Histone deacetylases in Duchenne muscular dystrophy: a role in the mechanism of disease and a target for inhibition
Mariarita Bertoldi, Emilio Albamonte, Luca Bello, Adele D’amico, Riccardo Masson, Vincenzo Nigro, Marika Pane, Chiara Panicucci, Maria Sframeli, Federica Ricci

TL;DR
This paper explores how overactive histone deacetylases (HDACs) contribute to Duchenne muscular dystrophy and suggests inhibiting HDACs as a potential treatment.
Contribution
The paper identifies HDACs as a novel therapeutic target in Duchenne muscular dystrophy by linking their overactivity to multiple disease mechanisms.
Findings
DAPC disassembly in DMD leads to HDAC overactivity and disrupted muscle signaling.
Increased HDAC activity causes chromatin tightening and suppressed muscle homeostasis genes.
HDAC inhibition is proposed as a strategy to counteract DMD-related muscle and immune pathologies.
Abstract
Aberrant activity of histone deacetylases (HDACs) is a pathological phenomenon in several diseases, including Duchenne muscular dystrophy (DMD). In DMD, the upregulation of HDACs is driven by the disassembly of the dystrophin-associated protein complex (DAPC), which, under normal physiological conditions, provides mechanical stability to muscle fibres and acts as a signalling hub anchoring signalling proteins and molecules to their functional sites. In dystrophic muscle, DAPC disassembly causes delocalisation of signalling proteins and, therefore, disrupts signalling pathways. Displacement of epigenetic signalling molecules leads to the uncontrolled activity of HDACs and excessive removal of acetyl groups from histone proteins. Consequently, chromatin becomes tightly bound, preventing the expression of genes involved in muscle homeostasis. The pathological consequences of increased HDAC…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
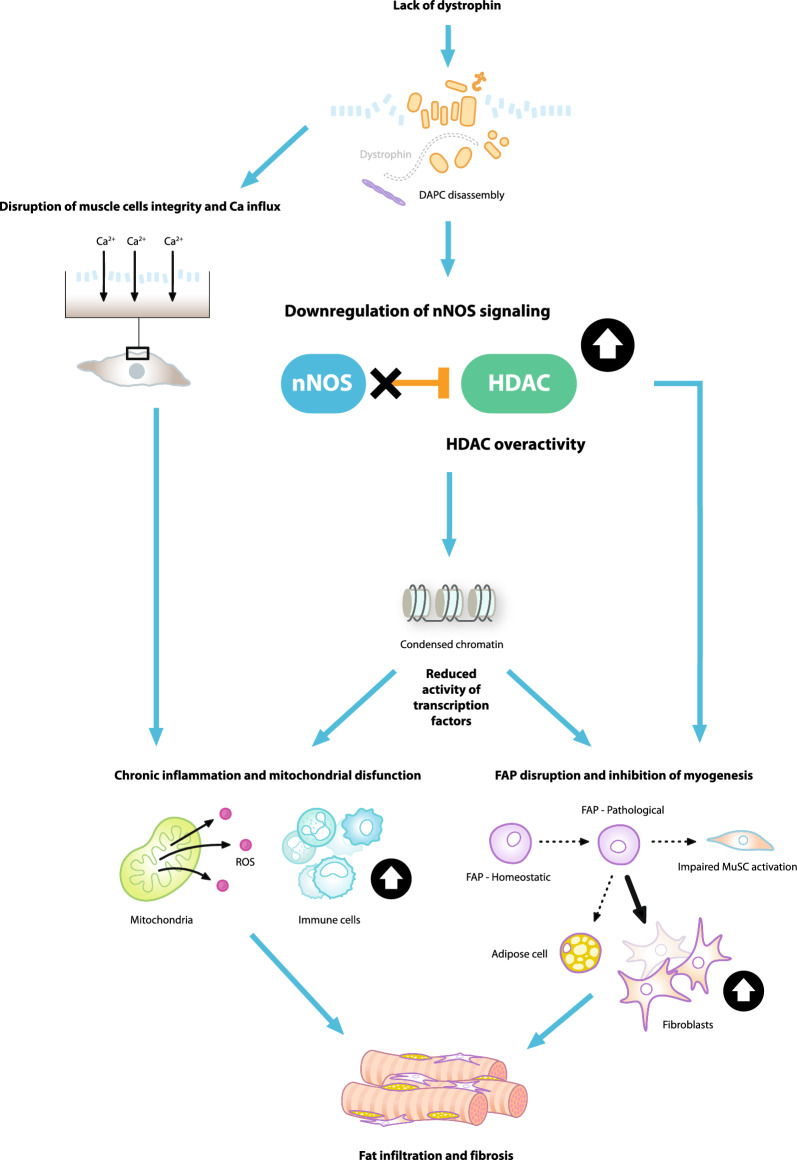 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMuscle Physiology and Disorders · Histone Deacetylase Inhibitors Research · Sirtuins and Resveratrol in Medicine
Introduction
Duchenne muscular dystrophy (DMD) is a severe disease characterised by progressive loss of muscle tissues [1]. The earliest symptoms present after the first years of life, with loss of ambulation typically occurring in the early teens [2] and the need for assisted ventilation at approximately 20 years of age [3]. Premature death occurs in most patients from 20 to 40 years of age from cardiac and/or respiratory failure [4].
