was conducted on the aforementioned transcripts and encoded proteins, resulting in the identification of numerous genes implicated in the biosynthesis of galloylated catechins. These include LAR, DFR, F3H and SCPL1A (Supplementary Table S5).
Figure 2. Annotated agave full-length genes in public databases (A) and species distribution based on Nr annotation (B).
Figure 3. GO classifications of annotated agave full-length genes (A) and KOG classes of annotated agave full-length genes (B).
Identification and protein structure analysis of CpSCPL1A genes
We used DIAMOND and HMMER comparison tools to ensure accurate sequence extraction. Seventeen CpSCPL1A genes with complete coding sequences (CDS) and open reading frames (ORF) were identified in the full-length transcriptome of C. perpetua and were named CpSCPL1A1 to CpSCPL1A17 (Table 2). The analysis of the physicochemical properties revealed minimal variation in the protein sequences encoded by the CpSCPL1A gene family. Their protein lengths ranged from 427 to 506 amino acids, with theoretical pI values between 4.84 and 6.96, indicating their acidic nature. Instability indexes ranged from 33.70 to 45.90, with CpSCPL1A9 being the highest at 45.90 and CpSCPL1A6 being the lowest at 33.70, suggesting general stability in CpSCPL1A proteins. Bioinformatic analysis of subcellular localization revealed that all proteins located in vacuoles, except for CpSCPL1A3 and CpSCPL1A7, which located outside the oxidoreductase. The secondary structure prediction for each member of CpSCPL1A gene family showed that the proportion of α-helix ranges from 28.78% to 33.72%, β-turns from 4.68% to 7.49%, and the remainder were composed of extended strands and random coils, indicating that the CpSCPL1A gene family exhibited a degree of conservation in their secondary structure. In addition, Using the gfnaoo software, we identified 13 4CL genes, 10 ANR genes, 5 CHS genes, 11 DFR genes, 11 PAL genes, and 1 UGT84 gene within the transcriptome dataset. These genes are known to play roles in the biosynthesis of diverse flavonoid compounds, including catechins.
**:
Motif analysis of CpSCPL1A proteins
To explore the evolutionary relationships and conserved motif distribution within the CpSCPL1A proteins in C. perpetua, we employed the MEME-Suite to analyze the conserved domains of 17 CpSCPL1A proteins. Ten conserved motifs are named motif 1–10 (Figure 4, Supplementary Table S6). Motif1, motif4, motif5, motif6 and motif8 were notably present in all CpSCPL1A proteins. These motifs were contiguous in the sequence, located near the N-terminus, suggesting their crucial role in CpSCPL1A protein function. Additionally, motif3 was found in all CpSCPL1A proteins except CpSCPL1A 10 and CpSCPL1A 17, whereas motif9 was absent only in CpSCPL1A 8 and CpSCPL1A 9. Phylogenetic analysis revealed that certain motifs are unique to phylogenetic groups, for instance, motif7 was exclusively found in all group I members, whereas motif10 was unique to group II (Figure 4). Compared to groups I and II, group III members had fewer motifs, specifically lacking motif2, motif7, motif9 and motif10, suggesting they may have some distinct biological functions.
Figure 4. Analysis of phylogenetic relationships and conserved motifs of 17 CpSCPL1A proteins.
Phylogenetic analysis of CpSCPL1A proteins
To elucidate the phylogenetic relationship within the CpSCPL1A proteins of C. perpetua, we constructed NJ tree including 17 CpSCPL1A proteins of C. perpetua and 22 CsSCPL1A proteins from C. sinensis (Supplementary Table S2). Phylogenetic analysis showed that all SCPL1A proteins could be categorized into Group I~V (Figure 5). Specifically, Group I comprised 11 CpSCPL1A proteins (CpSCPL1A 1–11) and 1 CsSCPL1A protein, while Group II included 4 CpSCPL1A proteins (CpSCPL1A 1–14, CpSCPL1A 16) and 6 CsSCPL1A proteins. Group III contains 2 CpSCPL1A proteins (CpSCPL1A 15 and CpSCPL1A 17) and 13 CsSCPL1A proteins. Groups IV and V consist of TEA023451 and TEA034056, respectively. Notably, proteins within the same group exhibited high sequence homology, suggesting they may shared similar functions.
