Nutritional approaches in combating therapeutic resistance and enhancing treatment efficacy in cancer: the impact of ketogenic diets
Lei Wang, Pezhman Shafiei Asheghabadi, Saba Mashhadikhan, Sevda Nasirzade, Yeganeh Ettehad, Sepideh Gholamrezaie, Fatemeh Jafari, Maryam Rahmani, Shaghayegh Mehdizadeh, Neda Zali, Najma Farahani, Russel J. Reiter, Maliheh Entezari, Mina Alimohammadi, Afshin Taheriazam

TL;DR
This paper reviews how ketogenic diets may help fight cancer by targeting cancer cell metabolism and improving treatment effectiveness.
Contribution
The paper provides a review of the molecular mechanisms and translational potential of ketogenic diets in cancer therapy.
Findings
Ketogenic diets may suppress the PI3K/Akt/mTOR pathway and reduce IGF-1 signaling in cancer cells.
Combining KD with standard therapies could enhance cancer cell susceptibility to treatment.
Current evidence is limited to preclinical and small clinical studies, with a need for large RCTs.
Abstract
Cancer remains a major global health challenge, with therapeutic resistance significantly limiting treatment success. Traditional approaches, including surgery, chemotherapy, and radiotherapy, often encounter barriers such as metastasis and adverse effects on healthy tissues. The Warburg effect, which describes cancer cells’ reliance on glucose fermentation despite oxygen availability, has prompted investigations into alternative metabolic interventions. The ketogenic diet (KD), characterized by high fat intake and low carbohydrate intake, induces a metabolic shift that may selectively disadvantage malignant cells while preserving normal tissue function. While emerging evidence, including preclinical studies, early-phase trials, and limited clinical series, suggests that KD may help overcome treatment resistance by suppressing the PI3K/Akt/mTOR pathway, reducing insulin-like growth…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
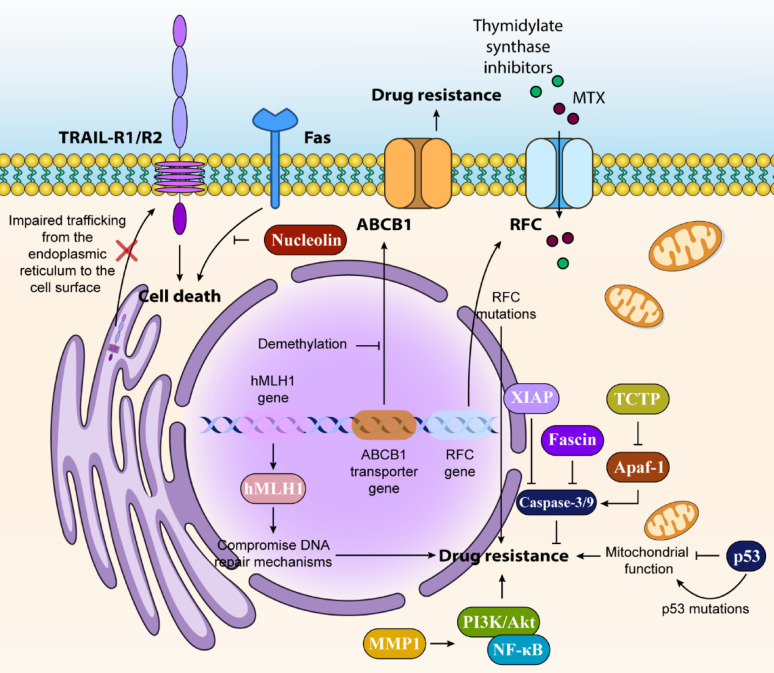 Figure 1
Figure 1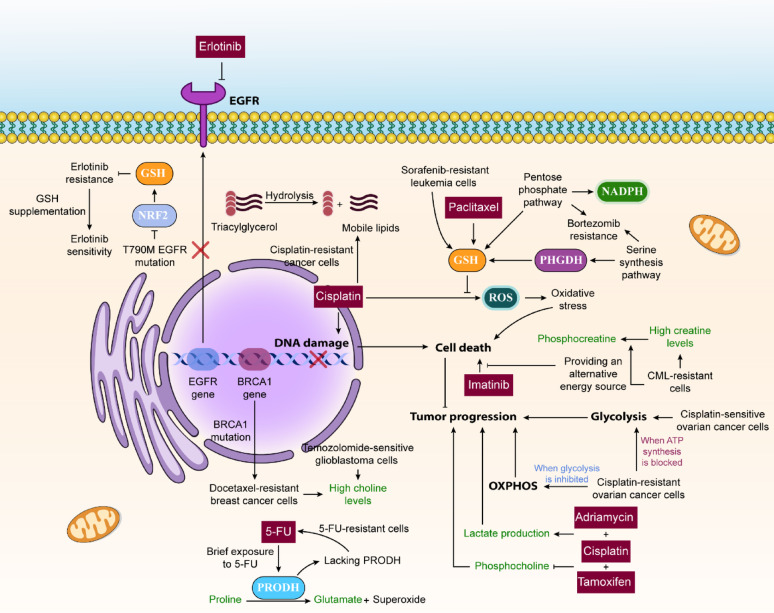 Figure 2
Figure 2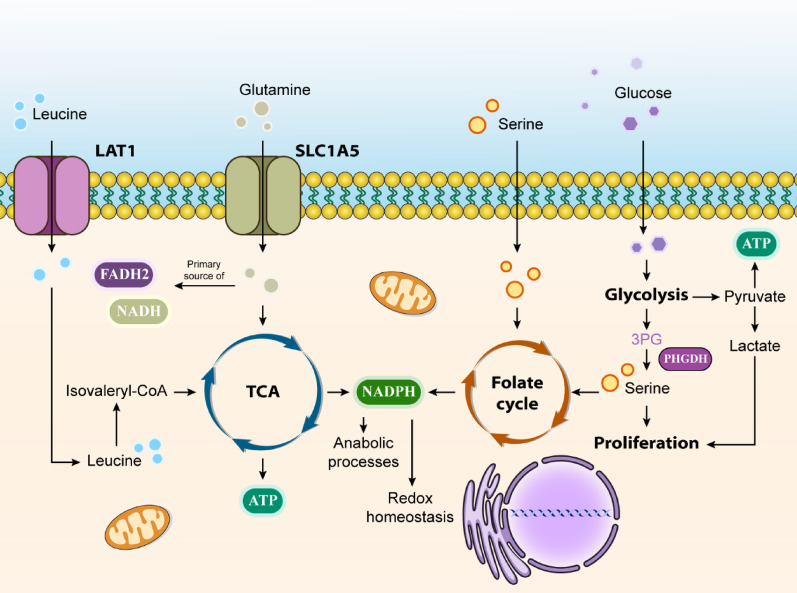 Figure 3
Figure 3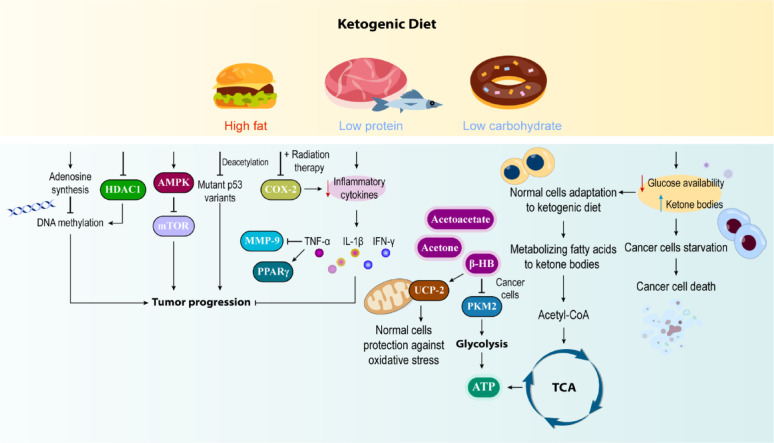 Figure 4
Figure 4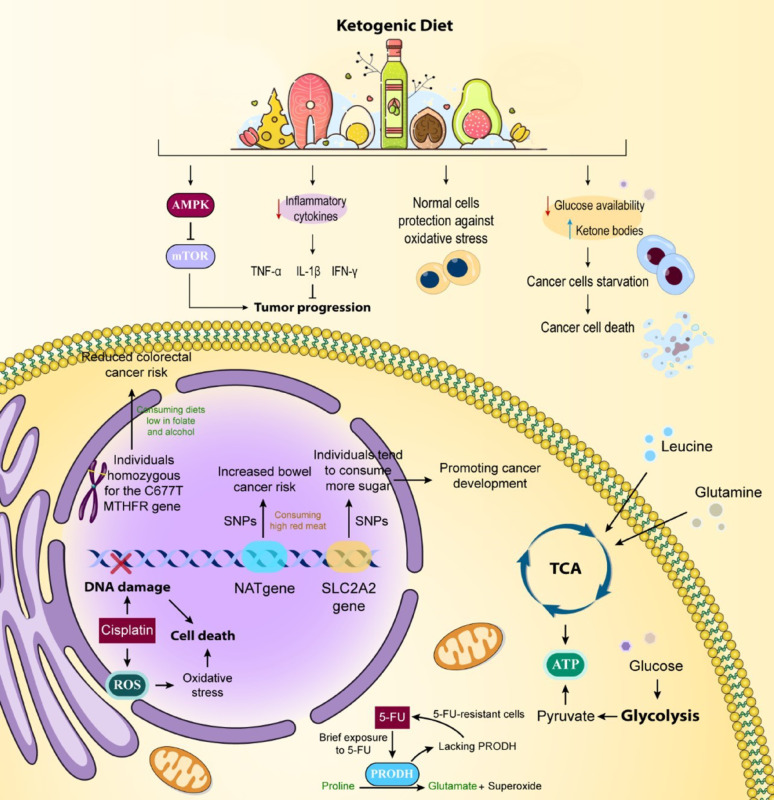 Figure 5
Figure 5Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDiet and metabolism studies · Dietary Effects on Health · Gut microbiota and health
Introduction
Cancer continues to be one of the most significant health challenges of our time, emerging as a leading cause of death worldwide, particularly as treatment resistance grows and incident rates increase [1]. Statistical data from 2020 reveals concerning figures in the United States: over 1.8 million new cancer diagnoses and approximately 606,000 deaths, with young people accounting for 89,500 cases and 9,270 fatalities annually [2]. While traditional treatment methods such as surgical procedures, chemotherapeutic drugs, radiotherapy, and targeted therapies like monoclonal antibodies and tyrosine kinase inhibitors have shown success in tumor control and elimination, they face significant limitations [3]. A breakthrough in understanding cancer cell metabolism came in the 1920 s when Otto Warburg identified the phenomenon now known as the Warburg effect [4]. This discovery revealed that cancer cells, unlike normal cells, predominantly rely on fermentation for energy production, even in oxygen-rich conditions, due to compromised mitochondrial function [5]. This metabolic adaptation supports the rapid production of building blocks necessary for cell division [6]. Based on this understanding, scientists have begun exploring nutritional approaches, particularly the ketogenic diets (KDs) and intermittent fasting protocols, as complementary therapeutic strategies that target cancer’s unique metabolic requirements [7–9].