DMD is an X-linked recessive disorder caused by mutations in the dystrophin gene that lead to a complete or near-complete loss of functional dystrophin protein expression [1]. This loss of functional dystrophin leads to disassembly of the transmembrane dystrophin-associated protein complex (DAPC) [5, 6]. The disassembly of this complex triggers a cascade of pathological events that include chronic inflammation and failed regeneration [5, 6]. One key consequence of dystrophin loss and DAPC disassembly is increased histone deacetylase (HDAC) enzyme activity [7, 8].
HDACs are evolutionarily conserved enzymes involved in the epigenetic regulation of gene expression that remove acetylated groups from the lysine of histone proteins [9]. HDACs have been implicated in skeletal muscle remodelling, both in physiological and pathological conditions [10]. HDAC hyperactivity has been observed in DMD animal models and patients with DMD [7, 11–13]. In particular, DMD muscles display an aberrant, constitutive activation of HDAC2 [7]. As such, the inhibition of HDAC activity has received attention as a therapeutic approach for treating muscular dystrophies [14].
DAPC disassembly leads to HDAC upregulation
Multi-functional role of DAPC
The dystrophin protein is an essential component of the DAPC, a highly organised multiprotein complex that plays a pivotal role in muscle fibre structural integrity [15]. The DAPC comprises several proteins, including dystrophin, syntrophins, dystroglycans, sarcoglycans and neuronal nitric oxide synthase (nNOS) [15]. Dystrophin acts as a bridge to connect the intracellular cytoskeletal filaments to the sarcolemma, stabilising the sarcolemma during muscle fibre contraction and relaxation. However, the DAPC also plays a key role in cell signalling, acting as a molecular scaffold to anchor a variety of signalling molecules [16].
DAPC signalling regulates HDAC activity
The muscle-specific nNOS enzyme, which produces intramuscular nitric oxide (NO) is anchored to the sarcolemma by the syntrophin subcomplex of the DAPC [17–19]. NO is a critical signalling molecule involved in regulating a variety of cellular processes involved in muscle tissue maintenance and repair [20]. For example, NO activates transient receptor potential cation channel, subfamily V, member 1 (TRPV1), increasing intracellular calcium and subsequently activating mammalian target of rapamycin (mTOR)-mediated signalling to drive muscle fibre growth [21].
Another key role of NO is to mediate the S-nitrosylation of HDACs via a post-translational modification where NO attaches a nitrosyl group to a cysteine residue in HDACs [7]. This modification inhibits the function of HDACs and plays a crucial role in regulating gene expression in muscle cells [7]. This gene regulation is dependent on the balance between the activities of histone acetyltransferases (HATs) and HDACs to essentially “switch on and off” target genes. HATs catalyse the addition of acetyl groups to histone proteins, inducing chromatin unravelling and triggering the transcription of genes coding for muscle repair factors. In contrast, HDACs remove acetyl groups from histones, rendering chromatin more compact and thereby inaccessible for transcription [20]. For example, HDAC2 inhibits follistatin gene transcription in mdx (dystrophin-deficient) muscle cells, which in turn blocks myostatin, an inhibitor of muscle growth [7, 22, 23]. It has also been suggested that HDAC2 effects on muscle could occur via epigenetic modulation of microRNA genes [24]. Decreased production of NO, therefore, leads to increased activity of HDACs, reductions of histone acetylation, more condensed chromatin structure and ultimately repressed expression of selected genes [20].
DAPC disassembly triggers aberrant HDAC activity
In DMD, DAPC disassembly disrupts the physical connection between the sarcolemma, cytoskeleton and the extracellular matrix. This causes the sarcolemma to become leaky and highly susceptible to injury [6, 25]. DAPC disassembly also leads to the mislocalisation of nNOS and the subsequent depletion of NO [17, 18]. The reduction in NO generation prevents NO-mediated S-nitrosylation and inhibition of HDACs, leading to an aberrant, constitutive HDAC hyperactivation [7].
Patients with DMD exhibit selective alterations of nNOS and catalytic activity from muscle membranes, contributing to muscle fibre degeneration [17]. In dystrophic (mdx) mice, these alterations have been shown to impair contractile function and cause muscle fatigue [26, 27]. Additional effects of the disassembly of DAPC include elevated reactive oxygen species, mitochondrial dysfunction and calcium dysregulation [25, 28].
