Differences in metavirome among Aedes albopictus, Culex tritaeniorhynchus, and Anopheles sinensis in Jiangxi Province, China
Xin Ran, Dajin Xiao, Yangbowen Wu, Yong Shi, Shiwen Liu, Yu Bai, Qiang Zhang, Lan Liu, Qian Liu, Jianxiong Li, Minghui Zhao

TL;DR
This study compares the virus communities in three mosquito species in Jiangxi Province, China, identifying differences in the types and abundance of viruses present.
Contribution
The study provides a detailed comparative analysis of metaviromes in three mosquito species using next-generation sequencing.
Findings
86 viruses were identified, with 49 belonging to established families and 37 unclassified.
Significant compositional differences were observed in the top 30 most frequently detected viruses among the three mosquito species.
The study highlights the need for further validation of the biological roles of identified viruses in host-virus interactions.
Abstract
Mosquito metavirome research aims to comprehensively characterize the diversity of mosquito-associated viruses, particularly focusing on insect-specific viruses (ISVs) and their potential interactions with arboviruses of public health concern. Advances in next-generation sequencing (NGS) have significantly expanded our understanding of the viromic complexity within mosquito populations, revealing numerous novel viral species and genera. These studies not only contribute to viral taxonomy and evolutionary biology but also provide critical insights into the