A KDary approach, which consists of predominantly fats (80–90%), sufficient protein, and severely restricted carbohydrates (2–8%), promotes a metabolic transition from glucose-dependent energy production to mitochondrial fat oxidation [10]. This metabolic shift shows promise in cancer management by targeting malignant cells while preserving healthy tissue function. Research has shown that this dietary intervention reduces insulin-like growth factor-1 (IGF-1) and increases ketone body production, with studies in animal models demonstrating that limiting carbohydrate intake can impede cancer progression [11, 12].
Recent research indicates that KDs might help address the challenge of drug resistance in cancer treatment through various molecular mechanisms [13]. By restricting glucose availability and forcing metabolic adaptation, this dietary approach could potentially resensitize resistant cancer cells to chemotherapy agents [13]. Additionally, the diet’s suppressive effect on the PI3K/Akt/mTOR pathway might enhance the effectiveness of targeted therapies [14]. The diet-induced reduction in insulin and IGF-1 signaling could also help counteract survival pathways that typically contribute to treatment resistance [15]. While these mechanisms suggest promising potential for KDs in overcoming therapeutic resistance, definitive clinical evidence is still being gathered through ongoing research. This comprehensive review explores the KD’s development over time, examines cancer metabolism through Warburg’s pioneering insights, details the diet’s anti-cancer mechanisms, and evaluates both laboratory and clinical evidence supporting its use alongside conventional cancer treatments.
Therapeutic resistance in cancer: mechanisms and implications
Overview of therapeutic resistance
Cancer cells exhibiting drug resistance display distinctive epigenetic markers on their DNA, histones, and various regulatory elements involved in gene expression; research has identified specific epigenetic modifications, such as DNA hypermethylation patterns associated with tumor suppressor gene inactivation, as crucial indicators of cancer development [16, 17]. An illustrative example is the demethylation of the ABCB1 transporter gene, which reduces chemotherapy drug retention within cancer cells, resulting in multiple drug resistance [18]. Additionally, epigenetic modifications can compromise DNA repair mechanisms, as seen in the hypermethylation of hMLH1, contributing to colorectal cancer progression and treatment resistance [19].
The Food and Drug Administration (FDA) has authorized two categories of epigenetic-modifying medications: DNA methylation inhibitors (including 5-azacitidine and Decitabine) and histone deacetylase inhibitors (such as Vorinostat, Belinostat, Romidepsin, and Panobinostat) [20, 21]. While Decitabine may not directly inhibit tumor growth, its ability to block DNA methylation enhances tumor sensitivity to various chemotherapy agents, including carboplatin, cisplatin, and 5-FU [22]. In colorectal cancers, specific epigenetic signatures have been identified; early detection markers include methylation patterns in MDF1, SSTR2, and CMTM3, while CLDN11 hypermethylation correlates with metastasis and poor outcomes [23, 24]. TUSC3 silencing through promoter hypermethylation increases EGFR expression, promoting cancer cell survival [25]. Novel therapeutic approaches may include DNA methyltransferase inhibitors and histone deacetylase targeting drugs; recent research highlights the potential of new compounds like CUDC-101 as promising treatments for solid tumors [26]. MicroRNAs (miRNAs), short non-coding sequences of 19–25 nucleotides, regulate gene expression through post-transcriptional modifications. These epigenetic regulators significantly influence chemotherapy resistance development in various cancers [27]. Studies have revealed their impact on drug resistance genes, cell proliferation, cell cycle regulation, and apoptosis pathways, suggesting their potential as predictive markers for treatment response [27]. The catalog of cancer-associated miRNAs continues to expand.
Post-translational histone modifications (PTMs) are recognized by specialized protein domains: chromo-domains bind methylation, bromo-domains detect acetylation, BCRT domains recognize phosphorylation, and PHD domains interact with methylation marks. Chromatin remodeling complexes like SWI/SNF, ISWI, CHD, and INO80 further modulate chromatin structure. This interplay between DNA and histone modifications generates extensive non-genetic diversity, enabling precise biological control. Genomic analyses reveal frequent mutations in enzymes responsible for these modifications and remodeling factors, contributing to cancer development [28].
The establishment of tumor-specific epigenetic patterns not only triggers cancer formation but also enables the accumulation of additional genetic and epigenetic alterations, particularly those that enhance tolerance against therapeutic agents [29]. Introducing drug-free intervals may restore treatment sensitivity by allowing epigenetic resistance markers to revert. During these periods, non-genetically resistant cancer cells can be targeted using a combination of conventional treatments and epigenetic-modifying agents, such as HDACi, DNMTi, EPZ004777, and BET-I. This approach can transform drug-resistant epigenetic profiles back to drug-sensitive states, although genetically resistant cells may persist. A therapeutic strategy combining conventional and epigenetic drugs may effectively reduce tumor size while preventing the development of genetic resistance to targeted therapies [29, 30]. Cancer cells develop resistance to chemotherapy through various mechanisms, including enhanced drug efflux, reduced drug uptake, drug deactivation, modifications to drug targets, and resistance to cell death. Scientists are still uncovering the complex processes involved in how cancer cells transport chemotherapy drugs in and out of cells [31, 32]. One notable example involves the reduced folate carrier (RFC), which facilitates the uptake of drugs like methotrexate (MTX) and thymidylate synthase inhibitors. Research has shown that tumors with mutations affecting RFC function can become resistant to MTX. The impact of RFC on treatment outcomes is demonstrated in childhood acute lymphoblastic leukemia (ALL), where patients with the RFC genotype (80AA) show elevated plasma MTX concentrations, suggesting compromised drug uptake and increased mortality rates [33].
Cancer cells can resist treatment by circumventing programmed cell death mechanisms. Caspases, which are crucial enzymes in apoptosis, function by breaking down cellular components and activating secondary death pathways. The Bcl-2 protein family, comprising both pro-death and anti-death members, plays a vital role in cell survival decisions. In many cancers, anti-death proteins become overactive, enabling cells to survive chemotherapy exposure. Treatment resistance often stems from either increased anti-death signals or decreased pro-death protein function [34–37].
The translationally controlled tumor protein (TCTP) helps cancer cells survive by disrupting the Apaf-1 complex, thereby preventing caspase activation. High levels of TCTP often correlate with chemotherapy resistance due to its interference with cell death mechanisms, particularly in HeLa cells [38]. Another factor in treatment resistance involves the inhibitors of apoptosis proteins (IAPs), particularly X-linked IAP (XIAP), which shows the strongest anti-death effects through its ability to block caspase-9 activation [39]. Dysregulation of IAP proteins and NF-κB signaling in cancers contributes significantly to treatment resistance and poor outcomes, particularly in acute myeloid leukemia [40]. XIAP deficiency has been linked to X-linked lymphoproliferative syndrome type 2, suggesting that targeting IAPs with small-molecule inhibitors could potentially enhance cancer cell death [41].