The pathological consequences of HDAC hyperactivity
HDAC upregulation and consequent hyperactivity have been observed in a wide range of cell types and have a wide range of pathological consequences for muscle damage repair [10].
Mitochondria and muscle fibres
Mitochondrial dysfunction is one of the earliest deficits in mouse models of DMD [29, 30]. It is associated with a progressive impairment of mitochondrial biogenesis caused by epigenetic modifications and increased histone deacetylation of the peroxisome proliferator-activated receptor-gamma coactivator 1 α (PGC-1α) promoter [31]. The progressive increase in HDAC activity in DMD may explain the progressive deacetylation of the PGC-1a promoter [31]. Administration of the HDAC inhibitors givinostat and trichostatin A was shown to restore the physiological epigenetic profile on the PGC-1α promoter, improving mitochondrial biogenesis and enabling a positive fibre-type switch towards a more oxidative phenotype [31].
Fast-twitch fibres are preferentially affected in DMD, while slow-twitch (oxidative) fibres are relatively spared [32], suggesting that shifting skeletal muscle towards a slower, more oxidative phenotype may protect dystrophic muscles. Muscle fibre-type shifts are regulated by the HDAC-myocyte enhancer factor 2 (MEF2)-PGC-1α pathway [33, 34]. Class II HDACs act as repressors of MEF2, a key transcriptional activator for PGC-1α expression in skeletal muscle [33, 34]. Thus, HDAC inhibition may promote a transition to a slow/oxidative fibre type [33, 34].
Immune cells
Immune cell infiltration is a main feature of DMD and is strongly associated with disease severity [25]. In healthy muscle cells, pro-inflammatory macrophages clear damaged fibres after injury. These cell types are then replaced by anti-inflammatory macrophages that drive fibre regeneration [35]. The transition from a pro- to anti-inflammatory state is driven by regulatory T (Treg) cells (Foxp3 + Treg) [36]. In DMD, repeated injury causes the chronic inflammatory response to be maintained by overlapping pro-inflammatory and anti-inflammatory signalling, preventing the full resolution of inflammation [25].
HDACs are known to play a significant role in modulating immune responses, through epigenetic effects on various transcription factors. These include transcription factor nuclear factor kappa B (NF-κB), a master regulator of innate immune responses and essential for many macrophage functions [37–39], and Foxp3, the master transcription factor of Treg cells [39, 40]. Thus, the HDAC hyperactivity observed in DMD might be expected to affect the balance between pro-inflammatory and anti-inflammatory immune cell populations [41]. Of note, in addition to its role in modulating immune responses, the transcription factor NF-κB also plays a key role in various aspects of skeletal muscle biology [42], representing another means by which HDAC dysregulation may contribute to the pathophysiology of DMD.
Fibro-adipogenic cells/muscle satellite cells
Muscle satellite cells (MuSCs) are the principal contributors to the regeneration of injured muscles. MuSCs are skeletal muscle precursor cells that are activated in response to stress stimuli, such as injury [43, 44]. Activated MuSCs undergo asymmetric division and myogenic differentiation to repair muscle [43, 44]. HDACs regulate gene expression in MuSCs by controlling the activity of myogenic proteins such as MyoD and MEF2 [43, 45, 46], and so the hyperactivation of HDACs in DMD can impair the differentiation of satellite cells [43, 47].
Fibro-adipogenic cells (FAPs) also play an important role in muscle regeneration [48]. FAPs enhance the activation and differentiation of MuSCs to repair damaged muscle [49]. However, in DMD, FAPs differentiate primarily into fibroblasts or fat cells [50]. Studies in mdx mice have shown that HDACs determine the fate of FAPs via regulation of myogenic microRNAs [51]. In young mdx mice, HDAC inhibition was shown to induce the expression of core components of the myogenic transcriptional machinery, resulting in promyogenic differentiation while suppressing the fibro-adipogenic phenotype [51].
Therefore, both the altered differentiation process in the MuSCs and the loss of myogenic signals from FAPs contribute to making the MuSCs ineffective during the muscle repair and regeneration process in patients with DMD.
HDAC inhibition is a novel therapeutic approach in DMD
Although HDAC inhibitors have been in development for many years across several diseases, relatively few have been approved for use [14]. The discovery that the lack of dystrophin and DAPC disassembly results in constitutive HDAC activation in DMD muscles has led to efforts to explore HDAC inhibitors as a treatment strategy for DMD [14]. Considering that muscle damage and repair mechanisms are disrupted by overactive HDAC activity, suppressing this activity could enhance muscle repair and reduce the severity of DMD pathology.