Figure 5. Phylogenetic analysis of SCPL1A protein in C. sinensis and C. perpetua. The figure shows that different colors represent different groups, and the triangle size represents different bootstrap values.
The expression patterns of the CpSCPL1A gene at four flower development stages
Expression patterns of 17 CpSCPL1A genes in C. perpetua flowers at four developmental stages were analyzed (Figure 1). The results showed that all genes were expressed with significant variations in expression levels across different stages (Figure 6A, B). Two genes (CpSCPL1A4, CpSCPL1A16) were highly expressed at the Stage 1 (S1), while CpSCPL1A11, CpSCPL1A12, CpSCPL1A13, CpSCPL1A14 were highly expressed the Stage 2 (S2). Notably, threel CpSCPL1A genes (CpSCPL1A7, CpSCPL1A9, CpSCPL1A17) were significantly expressed at the Stage 3 (S3), and CpSCPL1A2, CpSCPL1A3, CpSCPL1A5, CpSCPL1A6, CpSCPL1A8, CpSCPL1A10 were significantly expressed at the Stage 4 (S4).
Figure 6. Relative expression levels of the CpSCPL1A gene at four flower development stages. Values represent the mean±SD, measured from at least three biological replicates. A: The relative expression of a single gene in different periods. B: Relative expression heat map of CpSCPL1A gene family at different developmental stages.
Combined analysis of RNAseq expression and metabolite data
We employed high-performance liquid chromatography (HPLC) to assess the contents of galloylated catechins across S1 to S4 (Figure 7A). The results showed that the contents of CG and EGCG initially increased, reached its peak at S3, and then decreased at S4. The content of ECG decreased at S2, increased at S3, and subsequently decreased at S4. The content of GCG increased from S1 to S2 but then continuously declined.
Figure 7. A: Relative content of 4 galloylated catechins at four flower development stages (The sample size of each stage is 25). B: Co-expression analysis of SCPL1A genes and 4 galloylated catechins. S1–S4, representing 4 stages of flower development.
To investigate the role of the CpSCPL1A gene family in galloylated catechins biosynthesis, correlation analysis was performed between the quantitative fluorescence results of 17 CpSCPL1A genes and the content of galloylated catechins (Figure 7B). ECG content was positively correlated with 5 genes (CpSCPL1A7, CpSCPL1A9 and CpSCPL1A15–17), while negatively correlated with CpSCPL1A6. GCG content was positively associated with 5 genes (CpSCPL1A1, CpSCPL1A11–14), whereas showed a negative relation with 3 genes (CpSCPL1A3, CpSCPL1A8 and CpSCPL1A10). Interestingly, CG and EGCG content exhibited same regulatory pattern, with both being positively correlated with 8 genes (CpSCPL1A1, CpSCPL1A9, CpSCPL1A12–17) and negatively correlated with 6 genes (CpSCPL1A2, CpSCPL1A3, CpSCPL1A5, CpSCPL1A6, CpSCPL1A8, CpSCPL1A10). No significant correlations were observed for the remaining genes.
Discussion
The transcriptomic profile serves as a vital bridge between the genomic blueprint and the biological activities governed by the proteome (Ayadi et al. 2018; Satam et al. 2023). Full length transcriptome sequencing, mainly through Oxford Nanopore Technology (ONT), compared to PacBio-SMRT sequencing technology, Oxford Nanopore Technologies (ONT) offers the advantage of generating exceptionally long read lengths, which enhances the detection of complex genomic features (Jain et al. 2016; Lu et al. 2016; Sigurpalsdottir et al. 2024). In this study, ONT sequencing was used to obtain the full-length transcriptome of C. perpetua flowers at four developmental stages, 38.30 Gb of clean data and a total of 7,972,574 full-length non-chimeric (FLNC) sequences were generated (Table 1). A comparative analysis with the full-length transcriptome of C. nitidissima petals using PacBio SMRT sequencing revealed that the number of unique transcripts obtained (45,372) was significantly lower than that (57,487) of C. perceptua. In C. nitidissima, 43,073 coding sequences (CDS) were identified, most ranging from 1,500 to 2,500 bp, in contrast, C. perpetua exhibited 41,961 CDS, primarily concentrated between 300 and 2,400 bp. This discrepancy may be attributed to differences in the sample material provided for sequencing.