In colon cancer cells, impaired trafficking of death receptors TRAIL-R1 and TRAIL-R2 from the endoplasmic reticulum to the cell surface creates resistance to TRAIL-induced death [42]. Numerous cancers exhibit dysfunction of TRAIL receptors, and alterations in decoy receptors provide another mechanism for evading cell death. For instance, gastric cancers often show elevated levels of the decoy receptor TRAIL-R3 [43]. Additionally, c-FLIP, a key anti-apoptotic regulator, can block caspase-8 recruitment and suppress chemotherapy-induced TRAIL-mediated cell death [44].
Various signaling molecules influence cell death pathways and alter cancer cells’ response to chemotherapy. The JNK and p38-MAPK pathways regulate multiple Bcl-2 family proteins, while JNK can activate PUMA and promote cell death in resistant cells through Akt/Fox03a signaling [45]. In breast cancer, the protein fascin promotes treatment resistance by increasing anti-apoptotic protein expression while suppressing pro-apoptotic proteins like caspases-3 and − 9 [46] (Fig. 1).
Fig. 1. Mechanisms and Implications of Cancer Therapeutic Resistance
Metabolic reprogramming in cancer and its role in resistance
Through metabolomic analysis, researchers have identified several key metabolic adaptations in resistant tumor cells responding to various chemotherapy treatments (Fig. 2). Cell survival and growth depend heavily on maintaining redox balance and antioxidant capabilities. Platinum-based drugs like cisplatin, widely used in cancer treatment, such as ovarian cancer, work by triggering cell death through DNA damage and oxidative stress from ROS [47–49]. Research shows that cisplatin-resistant lung cancer cells undergo metabolic reprogramming, leading to increased ROS production and greater reliance on oxidative metabolism rather than glycolysis. The elevated ROS levels and metabolic changes promote epithelial-mesenchymal transition (EMT) [47, 50, 51].
These resistant cells show increased glutamine consumption and vulnerability to glutamine restriction. They convert glutamine to glutamate for GSH synthesis, which helps reduce cellular ROS levels. Targeting glutamate metabolism specifically eliminates cisplatin-resistant cells [47]. While glutamine provides the glutamate and glycine components of GSH, cysteine comes from different sources. Cysteine, which provides GSH’s thiol group, requires both cellular uptake and limited internal production to maintain GSH availability. The thiol components from GSH, cysteine-glycine, and cysteine can neutralize platinum molecules, preventing them from damaging cellular components [52].
Cysteine plays a crucial role in cancer cell growth, survival, and metabolic adaptation. It influences how cancer cells, particularly ovarian cancer cells, adapt to low oxygen conditions, contributing to resistance against platinum-based chemotherapies. Controlling cysteine availability could potentially reverse both hypoxia and carboplatin resistance. Additionally, cysteine serves as an alternative pyruvate source for cancer cells, with its breakdown providing approximately 20% of cellular pyruvate [53].
Interestingly, in the case of erlotinib (an EGFR inhibitor), T790M EGFR mutation-induced resistance shows a different pattern. These resistant cells exhibit reduced GSH levels due to T790M mutation’s suppression of NRF2 activity, which controls GSH-synthesizing enzyme expression [54]. GSH supplementation restored erlotinib sensitivity in resistant cells, while GSH reduction in sensitive cells induced resistance. This unusual relationship between decreased GSH levels and EGFR tyrosine kinase inhibitor resistance warrants further investigation [55].
Research has demonstrated that bortezomib-resistant cells in multiple myeloma redirect glucose toward the pentose phosphate pathway and serine synthesis pathway, enhancing their antioxidant capabilities. The PPP generates NADPH, helping maintain GSH levels and cellular redox equilibrium. Additionally, elevated PHGDH activity in the serine synthesis pathway contributes to increased GSH production, promoting cell survival and bortezomib resistance [56, 57].
Regarding lipid metabolism adaptation, studies indicate significant metabolic reprogramming in chemotherapy-treated cancer cells, as lipid metabolism changes were observed in cisplatin-resistant bladder cancer through lipidomic analysis. Bladder cancer cells resistant to cisplatin showed distinct metabolites, primarily involving phospholipids, fatty acids, amino acids, and energy metabolism components [58].
Higher choline levels were detected in docetaxel-resistant BRCA1-mutated mammary tumors compared to sensitive ones [59]. In contrast, temozolomide-sensitive glioblastoma cells showed elevated choline and phosphorylcholine levels compared to resistant cells [60]. Generally, increased phospholipid levels correlate with drug resistance, though exceptions exist.
Cancer cells exhibit distinctive energy metabolism patterns as a fundamental characteristic, employing bioenergetic adaptations to survive drug treatments. Studies show that while cisplatin-sensitive ovarian cancer cells primarily utilize glycolysis, their resistant variants demonstrate metabolic flexibility. These resistant cells can either increase glycolytic activity when ATP synthesis is blocked or enhance oxidative phosphorylation (OXPHOS) when glycolysis is inhibited, showing their ability to alternate between these energy pathways [61].
Regarding glucose metabolism, cancer cells typically favor aerobic glycolysis. Research on sorafenib-resistant leukemia cells revealed increased glucose requirements and reduced pentose phosphate pathway (PPP) activity [62]. Studies on imatinib-resistant CML cells have yielded conflicting results. Initial research indicated increased glucose uptake and glycolysis in resistant BCR-ABL-positive cells during imatinib treatment, while later studies found decreased glucose consumption and lactate production [63]. The reason for these contradictory findings remains unclear. Studies of flavopiridol-resistant prostate cancer cells also demonstrated enhanced glycolysis and reduced sensitivity to cisplatin and docetaxel [64].
In terms of polyamine metabolism, these compounds play vital roles in eukaryotic cell growth. Significant increases in polyamine synthesis were observed in platinum-resistant ovarian cancer cells [65]. Research on colorectal cancer revealed that tumor-associated macrophages (TAMs) release putrescine when exposed to 5-Fluorouracil (5-FU), contributing to treatment resistance. Researchers found that inhibiting ornithine decarboxylase, crucial for putrescine production, reduced resistance to 5-FU and enhanced its anti-tumor effects [66]. The progressive shift of adriamycin-sensitive cells toward metabolic patterns similar to resistant cells provides evidence for metabolic adaptation during chemoresistance development [67].
Metabolic adaptations to chemotherapy appear to be dynamic processes. For instance, brief exposure of gastric cancer cells to 5-FU results in decreased proline and elevated glutamate levels, potentially linked to increased proline dehydrogenase (PRODH) activity. However, 5-FU-resistant cells show minimal changes in these metabolites, lacking PRODH upregulation after treatment. PRODH, which converts proline to glutamate while generating superoxide, becomes more active when 5-FU disrupts nucleotide synthesis. This process, involving mitochondrial superoxide production, contributes to resistance development [68].
Furthermore, combination therapies can produce unique metabolic effects. In breast cancer cells, combining cisplatin with tamoxifen decreases phosphocholine levels, while cisplatin plus adriamycin increases lactate production [69]. Although precisely predicting these metabolic changes remains challenging, researchers are working to identify plasma biomarkers that could indicate drug resistance in human epithelial ovarian cancer (Table 1) [70].
Table 1. The metabolic adaptations of drug-resistant cancer cellsMetabolic adaptationKey findingsDrug involvedCancer typeReferencesOxidative Stress AdaptationIncreased glutathione (GSH) synthesis to cope with oxidative stress.CisplatinOvarian, Lung Cancer [47, 51, 71]Cysteine uptake critical for GSH bioavailability and platinum resistance.CarboplatinOvarian cancer [53, 72, 73]Increased GSH levels in paclitaxel-resistant cells.Paclitaxel (Taxol)Triple Negative Breast Cancer [74]Downregulated GSH levels in erlotinib-resistant EGFR-mutated cells.ErlotinibEGFR-driven Cancers [54]Lipid Metabolism AdaptationHigher basal lipid content in cisplatin-resistant cells.CisplatinOvarian and Bladder Cancers [58, 71]Altered choline phospholipid metabolism in erlotinib-resistant cells.ErlotinibPancreatic Cancer (HPAC cells) [75]Increased choline levels in docetaxel-resistant BRCA1-mutated tumors.DocetaxelBreast Cancer [59]Upregulation of choline in temozolomide-sensitive GBM cells.TemozolomideGlioblastoma (GBM) [60]Bioenergetic AdaptationSwitch between glycolysis and oxidative phosphorylation (OXPHOS).CisplatinOvarian Cancer [61]Elevated creatine and phosphocreatine levels in imatinib-resistant cells.ImatinibChronic Myelogenous Leukemia [76]Dependence on Glucose/GlycolysisIncreased glucose demand and reduced PPP flux in sorafenib-resistant cells.SorafenibLeukemia [77]Contradictory glycolysis changes in imatinib-resistant CML cells.ImatinibChronic Myelogenous Leukemia [76, 78]Enhanced glycolysis in flavopiridol-resistant prostate cancer cells.FlavopiridolProstate Cancer [64]Polyamine SynthesisIncreased polyamine synthesis in platinum-resistant cells.PlatinumOvarian Cancer [49]Putrescine secretion by TAMs induces 5-FU resistance.5-Fluorouracil (5-FU)Colorectal Cancer [66]
Fig. 2A Schematic Representation of the Role of Metabolic Reprogramming in Cancer Drug Resistance
Warburg effect and cancer metabolism
The metabolic differences between cancer cells and normal tissue cells represent a fundamental characteristic of cancer. These extensive metabolic modifications are primarily triggered by oncogenic signaling pathways and modified metabolic enzymes, enabling cancer cells to meet their growth requirements in environments with variable nutrient availability. However, this adaptation creates a dependency on continuous nutrient and energy supplies, beyond the well-documented glucose metabolism alterations, leading to enhanced production of amino acids and fatty acids to support tumor growth [79, 80].