Early studies in rodent and human muscle cells indicated that HDAC inhibitors could enhance muscle differentiation through follistatin expression and the recruitment and fusion of myoblasts into preformed myotubes [52, 53]. A subsequent seminal study showed that HDAC inhibitors increased the size of myofibres in dystrophin-deficient (mdx) mice by inducing the expression of the myostatin antagonist follistatin in satellite cells [23]. These studies inspired the clinical translation of HDAC inhibitors for the treatment of DMD.
Givinostat is a class I and II HDAC inhibitor that counteracts the pathogenic events downstream of dystrophin deficiency [54]. It has been approved in the United States, United Kingdom and Europe for the treatment of DMD in patients aged 6 years and older, regardless of the underlying genetic mutation [55–57]. Its potential therapeutic effects were first established in various preclinical studies. Givinostat was shown to reduce the production of pro-inflammatory cytokines in human peripheral blood mononuclear cells, and to exhibit anti-inflammatory effects in a mouse model of inflammation [58]. In mouse models of DMD, givinostat promoted the formation of muscles with increased cross-sectional area and reduced fibrosis and fatty infiltration, leading to improvements of muscle function as evaluated by treadmill and grip strength tests [59, 60].
Givinostat has shown a beneficial effect in patients with DMD over the course of several clinical trials. In a Phase 1 trial in healthy males, givinostat reduced the production of pro-inflammatory cytokines without affecting production of anti-inflammatory cytokines, with an accompanying dose-dependent, transient reduction in platelets [61]. In an open-label Phase 2 study investigating the effects of givinostat on histological disease progression in boys with DMD, treatment with givinostat significantly increased muscle fibre area fraction while reducing fibrosis, necrosis and fat deposition [62]. The EPIDYS Phase 3 study met its primary endpoint, with a significantly smaller decline in the time taken to perform the four-stair climb with givinostat (plus corticosteroids) compared with control (placebo plus corticosteroids), and showed favourable efficacy across secondary endpoints [63]. As measured by magnetic resonance spectroscopy, givinostat reduced new muscle fat fraction infiltration in the vastus lateralis by approximately 30% [63] and also significantly reduced new fat infiltration in all key muscle groups required for ambulation compared with control [64].
Studies of other HDAC inhibitors are ongoing. A recent study demonstrated beneficial effects of the HDAC6-selective inhibitor tubastatin A in the mdx mouse [65]. Inhibition of HDAC6 by tubastatin A was shown to increase muscle strength, improve microtubule and neuromuscular junction organisation and reduce muscle atrophy in mdx mice [65]. Rather than directly affecting the expression of individual genes as shown for other HDACs, HDAC6 acts on transforming growth factor-β signalling by targeting Smad3 in the cytoplasm [65]. Epigenetic small molecule screening in dmd zebrafish has also identified HDAC inhibitor candidates for further evaluation in DMD [66, 67].
Conclusions
DMD is an irreversible, progressive muscle-wasting disease caused by mutations in the dystrophin gene. Dystrophin is a core component of the DAPC, connecting actin filaments within muscle cells to the sarcolemma and acting as a shock absorber that stabilises the membrane during muscle contraction. Dystrophin regulates muscle repair, at least in part, via HDAC activity control. HDAC upregulation or hyperactivity is a major contributor to the pathology of DMD and, therefore, represents a therapeutic avenue in the treatment of DMD and possibly other neuromuscular diseases. HDAC inhibition influences pathophysiological and epigenetic mechanisms contributing to DMD and can have an important role structurally in recovering muscle fibre integrity, and biochemically in restoring signalling pathways.
For these reasons, the inhibition of HDAC offers a novel approach to slow down the progression of the disease with the aim to: (i) re-establish the production of anti-inflammatory proteins to prevent the immune system from being constantly activated and, thus, reduce chronic inflammation; (ii) reduce fibrosis and fat deposition by redirecting FAPs differentiation towards a myogenic path rather than differentiating into fibroblasts and adipose cells; (iii) promote muscle regeneration by restoring the ability of FAPs to produce growth factors needed to activate muscle stem cells which transform into new mature muscle fibres or repair damaged fibres. An improved understanding of the pathophysiological role of HDAC in DMD will provide insights into how pharmacological treatments can counteract the multiple and complex pathological events in DMD.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1US Food and Drug Administration. DUVYZAT (givinostat) oral suspension, Label. Available at https://www.accessdata.fda.gov/2024.
- 2European Medicines Agency. Duvyzat oral suspension, Summary of Product Characteristics. Available at https://www.ema.europa.eu/en/medicines/human/EPAR/duvyzat 2025.
- 3UK Medicines and Healthcare products Regulatory Agency. Duvyzat oral suspension, Summary of Product Characteristics. Available at https://products.mhra.gov.uk/2024.