After eliminating redundancies, we obtained 57,487 unique full-length transcripts (Table 1). Further analysis of Gene Ontology (GO) and Clusters of Orthologous Groups (COG) (Figure 3) annotations revealed a significant enrichment of genes associated with specialized metabolite biosynthesis. Delving deeper into the transcripts pertinent to these categories, we pinpointed 147 pivotal enzyme-encoding genes that contribute to the galloylated catechins biosynthesis pathway, including 17 CpSCPL1A genes termed CpSCPL1A1 to CpSCPL1A17 (Table 1). Most CpSCPL1A genes in C. perpetua exhibit analogous conserved motifs (Figure 4), such as motifs 1, 4, 5, 6 and 8 detected in all members, suggesting a highly conserved protein structure. Nonetheless, the precise functions of many of these conserved motifs remains unknown. A conserved motif of the plant gene family is a DNA sequence with specific functions highly preserved across various family members and intimately associated with gene expression regulation, protein structure, and function. The presence of a conserved motif may facilitate the RNA spliceosomes in recognizing splice sites, thus ensuring the accurate splicing of RNA (Liu et al. 2018).
Varying counts of CpSCPL1A genes have been identified in different plant species, such as 19 genes found in Arabidopsis (Swarbreck et al. 2008) and 16 in maize (Li et al. 2015) (Supplementary Table S1). The Whole Genome Duplication (WGD) event led to the widespread expansion of the SCPL1A gene family in the C. sinensis. Among the 22 SCPL1A gene families identified in the C. sinensis genome, 4 originated from WGD events that occurred approximately 30 to 40 million years ago. At the same time, 15 were generated through recent, species-specific tandem repeat amplification, a critical factor contributing to the diversity of the galloylated catechins biosynthetic pathway in C. sinensis. The phylogenetic analysis of the SCPL1A gene family revealed that CpSCPL1A12, CpSCPL1A13, and CpSCPL1A16 in C. perpetua were closely related to the highly expressed genes TEA023444 and TEA023432 in C. sinensis flowers (Figure 5). CpSCPL1A1, CpSCPL1A2, CpSCPL1A4, CpSCPL1A5, CpSCPL1A6, CpSCPL1A10, and CpSCPL1A11 shared a close relationship with the highly expressed gene TEA034031 in C. sinensis apical buds, while CpSCPL1A17 was closely correlated to the highly expressed gene TEA034033 in C. sinensis apical buds and young leaves (Figure 5). Phylogenetic analysis offers a reliable approach to examining the correlation between sequence similarity and the functional attributes of proteins belonging to the same clade, and the proteins in the same clade often exhibit similar functions (Wani et al. 2021). The results of phylogenetic analysis in this study suggested that these 11 genes have potential significance as key genes correlated with galloylated catechins synthesis, and urther research is needed to determine their tissue expression patterns in C. perpetua.
Compared to other Camellia species, flowers of Sect. Chrysantha exhibits higher levels of flavonoids and their derivatives. These plant-derived flavonoids, including quercetin, daidzein, and catechins, are instrumental in plant growth and development. Flavonoid metabolites are intricately involved in regulating tissue interactions, defense mechanisms and adaptive responses to environmental changes (Dias et al. 2021). Galloylated catechins is at the forefront of flavonoid research, there are many gene families involved (Huang X et al. 2022). Among them, SCPL1A are key enzymes in synthesizing the galloylated catechins monomers EC, EGC, ECG, and EGCG. Notably, the downregulation of SCPL expression has been observed to reduce the contents of these galloylated catechins monomers in C. sinensis leaves (Falcone Ferreyra et al. 2012; Yao et al. 2023). Correlation analysis between structural genes involved in metabolic pathways and their respective metabolic products has emerged as a potent strategy for identifying key genes. We utilized Pearson’s correlation analysis to explore the association between galloylated catechins content and CpSCPL1A gene expression levels (Figure 7A, B). CG and EGCG content were both positively correlated with 8 genes (CpSCPL1A1, CpSCPL1A9, CpSCPL1A12–17), ECG content was positively related with 5 genes (CpSCPL1A7, CpSCPL1A9, and CpSCPL1A15–17), and GCG content was positively associated with 5 genes (CpSCPL1A1, CpSCPL1A11–14) (Pearson correlation coefficient >0.9, p<0.05, Figure 7B), indicating that gene expression of these 11 CpSCPL1A genes might be responsible for the uniquely high contents of production of galloylated catechins. Functional experimentation is needed to determine whether some or all of these SCPL genes are involved in catechins galloylatedification. In addition, transcription factors can regulate gene expression by binding to DNA (Han et al. 2016; Wang et al. 2018; Xu et al. 2018), which may further regulate the production of galloylated catechins in C. perpetua flowers. However, in C. perpetua flowers, the interactions between transcription factors, structural genes, and metabolite biosynthesis are complex, so more research is needed to explore their mechanisms of action.