Cancer cells characteristically demonstrate elevated glucose consumption to generate intermediates essential for synthesizing lipids, proteins, and nucleic acids. They also maintain TCA cycle intermediate levels through increased glutamine uptake and metabolism. This heightened biosynthetic activity necessitates greater NADPH production, which supports both anabolic processes and cellular redox homeostasis. The transformation of cells during cancer development involves epigenetic changes dependent on various metabolites: acetyl-CoA (acetylation), NAD (deacetylation), SAM (methylation), α-ketoglutarate (demethylation), and UDP-GlcNAc (glycosylation). Modern cancer research has revealed that understanding genetic mutations alone is insufficient; cancer cells exist within complex tumor tissues, interacting with their microenvironment and developing characteristics that enhance their survival, growth, and spread [81–84] (Fig. 3).
Regarding glutamine metabolism, this most abundant plasma amino acid serves crucial roles in cancer cell proliferation. It provides nitrogen for nucleotide, amino acid, and hexosamine synthesis, while contributing both nitrogen and carbon to various cellular reactions. Glutamine is particularly vital as it acts as a precursor for non-essential amino acids (NEAAs) and fatty acid synthesis [85, 86].
The non-essential amino acid serine plays vital roles in cellular proliferation and survival. Cells can either synthesize serine from glycolysis-derived 3-phosphoglyceric acid (3PG) or acquire it from external sources [87]. As the third most utilized metabolite by cancer cells, after glucose and glutamine, serine provides one-carbon units to the folate cycle, supporting nucleotide synthesis, methylation processes, and NADPH generation for antioxidant defense [88]. Given cancer cells’ strong dependence on serine, therapeutic strategies targeting either de novo serine synthesis or external serine availability show promise. This has led to increased interest in phosphoglycerate dehydrogenase (PHGDH), a key enzyme in serine synthesis, as a potential therapeutic target [89].
Leucine, an essential branched-chain amino acid (BCAA), must be obtained through protein-rich foods such as meat, dairy, and legumes. This amino acid serves crucial functions in protein synthesis and various metabolic processes, including blood sugar regulation, muscle and bone tissue maintenance, and growth hormone production. As a ketogenic amino acid, leucine breaks down into ketone bodies - acetoacetate and β-hydroxybutyrate. Through its activation of mTOR and S6K1, leucine can interfere with insulin signaling, resulting in decreased glucose utilization in skeletal muscle tissue [90–92].
Leucine serves as an energy source when enzymatically converted to isovaleryl-CoA and utilized in the TCA cycle for ATP production. During tumor progression, cancer cells require alternative energy sources to sustain their rapid growth. Studies have documented elevated BCAA levels, including leucine, in the plasma of patients with pancreatic cancer and melanoma. Research indicates that leucine activates mTORC1 by providing acetyl-CoA to the EP300 acetyltransferase, which in turn influences Raptor acetylation at K1097. Many cancers rely on mTOR activity to maintain cellular growth and division [93–95].
Research indicates that limiting leucine availability may inhibit cell growth, promote apoptosis, and reduce FASN expression in breast cancer. The L-type amino acid transporter (LAT1) facilitates cellular leucine uptake; therefore, LAT1 inhibition leads to reduced mTOR signaling and tumor growth suppression. However, research also demonstrates the benefits of leucine-rich diets for cancer patients experiencing malnutrition and cachexia [96, 97].
Fig. 3. Cancer Metabolism: The Crosstalk between Metabolism of Organic Macromolecules and Cancer Progression
KDs: composition and biological impact
The KD, high in fat and low in protein and carbohydrates, shows therapeutic potential as a safe, cost-effective, and easy-to-implement strategy in cancer treatment. It can be used alone or alongside conventional therapies to enhance chemotherapy efficacy, protect healthy tissue, reduce inflammation, and modulate proteins and factors like MMPs, HDACs, AMPK, PK, and P53. Evidence from human and animal studies supports KD’s role in reducing tumor growth and improving patient outcomes [98]. Malignant cells exhibit altered metabolism, heavily relying on glucose via the Warburg effect and producing lactate even in oxygen-poor conditions. This dependence makes them vulnerable to the KD, which lowers glucose and raises ketone bodies that cancer cells cannot utilize, effectively starving them. In contrast, healthy cells adapt by metabolizing fatty acids into ketones for efficient energy production, preserving normal function [99]. It should be considered that if KD were a low protein diet, limitations would occur in the context of malnutrition and sarcopenia. Regarding the heterogeneity of KD protocols, Table 2 provides details of varied KD composition according to clinical studies and RCTs.
Table 2. Ketogenic diet composition in cancer clinical studiesCancer typeFat %Protein %Carb %Ketogenic ratio (Fat: Protein + Carb)NotesReferenceOvarian/Endometrial Cancer70255~ 2.8:1Protein notably higher than theory, carbs restricted [100]Breast Cancer80–8516–182–41.6:1–2:1Closer to classical KD but with increased protein [101]Pancreatobiliary Cancer8015–203–61.75:1Moderate protein [102]Breast Cancer55196~ 1.4:1Use of MCT; higher protein, lower fat than standard KD [103]Pancreatobiliary Cancer post-pancreatectomy70–8015–253–61.05:1–1.75:1Moderate protein range [104]
KD and Cancer-related mechanisms, genes, and/or proteins
Inflammation is a complex response in vascular tissues after injury or during chronic disease, involving cytokines, chemokines, and transcription factors, and is closely linked to cancer. In some cancers, inflammation precedes malignancy, while in others it promotes tumor growth, spread, and therapy resistance. Studies show that KD reduces inflammatory cytokines (TNF-α, IL-1β, IFN-γ), potentially improving cancer treatment and prevention [105]. TNF-α can trigger IL-8 release and activate NF-kB transcription factors, crucial elements in cancer progression [106].
Research focusing on GBM has revealed that combining KD with DON (a glutamine antagonist) significantly reduces tumor growth, enhances survival rates, and decreases inflammation [107]. In breast cancer, TNF-α serves multiple functions in disease progression and metastasis. A randomized control trial by Khodabakhshi et al. showed significant TNF-α reduction in breast cancer patients after 12 weeks of KD, attributed to MMP-9 suppression and PPARγ activation [103].
Cyclooxygenase (COX), existing in two isoforms (COX-1 and COX-2), plays a crucial role in prostaglandin and eicosanoid production from arachidonic acid. COX-2 overexpression has been documented in various cancers, including colon, breast, and lung cancers. Given COX-2’s significant influence on tumor development stages, selective COX-2 inhibitors show promise in cancer prevention and treatment. In vitro studies using mouse glioma models have shown that KD significantly reduces COX-2 expression, while combination studies with radiation therapy suggest decreased expression of COX-2 and other inflammatory markers. However, additional research is needed to definitively establish KD’s effectiveness in COX-2 expression modulation [108, 109].
Matrix metalloproteinases (MMPs), which function as zinc-dependent endopeptidases, play an essential role in breaking down the extracellular matrix (ECM). The ECM is vital for cellular cohesion and maintaining bodily structure. Beyond ECM degradation, these enzymes significantly influence various aspects of cancer development, including its spread, invasion, and blood vessel formation. Research has identified elevated levels of several MMPs across multiple cancer types, including colorectal cancer, glioblastoma, and gastric cancer. A notable member, MMP-9, belongs to the gelatinase subfamily and originates from inflammatory cells, tumor-adjacent stromal cells, or the cancer cells themselves. Research suggests that implementing a KD alongside traditional cancer treatments may significantly inhibit MMP-9 expression in mouse models of colon cancer [110–113].
HDACs represent a crucial protein family that governs gene transcription and protein function by removing acetyl groups from specific lysine residues in core histone proteins bound to DNA. These enzymes are fundamental in regulating cellular processes closely associated with cancer development and progression. HDAC inhibitors have demonstrated the ability to modify gene expression patterns and influence various cellular processes, including growth inhibition, differentiation, cell death, and apoptosis activation. While multiple correlative studies have demonstrated connections between KDs, ketone bodies, and HDAC suppression in human cancers, this area requires further investigation [114–116].