ecological dynamics between mosquitoes and their viromes. NGS was employed to characterize the metavirome of three epidemiologically significant mosquito vectors, Aedes albopictus, Culex tritaeniorhynchus, and Anopheles sinensis in Jiangxi Province, China. This study integrated bioinformatic workflows to conduct comparative analyses…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
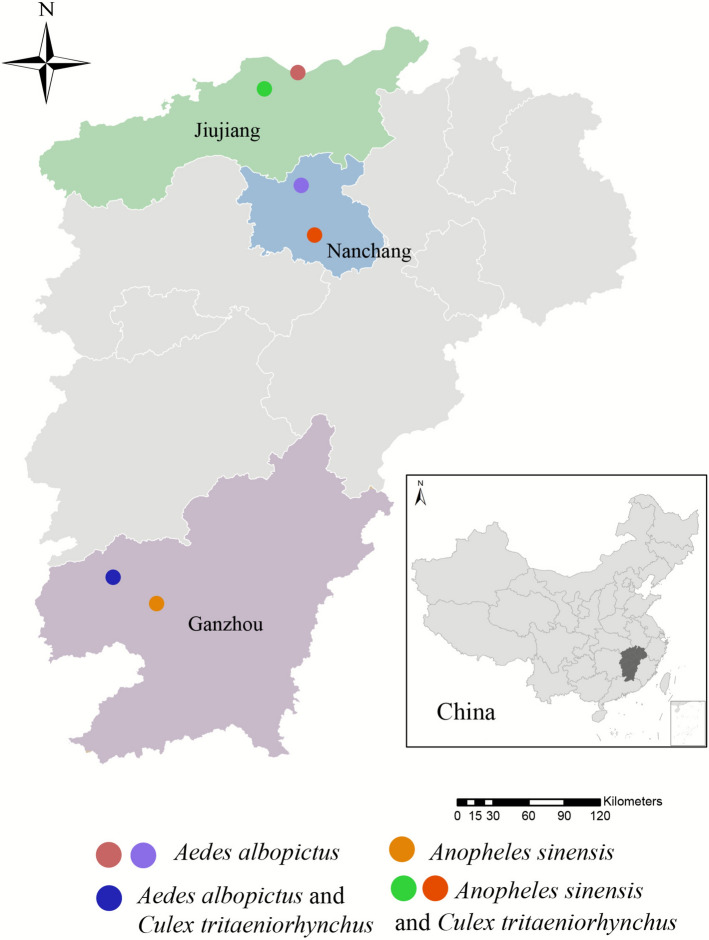 Figure 1
Figure 1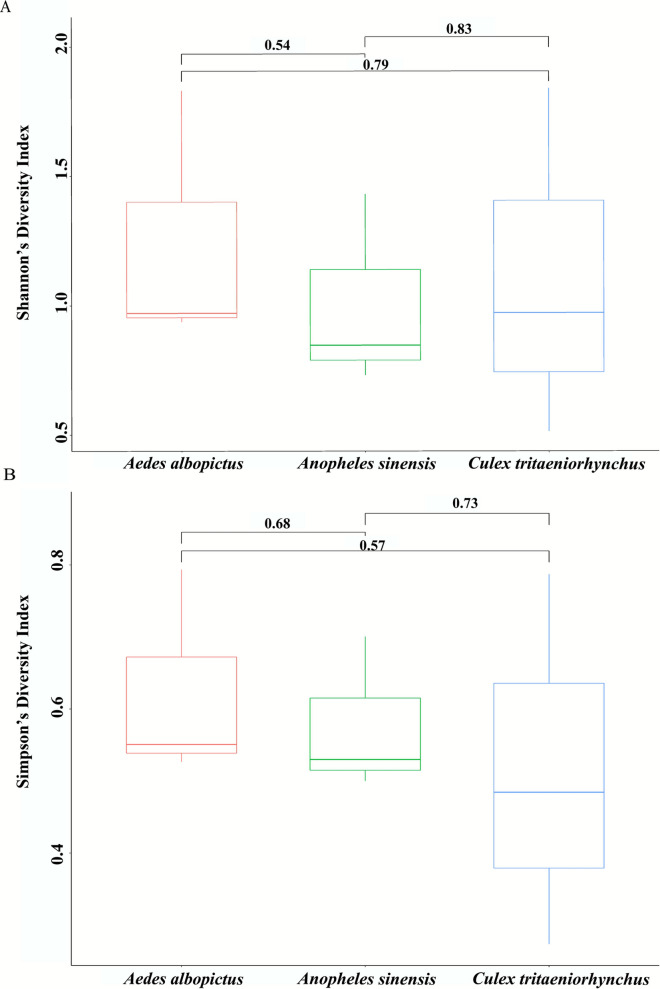 Figure 2
Figure 2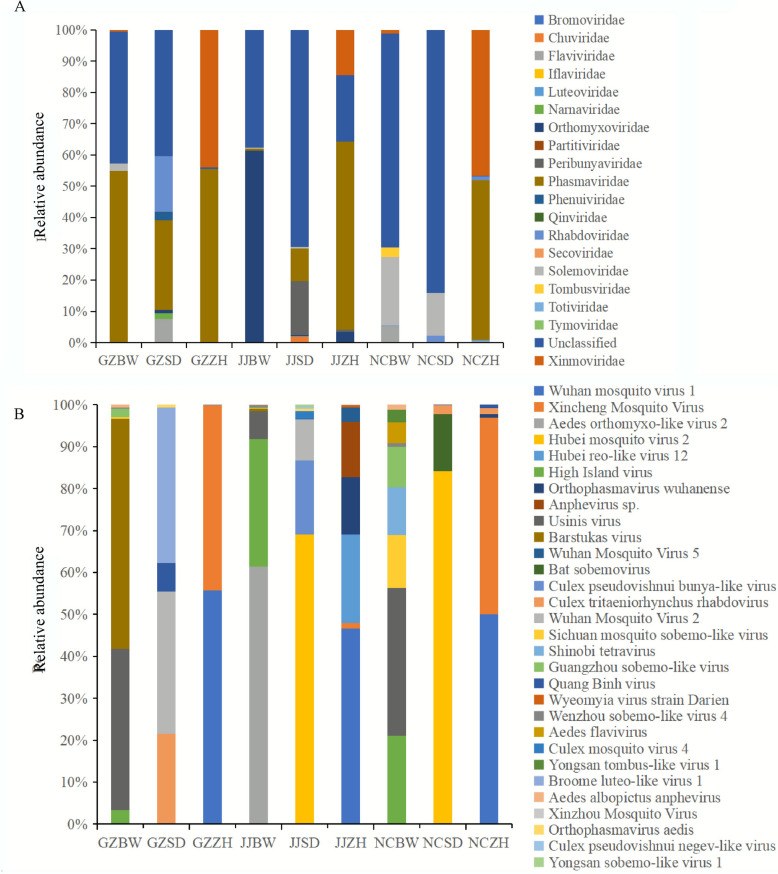 Figure 3
Figure 3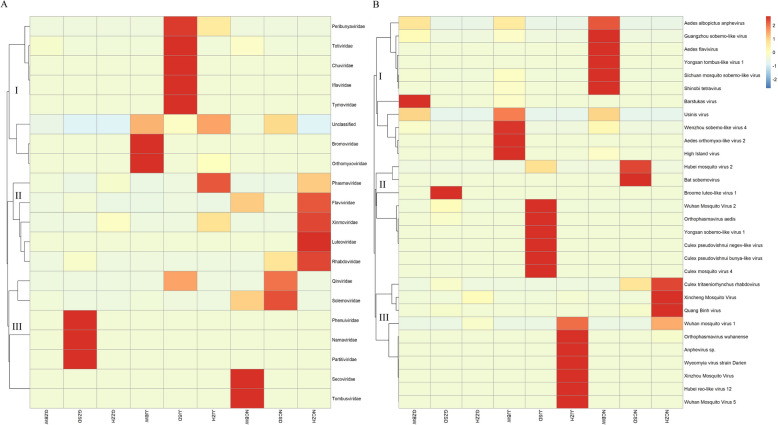 Figure 4
Figure 4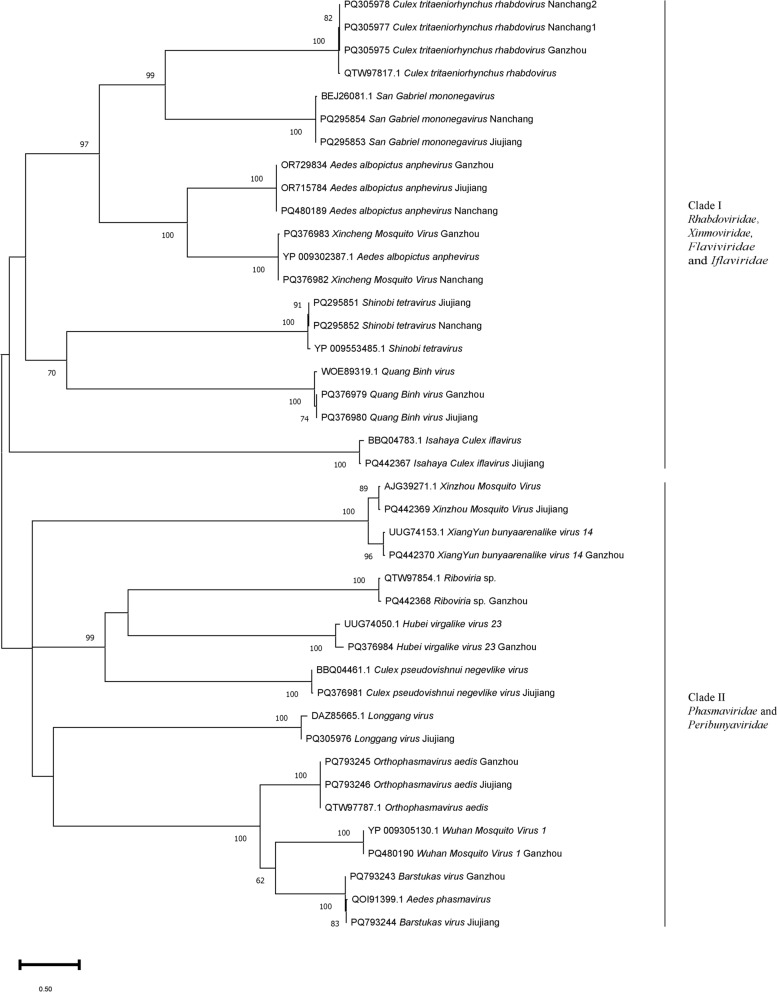 Figure 5
Figure 5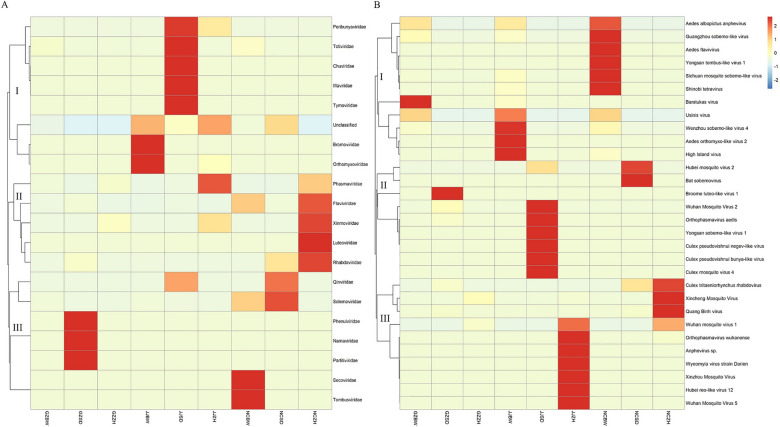 Figure 6
Figure 6- —Key Scientific and Technological Innovation Projects of the Jiangxi Provincial Health Commission
- —Nanchang Customs Technology Project
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Plant Virus Research Studies · Insect symbiosis and bacterial influences
Background
Situated in the subtropical humid monsoon climate zone along the southern bank of the middle-lower Yangtze River, Jiangxi Province provides optimal thermohydrometric conditions for mosquito proliferation and the transmission cycles of arboviruses [1, 2]. Aedes albopictus, Cx. tritaeniorhynchus, and An. sinensis are common vector mosquitoes in the province. Aedes albopictus serves as a vector for the dengue virus (DENV), Zika virus (ZIKV), and Chikungunya virus (CHIKV), which