Conclusions
In this study, we constructed the first full-length transcriptome of C. perpetua flowers, resulting in 38.30 Gb of sequencing data and the identification of 57,487 transcripts, including 42,883 simple sequence repeat (SSR) sites, 2,602 lncRNAs and 41,961 protein-coding sequences. Subsequently, 17 members of the CpSCPL1A gene family were identified, all containing conserved motifs 1, 4, 5, 6 and 8. Subcellular localization predictions suggest that CpSCPL1A proteins are localized in the vacuole and peroxisome. By the correlation analysis between the gene expression of 17 CpSCPL1A genes and the content of galloylated catechins at four developmental stages of C. perpetua flowers, we identified 11 key genes (CpSCPL1A1–2, CpSCPL1A4–6, CpSCPL1A10–13, CpSCPL1A17) involved in the biosynthesis of galloylated catechins. The findings of this study expanded the transcriptomic resources available for C. perpetua and provided valuable genetic information for the future identification of genes involved in the biosynthesis of galloylated catechins compounds in this species.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Ayadi L, Motorin Y, Marchand V (2018) Quantification of 2′-O-Me residues in RNA using next-generation sequencing (Illumina Ribo Meth Seq Protocol). In: Gaspar I (ed) RNA Detection. Methods in Molecular Biology. Humana Press, New York, pp 164910.1007/978-1-4939-7213-5_229130188 · doi ↗ · pubmed ↗
- 2Baranwal A, Aggarwal P, Rai A, Kumar N (2022) Pharmacological actions and underlying mechanisms of catechin: A review. Mini Rev Med Chem 22: 821–83334477517 10.2174/1389557521666210902162120 · doi ↗ · pubmed ↗
- 3Beier S, Thiel T, Münch T, Scholz U, Mascher M (2017) MISA-web: A web server for microsatellite prediction. Bioinformatics 33: 2583–258528398459 10.1093/bioinformatics/btx 198PMC 5870701 · doi ↗ · pubmed ↗
- 4Berardini TZ, Reiser L, Li D, Mezheritsky Y, Muller R, Strait E, Huala E (2015) The Arabidopsis Information Resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–48526201819 10.1002/dvg.22877 PMC 4545719 · doi ↗ · pubmed ↗
- 5Buchfink B, Xie C, Huson D (2015) Fast and sensitive protein alignment using DIAMNOND. Nat Methods 12: 59–6025402007 10.1038/nmeth.3176 · doi ↗ · pubmed ↗
- 6Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T (2009) trim AL: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25: 1972–197319505945 10.1093/bioinformatics/btp 348PMC 2712344 · doi ↗ · pubmed ↗
- 7Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R (2020) Tbtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant 13: 1194–120232585190 10.1016/j.molp.2020.06.009 · doi ↗ · pubmed ↗
- 8Chen X, Li J, Wang X, Zhong L, Tang Y, Zhou X, Liu Y, Zhan R, Zheng H, Chen W, et al. (2019) Full-length transcriptome sequencing and methyl jasmonate-induced expression profile analysis of genes related to patchoulol biosynthesis and regulation in Pogostemon cablin. BMC Plant Biol 19: 26631221095 10.1186/s 12870-019-1884-x PMC 6585090 · doi ↗ · pubmed ↗