Research findings have demonstrated that KDs play a significant role in suppressing mutant p53 variants through β-hydroxybutyrylation and deacetylation processes and promoting cellular death. This decrease in mutant p53 levels contributes to extended survival. Additionally, glucose-restricted dietary approaches facilitate p53 mutant deacetylation and breakdown; consequently, KDs either inhibit p53 mutant function or suppress gene expression throughout cancer development and advancement [117].
The AMP-activated protein kinase (AMPK) represents a serine/threonine kinase enzyme present across multiple cell types. AMPK modulation presents a promising therapeutic approach for treating diverse malignancies, including colorectal, pulmonary, and hepatic cancers. AMPK stimulation correlates with tumor suppressor pathways involving p53 and LKB1, cellular proliferation inhibition, inflammatory response reduction, growth restriction, and cell cycle disruption; therefore, AMPK serves a critical function in cancer prevention. Energy shortage conditions, such as glucose limitation and oxygen deprivation, trigger AMPK activation. Furthermore, pharmaceutical agents, including metformin, curcumin, quercetin, and certain anti-inflammatory medications, can stimulate AMPK activity. The metabolic shift from glucose utilization to ketone body consumption in malignant cells following KD implementation has been associated with enhanced AMPK stimulation [118–122].
miRNAs are known for their ability to modulate gene expression through target mRNA binding to control protein production or mRNA breakdown. miRNAs can influence tumors at every stage, encompassing initiation, advancement, apoptosis, blood vessel formation, proliferation, and cellular differentiation [123].
In this context, miR-21 regulates numerous biological mechanisms, including inflammatory responses, fibrotic processes, and carcinogenesis. Specifically, it has been found to regulate transforming growth factor β (TGF-β) through SMAD signaling networks in renal fibrosis and inflammation. Additionally, miR-21 facilitates colorectal cancer progression by targeting tumor suppressor messenger RNAs, including tropomyosin 1, programmed cell death 4 (PDCD4), and phosphatase and tensin homolog (PTEN) [124, 125].
Alterations in miRNAs, including Let-7, miR-21, and miR-145, impact colorectal cancer mechanisms. Let-7 miRNA acts as a tumor suppressor by modulating Ras, c-myc, and p53, essential for colorectal cancer initiation and progression. Oncogenic miR-21 is upregulated in tumors and targets tumor suppressors such as PTEN and PDCD1. The role of miR-145 is controversial; while previously seen as tumor-suppressive, recent studies link its upregulation to enhanced cancer cell migration and invasion. Environmental and lifestyle factors, including diet, influence miRNA expression in colorectal cancer [126–129].
Additional comprehensive mechanistic studies are required to establish the potential relationship between miRNA function and KD implementation in cancer development (Table 3).
Table 3. The relationship between inflammation, cancer, and the KDTopicKey findingsMechanism/EffectReferencesTNF-α & CancerKD/DON combination reduces TNF-α expression in GBMGlutamine antagonism [130]KD suppresses TNF-α in breast cancer via MMP-9 and PPARγPPARγ activation [131]MMPs & CancerKD reduces MMP-9 expression in cancerAnti-metastatic effects [113]PKM2 & CancerKD attenuates PKM2, reducing glucose uptake and lactate productionMetabolic reprogramming [13]Ketone bodies (particularly BHB) inhibit PKM2, reducing ATP and promoting apoptosisGlycolysis inhibition [132]p53 & CancerKD (p53 β-hydroxybutyrylation) induces deacetylation and degradation of mutant p53Epigenetic regulation [117]Epigenetics & CancerKetone bodies (BHB) modulate DNA methylation and histone modifications.β-hydroxybutyrylation [133, 134]
Numerous studies support KDs or high-fat, low-carbohydrate, adequate-protein nutrition as effective cancer therapies or preventive measures, alone or combined with drugs. These dietary approaches influence pathways like insulin signaling, PI3K, AKT, mTOR, ketone metabolism, adiponectin, leptin, and IGF-1. Both preclinical and clinical research confirm KD’s anti-aging and anticancer benefits. Combined with physical activity, KD reduces cancer risk across multiple malignancies by modulating metabolism and molecular signaling [135].
High-fat diets, especially those rich in saturated fats, can induce inflammation by mimicking lipopolysaccharide (LPS) actions, triggering inflammatory responses via receptors on macrophages and innate immune cells. In contrast, polyunsaturated fats like omega-3 fatty acids (EPA, DHA) exhibit anti-inflammatory effects. Chronic inflammation, characterized by elevated cytokines and nuclear factor kappaB (NF-κB) signaling, is a cancer hallmark. Low-carbohydrate, high-fat diets emphasizing unsaturated fats reduce tumor-associated macrophages, cytokines, NF-κB, and COX-2 expression, providing cancer prevention benefits. KD metabolism produces ketone bodies, with BHB activating mitochondrial uncoupling protein-2 (UCP-2), helping modulate inflammation and metabolism [136]. Conversely, its deficiency may cause excessive ROS, pro-inflammatory cytokine release, and sustained NF-κB activation. Consequently, ketone bodies serve essential functions in reducing oxidative stress and prolonging patient survival. Adiponectin and leptin represent peptide hormones synthesized by visceral white adipose tissue. Adiponectin demonstrates inverse correlation with leptin and other adipokines. Reduced adiponectin levels have been associated with type 2 diabetes, insulin resistance, metabolic syndrome, hypertension, cardiovascular disease, and cancer [137].
Finally, a direct relationship has been established between high-calorie diets and cancer risk, along with cancer prevention approaches through lifestyle modifications, including physical activity, exercise, and healthy diets rich in fruits, vegetables, and whole grains, while limiting red meat and saturated fat consumption. KDs may not completely prevent tumor development, but can delay tumorigenesis and enhance survival rates. Furthermore, KD demonstrates synergistic benefits for cancer treatment when combined with chemotherapy or alternative cancer therapies (Fig. 4).
Fig. 4KDs and Cancer-related Molecular Mechanisms: Mechanistic Overview
KDs and their role in overcoming therapeutic resistance
KDs are emerging as metabolic interventions to combat therapeutic resistance across multiple diseases. By shifting the body’s primary energy source from glucose to ketones, KDs disrupt pathological metabolic pathways and enhance treatment efficacy in drug-resistant conditions. Pre-clinical studies demonstrate that KDs sensitize tumors to chemotherapy and targeted therapies by exploiting metabolic vulnerabilities. In PI3K inhibitor-resistant cancer models, KDs reduced glucose availability to tumors, overcoming drug resistance and inhibiting growth. A meta-analysis of animal studies showed KD significantly extended survival time and reduced tumor weight [138]. While compliance remains challenging, emerging evidence positions KDs as adjuvant therapies to overcome resistance in oncology, as some examples have been discussed in the following.
Resistance to immune checkpoint blockade (ICB) therapy is a major issue in prostate cancer due to its immunosuppressive microenvironment. Recent research by Murphy et al. (2024) shows that the KD and its metabolite BHB may enhance ICB effectiveness in preclinical, ICB-resistant prostate cancer models. Their study tested combinations of anti-PD1 and anti-CTLA4 antibodies with vorinostat, a cyclic KD, or BHB supplementation using resistant PCa cell lines [139]. Cyclic KD (CKD) and BHB supplementation both delayed tumor growth as monotherapies in resistant prostate cancer models. CKD’s anti-tumor effects relied on BHB and a functional adaptive immune system. Combining ICB with HDAC inhibition, CKD, or BHB enhanced tumor control synergistically. These interventions increased tumor immunogenicity by upregulating MHC-I and improved the microenvironment by boosting CD8 + T cell infiltration, M1 macrophage polarization, monocyte differentiation, and lowering neutrophil presence [139].
KDs have been suggested to serve as effective metabolic adjuvants in cancer therapy by improving treatment response and addressing metabolic dysregulation in patients with advanced malignancies. The Keto-CARE trial (NCT03535701) investigated the impact of a well-formulated KD (WFKD) in individuals with stage IV metastatic breast cancer undergoing chemotherapy. The study demonstrated that participants were able to achieve and sustain nutritional ketosis (β-hydroxybutyrate >0.5 mM) throughout the intervention, accompanied by significant metabolic improvements [140]. Over a period of three months, the KD led to a 16% reduction in fasting plasma glucose and a 54% decrease in insulin levels (p < 0.01). Additionally, there was a 67% reduction in HOMA-IR scores, indicating a marked improvement in insulin sensitivity. Participants also experienced a 10% loss in body weight, primarily attributable to reductions in fat mass, as body fat percentage decreased from 45% to 37%. Nutritional ketosis was maintained for six months, facilitated by a phased approach to dietary support. The intervention was implemented in two phases to optimize adherence. In the initial three months, participants received daily ketogenic meals and intensive dietary coaching, resulting in a mean β-hydroxybutyrate level of 0.8 mM. In the subsequent three months, participants transitioned to preparing their own ketogenic meals with ongoing monitoring, successfully sustaining a mean β-hydroxybutyrate level of 0.7 mM [140].
The carbohydrate restriction (20–50 g/day) inherent in the KD effectively disrupted cancer-associated hyperglycemia while preserving lean body mass. This metabolic reprogramming may mitigate chemotherapy-induced insulin resistance and inhibit activation of the PI3K/mTOR pathway, both of which are implicated in therapeutic resistance. Importantly, the study reported no severe adverse events despite the concurrent administration of chemotherapy. The individualized dietary approach, emphasizing a variety of whole foods such as non-starchy vegetables, high-quality proteins, and satiating fats, contributed to a 75% retention rate at six months, surpassing typical adherence rates observed in dietary interventions within oncology populations [140].
Thus, this preliminary evidence proposes that structured KD protocols can safely and effectively modulate host metabolism during advanced cancer treatment. While further large-scale, controlled studies are warranted, the results of the Keto-CARE trial support the potential role of KDs as adjunctive therapies for improving metabolic parameters and enhancing treatment resilience in patients with metastatic cancer.
A preclinical performed by Talib highlighted the promising role of combining a KD with melatonin in addressing chemotherapy resistance in breast cancer. The KD exerts its effects by altering cellular metabolism, notably reducing ATP availability through a metabolic shift from glycolysis to ketolysis. This metabolic reprogramming potentially impairs the function of ATP-dependent drug efflux pumps, such as P-glycoprotein, which are commonly implicated in the development of multidrug resistance. Additionally, the KD influences tumor metabolism by lowering blood glucose levels and increasing concentrations of ketone bodies, including β-hydroxybutyrate and acetoacetate [141]. Melatonin, when administered alongside the KD, further enhances anti-tumor efficacy. It promotes apoptosis, as demonstrated by a 2.8-fold increase in caspase-3 activity compared to control groups. Moreover, melatonin downregulates key resistance-related genes, resulting in a 41% reduction in P-glycoprotein expression and a 37% decrease in glutathione S-transferase (GST) levels. The combination also significantly suppresses angiogenesis, evidenced by a 56% reduction in vascular endothelial growth factor (VEGF) expression in resistant breast cancer cell lines [141].
Experimental findings from this study show that melatonin exhibits an IC₅₀ of 8 mM in parental breast cancer cells and 12 mM in resistant lines. The resistance index (RI) in cisplatin-resistant cells was reduced from 4.2 to 1.8 following combination therapy, while vincristine-resistant cells showed a decrease in RI from 3.7 to 2.1. In vivo, the combined intervention led to a 68% reduction in tumor volume in cisplatin-resistant models and a 63% reduction in vincristine-resistant models. Notably, 70% of mice receiving the combination therapy achieved complete remission, compared to only 20% in those treated with chemotherapy alone [141]. A dual metabolic approach using the KD and melatonin may effectively overcome multidrug resistance in breast cancer. Melatonin inhibits the PI3K/AKT pathway by 34% and activates apoptosis, enhancing therapy. The 4:1 fat-to-carb KetoCal^®^ formulation offers a clinically relevant model for future trials, though further research is needed to confirm efficacy and optimize dosing in humans.
Healy et al. investigation into dietary influences on liver cancer development provided important insights into the KDKD’s potential role in modulating tumorigenesis. Although the study did not focus exclusively on KDs, the comparative evaluation of five different dietary regimens offers valuable information for understanding metabolic interventions in cancer management. The KD group exhibited the lowest tumor burden among the experimental diets, comparable to that observed with normal chow, despite the induction of obesity and glucose intolerance. Notably, tumor burden in this group showed no correlation with adiposity or fasting insulin levels, distinguishing its effects from those of sugar-rich diets. Furthermore, the KD was associated with significantly reduced hepatic triglyceride content compared to Western diets, with a 2.5-fold lower level observed in the KD group relative to the Western diet with lard [142]. Mechanistically, tumor burden positively correlated with postprandial insulin levels (r = 0.62, p < 0.01), hepatic interleukin-6 (IL-6) expression (r = 0.58, p < 0.05), and liver cholesterol accumulation (r = 0.71, p < 0.001). The KD also elicited higher hepatic expression of pro-apoptotic markers, including a 1.8-fold increase in cleaved caspase-3 compared to high-sugar diets and a 2.1-fold elevation in the tumor suppressor protein p21. Comparative analysis of dietary impacts revealed that tumor burden was substantially lower in the KD group (65 ± 18 mm³) compared to the Western diet (420 ± 95 mm³) and fructose diet (380 ± 88 mm³), with statistical significance (p < 0.05). Hepatic triglyceride levels were also reduced in the KD group (15 mg/g tissue) relative to the Western diet (38 mg/g tissue), while adiposity was highest in the ketogenic group (4.2 g) compared to the Western (3.8 g) and fructose (2.1 g) diets [142].
The findings suggest that dietary sugar intake, rather than fat content, serves as the primary driver of hepatocarcinogenesis in this model. The protective effects of the KD appear to be mediated through metabolic reprogramming that reduces substrate availability for lipogenesis and inflammatory pathways, enhanced apoptotic regulation via caspase-3 activation and p21-mediated cell cycle arrest, and modulation of the tumor microenvironment characterized by lower IL-6 and cholesterol-driven pro-tumor signaling. Importantly, the ketogenic formulation used (71% lard, 6% safflower oil) achieved these effects without exacerbating hepatic steatosis, distinguishing it from other high-fat diets [142]. Healy’s preclinical findings suggest carbohydrate restriction may benefit liver cancer treatment, but human trials are needed for validation. The disconnect between metabolic dysfunction and tumor suppression in KD-fed mice calls for further study of tissue-specific insulin signaling.
Zhuang et al. elucidated the synergistic relationship between metabolic interventions such as the KD and cancer therapeutics, specifically highlighting how glucose restriction potentiates metformin’s anti-tumor activity through multifaceted mechanisms. In vitro experiments revealed that low glucose conditions (0–5 mM) significantly amplified metformin’s cytotoxicity in breast (MCF7, MDAMB231, SKBR3) and ovarian (OVCAR3, PA-1) cancer cell lines. ATP depletion under low glucose, fructose, or galactose media (2.5–25 mM)—but not high glucose (25 mM)—indicated impaired glycolytic compensation as a critical factor. Furthermore, mTOR pathway inhibition, evidenced by reduced phosphorylation of AKT and S6K, occurred independently of AMPK activation, suggesting dual targeting of cancer survival pathways [143]. In vivo validation using a murine model demonstrated that a low-carbohydrate KD (93% fat, 2% carbohydrates) induced a 30% calorie restriction alongside sustained reductions in serum glucose. This dietary regimen enhanced metformin-mediated suppression of 4T1 breast tumor growth compared to standard chow, without significant weight loss in the subjects. These findings underscore the diet’s feasibility as an adjunct to pharmacotherapy [143]. The study posits a tripartite mechanistic framework for KD’s role in cancer therapy. First, glycolytic suppression reduces systemic glucose availability, limiting tumors’ access to their primary energy substrate. Second, hypoglycemia prevents metabolic compensation by cancer cells, sensitizing them to metformin-induced oxidative phosphorylation (OXPHOS) inhibition. Third, low glucose amplifies metformin’s disruption of mTOR-driven proliferative and survival signaling. Collectively, these preclinical insights suggest that KDs may counteract therapeutic resistance associated with high-glucose tumor microenvironments. However, translational validation through human trials remains essential to confirm clinical applicability. The work emphasizes the critical interplay between dietary context and metabolic drug efficacy, advocating for integrated metabolic-dietary strategies in oncology research [143].
Accumulating evidence supports KDs as versatile metabolic adjuvants in oncology, targeting cancer vulnerabilities to boost therapy. KDs act via glycolytic suppression, metabolic sensitization, and pathway modulation. Notably, KDs enhance conventional treatments, showing synergy with metformin (68% tumor suppression), chemotherapy (70% remission in resistant models), and immunotherapy (delayed progression in resistant cancers).
Overall, the Keto-CARE trial demonstrates that sustained nutritional ketosis is feasible during treatment, yielding a 16% fasting glucose reduction and a 67% decrease in insulin resistance without compromising safety. KD may reverse therapeutic resistance by downregulating PI3K/AKT signaling (34% reduction) and boosting immunogenicity via MHC-I. While preclinical data show tumor reduction and improved survival, human trials are limited. Challenges include adherence and personalized macronutrient balance. Future research should focus on randomized trials, biomarker stratification, and combining KDs with metabolic therapies like β-hydroxybutyrate to establish KD as a standard cancer treatment (Table 4).
Table 4. The key findings from the studies on KDs and their role in overcoming therapeutic resistance in cancerStudy focusKey findingsMechanism/effectCancer typeReferences KDs & Immune Checkpoint Blockade (ICB) Resistance CKD and BHB delay tumor growth in ICB-resistant prostate cancer.MHC-I upregulation, CD8 + T cell infiltrationProstate Cancer [139]HDAC inhibition and ketogenesis enhance ICB efficacy.M1 macrophage polarizationProstate Cancer [139] Keto-CARE Trial (Metastatic Breast Cancer) WFKD reduces fasting glucose (16%), insulin (54%), and HOMA-IR (67%).Improved insulin sensitivityMetastatic Breast Cancer [140]WFKD preserves lean body mass and reduces fat mass (45% to 37%).Metabolic reprogrammingMetastatic Breast Cancer [140] KD + Melatonin in Breast Cancer KD reduces ATP availability, impairing drug efflux pumps (P-glycoprotein).Metabolic shift to ketolysisResistant Breast Cancer [141]Melatonin enhances apoptosis (2.8-fold increase in caspase-3 activity).Downregulation of resistance genesResistant Breast Cancer [141]Combination therapy reduces tumor volume by 68% (cisplatin-resistant models).Angiogenesis suppression (56% VEGF reduction)Resistant Breast Cancer [141] KD in Liver Cancer KD reduces tumor burden (65 mm³ vs. 420 mm³ in Western diet).Reduced IL-6, cholesterol signalingLiver Cancer [142]KD increases pro-apoptotic markers (1.8-fold caspase-3, 2.1-fold p21).Apoptotic regulationLiver Cancer [142] KD + Metformin in Breast/Ovarian Cancer Low glucose amplifies metformin’s cytotoxicity (ATP depletion).Glycolytic suppressionBreast, Ovarian Cancer [143]KD enhances metformin’s mTOR pathway inhibition.OXPHOS inhibitionBreast Cancer [143]
Emerging evidence from preclinical and clinical studies on kd’s anticancer potentials
Preclinical evidence
Low-carbohydrate, high-fat KDs may prevent tumor progression and serve as supportive therapy, though limited studies have focused on colorectal cancer. Using a ketogenic infant formula (KF), researchers found that colon tumor-bearing mice fed KF exhibited preserved body and muscle mass, reduced tumor weight, and lower plasma IL-6 levels compared to controls. KF mice showed elevated blood ketone levels inversely correlated with tumor size, suggesting KF suppresses cancer progression and systemic inflammation without adverse effects on weight or muscle mass, potentially preventing cancer cachexia. This preclinical evidence supports further investigation of ketogenic interventions in colorectal cancer management [144]. KD can also exert opposite effects on tumor growth depending on a cancer’s capacity to metabolize ketone bodies. In 33 human cancer cell lines, high expression of ketolytic enzymes BDH1 and OXCT1 was associated with KD resistance, as seen in HeLa xenografts where KD accelerated growth. In contrast, PANC-1 tumors with low enzyme expression showed marked growth inhibition under KD. β-hydroxybutyrate promoted proliferation only in high-expressing cells, and enzyme downregulation sensitized resistant tumors to KD. Low ketolytic enzyme expression may serve as a predictor of KD responsiveness [145]. Notably, Maurer and colleagues demonstrated that glioma cells exhibit a metabolic disadvantage under glucose restriction, as they are unable to efficiently utilize ketone bodies for energy, unlike normal neurons. Their work combined in vitro assays (testing 3-hydroxybutyrate on rat hippocampal neurons and five glioma cell lines) with in vivo orthotopic xenograft mouse models. However, an unrestricted KD alone failed to control tumor growth, underscoring the need for combination approaches such as glycolysis inhibitors or antiangiogenic agents targeting non-oxidative pathways [146].
In a mouse model of peritoneal dissemination using colon 26 cells, a KD improved survival, health status, and anemia-related parameters compared to a regular diet, despite no significant reduction in tumor weight. These findings suggest the KD may alleviate systemic disease burden and could serve as a supportive or preventive approach against peritoneal dissemination [147]. Despite these promising findings, some preclinical models have also reported paradoxical pro-tumor effects or significant adverse outcomes, such as exacerbating metastasis and cachexia, under specific conditions, highlighting context-dependent responses [148, 149].
Clinical evidence
In contrast to robust preclinical data, clinical investigations of KDs in oncology remain limited in scope and scale. Most human trials to date have focused on feasibility, safety, and tolerability rather than antitumor efficacy. These studies consistently demonstrate that KDs can successfully induce ketosis, moderately reduce blood glucose levels, and are generally feasible for motivated patients, often improving quality of life. Individual case reports and small observational studies suggest potential antitumor activity, proposing that KDs may exploit the Warburg effect by restricting glucose availability thereby impairing cancer cell glycolysis while promoting ketone metabolism that preferentially supports normal cells [150]. Proposed clinical benefits include mitigation of cancer cachexia, reduced muscle wasting and fatigue, lowered insulin and growth-promoting hormone levels, enhanced immune modulation, and decreased toxicity from chemotherapy and radiation [150, 151].
However, no large-scale randomized controlled trials have yet confirmed KDs as an effective standalone or adjuvant therapy in humans. While triple-negative breast cancer and other aggressive malignancies with poor prognoses underscore the urgent need for novel therapeutic strategies, current clinical evidence remains insufficient to support routine clinical adoption outside of research settings.
The KD is gaining attention as a low-cost, minimally toxic, and easily implementable nutritional intervention with biologically plausible anticancer mechanisms including metabolic targeting, anti-inflammatory effects, epigenetic modulation, and microenvironmental remodeling. Preclinical data robustly support its role in tumor suppression and synergy with conventional therapies, whereas clinical evidence is still preliminary, primarily demonstrating safety and feasibility. Patient adherence remains a key barrier to implementation. Future priorities include rigorous clinical trials to evaluate efficacy across cancer subtypes and the development of standardized combination protocols integrating KDs with established anticancer treatments.
Challenges and controversies
The nutritional strategies demonstrate optimal potential when implemented as a complementary intervention alongside other treatment modalities. Despite limited patient sample sizes in various trials, researchers have presented compelling evidence suggesting ketogenic nutrition is safe, practical, and capable of improving outcomes for advanced cancer patients [152].
However, several significant challenges necessitate careful consideration. Dietary complexity and preparation require meticulous food measurement and a time-intensive process, making it challenging for families with demanding schedules. Additionally, the lack of a standardized protocol and composition variations may yield inconsistent results. Researchers recommend that future studies focus on developing a standardized KD treatment strategy, including precise duration and regimen, to minimize adverse effects [153].
Patient compliance challenges remain a critical limitation, as long-term adherence to rigid dietary restrictions proves difficult during cancer treatment. Younger patients may find the diet overly restrictive and unappealing. Addressing these concerns requires enhanced patient education, comprehensive support systems, and improved dietary flexibility [154].
Potential medical complications associated with KDs include frequent physiological challenges such as initial dehydration during fasting and gastrointestinal disruptions like nausea, vomiting, diarrhea, and constipation. More serious complications encompass lipid profile alterations, acute pancreatitis, cardiomyopathy, metabolic disturbances, including hyperuricemia, hyponatremia, hypoproteinemia, persistent acidosis, and hypomagnesemia, and bone and mineral metabolism issues such as hypercalciuria, hypocitraturia, kidney stone formation, and urine acidification. Recent research suggests the methodology remains tolerable as an adjuvant treatment [153].
An animal study in 2024 has raised concerns about KD potentially promoting metastasis under certain conditions. One study found that while KD reduced primary tumor growth, it also promoted tumor metastasis in the lungs of mice [148]. This highlights the complex and possibly heterogeneous tumor metabolic responses to ketones, which vary by tumor type and metabolic context.
Regarding long-term safety of the KD in cancer patients, Recent longitudinal data from a cohort of 55 advanced cancer patients followed between 2013 and 2023 provide important insights into the chronic tolerability and outcomes of KD in clinical practice. Among the 37 patients who adhered to the KD for at least three months, the median follow-up was 25 months, with a 5-year survival rate of 23.9%. Notably, patients who maintained the KD for ≥ 12 months (n = 21) demonstrated a markedly superior median overall survival (OS) of 55.1 months compared to 12 months in those who discontinued earlier (n = 32). After adjusting for background factors using inverse probability of treatment weighting, the difference remained statistically significant, suggesting a robust association between long-term dietary adherence and improved prognosis [155]. From a safety standpoint, these findings underscore that extended KD administration—up to 99 months in certain patients—appears feasible and well tolerated in a clinical oncology setting. No major diet-related toxicities necessitating discontinuation were reported, reinforcing earlier observations that KD can be safely integrated into supportive cancer care under medical supervision. Commonly monitored parameters such as renal and hepatic function, lipid profile, and body weight remained within manageable ranges, consistent with previous shorter-term studies demonstrating acceptable tolerability and metabolic stability. This long-term dataset adds to growing evidence that concerns regarding severe nutritional deficiencies or metabolic derangements under sustained KD are largely unsubstantiated when dietary protocols are properly monitored [155].
Importantly, these results also highlight that dietary adherence and patient selection critically influence outcomes. Patients who maintained KD for longer durations likely benefited from greater metabolic adaptation and improved treatment synergy, whereas early discontinuation often reflected disease progression or intolerance to dietary restrictions. Thus, structured nutritional counseling, periodic metabolic monitoring, and individualized dietary adjustments are essential for maximizing both safety and therapeutic potential [155].
Overall, the available evidence supports the long-term safety and potential survival advantage of KD as a supportive intervention in advanced cancer, particularly when maintained beyond 12 months. Nonetheless, given the observational nature of the study and potential confounding variables, randomized controlled trials are needed to confirm causality and delineate which tumor types and patient populations derive the greatest benefit. Future research should also address the mechanisms underlying prolonged metabolic adaptation, optimal macronutrient composition, and integration with standard cancer therapies to refine the clinical utility of KD in oncology.
Psychological and social implications of KD interventions present additional challenges, including extensive meal preparation requirements, limited social dining opportunities, potential feelings of isolation, variable dietary costs, and psychological stress. Recommendations for future research include a comprehensive investigation of adverse effects, ensuring safety and efficiency, and developing more adaptable dietary protocols. Despite these challenges, the ketogenic approach continues to show promise as a potential complementary cancer treatment strategy, warranting further nuanced scientific exploration.
It is a misconception to assert that the KD entirely lacks therapeutic potential in oncology, leads to nutritional deficiencies, reduces patient well-being, or adversely affects survival outcomes in individuals with cancer. Although current clinical data supporting its effectiveness remain limited, there is a clear need for future randomized trials to reinforce the evidence base and identify which patient subgroups may derive the greatest benefit [156]. A recently proposed clinical framework advocates for integrating the KD with other metabolic interventions to advance both investigational and therapeutic strategies for aggressive gliomas and other malignancies [157]. In the absence of robust, high-quality data, recommending KDs broadly to cancer patients remains contentious, and many individuals are expected to continue with conventional dietary practices. However, for those who independently opt to adopt a KD, we align with the perspective of Zemer et al. noted in their narrative review, who emphasize that “clinicians need to be informed about KD, and offer support and medical supervision for patients who self-select to follow KD. This can ensure that within the boundaries of KD, patients will make good and healthy dietary choices and prevent clinical disengagement in extreme cases” [158]. In conclusion, when properly administered, the safety profile of KDs in cancer care should no longer be a matter of dispute.
A 2020 survey by Klassen et al. involving 57 Canadian medical oncologists found that the overwhelming majority (87%) did not recommend KD to their patients [159]. This lack of endorsement stemmed from beliefs that the diet was either ineffective (31%) or had unknown clinical impacts (69%). Nearly all surveyed oncologists (96%) expressed at least one concern about KD recommendation, with predominant worries including risk of weight loss and malnutrition, potential psychological distress including anxiety and food-related fear, and possibility of additional adverse effects [159].
The ‘Prevention and Integrative Oncology’ working group within the German Cancer Society and German Society for Nutritional Medicine published a critical review expressing methodological concerns [160]. Their conclusion stated that “methodologically high-quality studies are absent, resulting in insufficient reliable evidence for the ketogenic diet. Consequently, the statement’s authors conclude that currently, employing a low-carbohydrate or ketogenic diet for this indication should be discouraged”. Interestingly, no alternative dietary approach was proposed. The statement further emphasized that KDs represent “substantial dietary limitations,” “elevate malnutrition risk within days to weeks,” and therefore result in weight reduction and deteriorating prognosis [160].
These studies reveal three dominant skeptical positions against low-carbohydrate and ketogenic diets in cancer management: (i) KDs may demonstrate no anti-tumor activity; (ii) KDs impose excessive restrictions, generating risk of psychological harm, anxiety, and food-related fear; (iii) KDs trigger weight loss, thus deteriorating patient outcomes [156].
Flexible KD protocols, including modified or less restrictive forms (such as modified Atkins diet), are preferred in resource-constrained settings to balance efficacy, cost, and cultural acceptability. Studies have confirmed ketogenic diet implementation feasibility in middle-resource countries with ongoing challenges including supplement affordability, hospital resource allocation, and ensuring continuous clinical support. In summary, socioeconomic and cultural factors significantly affect KD implementation, and tailored, flexible approaches with local community engagement, education, and consideration of food accessibility are critical for successful equity in ketogenic diet therapies, especially in cancer or epilepsy treatment in resource-limited settings. This inclusion would strengthen the review by recognizing these real-world challenges and proposing adaptive strategies to improve access and adherence beyond theoretical dietary prescriptions.
In addition, adaptive and personalized KD strategies are particularly important in translating metabolic therapies from controlled clinical environments to real-world settings with variable healthcare infrastructure. In resource-limited contexts, individualized protocols that incorporate locally available food sources, community dietitian training, and culturally appropriate meal designs can substantially enhance dietary feasibility and patient adherence. Personalization informed by metabolic profiling, genomic insights, and biomarker monitoring may further optimize dietary response, allowing clinicians to adjust macronutrient ratios and caloric targets based on individual tolerance and treatment stage. Moreover, integration of digital health technologies, such as smartphone-based tracking apps, tele-nutrition platforms, and AI-driven adherence monitoring, offers scalable tools for remote supervision, early detection of metabolic imbalances, and continuous feedback loops between patients and clinical teams. Artificial intelligence can also assist in dynamically adjusting diet plans through predictive modeling of metabolic trends and automated alerts for non-compliance or adverse effects, improving both safety and long-term adherence. These adaptive and technology-assisted frameworks hold promise for making ketogenic and related metabolic interventions both accessible and sustainable in diverse healthcare settings, narrowing disparities in nutritional oncology care across socioeconomic boundaries.
Conclusions and future prospects
Nutritional care in oncology has advanced significantly in recent years, with greater awareness, wider implementation of clinical nutrition programs, and broader integration of multidisciplinary teams. Nevertheless, major gaps persist; nutrition specialists remain underrepresented in oncology teams, standardized malnutrition screening is still lacking, and economic as well as logistical barriers continue to limit equitable access to high-quality dietary support.
Emerging metabolic interventions such as the KD have generated substantial interest due to encouraging preclinical evidence and preliminary clinical findings. Modified, protein-sufficient KD protocols, distinct from classical epileptic versions, may target tumor cell metabolism, reduce glucose dependence, and potentially improve the efficacy of standard therapies. However, existing evidence is largely based on animal studies, pilot projects, and small non-randomized trials, while robust, large-scale RCTs remain limited. Current data mainly confirm feasibility and short-term safety, leaving questions of long-term clinical benefit unresolved.
Future progress will rely on both systemic reform and targeted research development. System-level priorities include policy initiatives to formally integrate medical nutrition into oncology care pathways, insurance coverage and reimbursement for dietitian-led interventions, routine inclusion of certified nutrition experts in cancer teams, and enhanced education programs for healthcare providers. On the research front, clearly defined priorities must guide the next phase of inquiry:
- I.Establishment of standardized protocols for nutritional assessment, intervention, and outcome measurement across cancer types.
- II.Stratification of patients by molecular, metabolic, and biomarker profiles to identify those most likely to benefit from KD and related dietary interventions.
- III.Implementation of large, multicenter, RCTs to determine clinical efficacy, long-term safety, and optimal therapeutic combinations with standard treatments.
- IV.Development of comprehensive registries and international data-sharing initiatives to promote reproducibility and guideline harmonization.
In parallel, mechanistic research should continue to elucidate the molecular and metabolic pathways modulated by nutritional interventions, with careful assessment of potential adverse effects such as enhanced metastatic potential in certain tumor contexts.
Overall, while nutritional oncology, including KD and other metabolic strategies, presents promising opportunities to improve patient outcomes and quality of life, these benefits can only be validated through coordinated, high-quality research and system-level integration. Advancing this field will require rigorous clinical investigation, biomarker-guided patient selection, and global collaboration to establish standardized, evidence-based nutritional care in oncology.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alidadi M, Banach M, Guest PC, Bo S, Jamialahmadi T, Sahebkar A, editors. The effect of caloric restriction and fasting on cancer. Seminars in Cancer Biology; 2021: Elsevier.10.1016/j.semcancer.2020.09.01032977005 · doi ↗ · pubmed ↗
- 2Van Dang C. Cancer metabolism: The known, unknowns. 2018. p. 1.10.1016/j.bbcan.2018.07.00630336841 · doi ↗ · pubmed ↗
- 3Li T, Le A. Glutamine metabolism in cancer. Heterogeneity Cancer Metabolism. 2018;1063:13–32.10.1007/978-3-319-77736-8_229946773 · doi ↗ · pubmed ↗
- 4Kassab AE. Recent advances in targeting COX-2 for cancer therapy: a review. RSC Med Chem. 202516:2974-300210.1039/d 5md 00196 j PMC 1208234040386345 · doi ↗ · pubmed ↗
- 5Ferrer M, Mourikis N, Davidson EE, Kleeman SO, Zaccaria M, Habel J et al. Ketogenic diet promotes tumor ferroptosis but induces relative corticosterone deficiency that accelerates cachexia. Cell Metab. 2023;35(7):1147-62.e 7.10.1016/j.cmet.2023.05.008PMC 1103750437311455 · doi ↗ · pubmed ↗