have caused widespread infectious disease outbreaks worldwide; such outbreaks affect the health and lives of hundreds of millions of people every year [3, 4]. Culex tritaeniorhynchus is the primary vector for Japanese encephalitis virus (JEV), an endemic disease pathogen in Asia. This vector is prevalent in tropical and subtropical regions of East Asia and causes nearly 70,000 infections of JEV each year, which results in a mortality rate of 20–30% [5, 6]. Although An. sinensis is recognized as the primary vector of malaria parasites in China, other pathogens have recently been detected in it, including ZIKV, JEV, and Getah virus [7–10]. The first local outbreak of dengue fever occurred in Zhanggong District, Ganzhou City, Jiangxi Province, in 2017, whereas a number of municipalities and counties saw local outbreaks of the same disease in 2019 [11]. For 2 months, a total of 81 cases had been reported. Although no fatalities were recorded, the outbreak caused serious public health concern for the local people [12]. In 2021, Wang et al. reported the first detection of the ZIKV in Cx. tritaeniorhynchus and An. sinensis mosquitoes collected in Jiangxi Province [7]. Currently, Ae. albopictus is the only vector species that carries the DENV and ZIKV in Jiangxi Province. No study reported Cx. tritaeniorhynchus and An. sinensis transmitting these viruses [13]. While the detection of ZIKV is crucial for control measures, it can also lead to public panic and misallocation of resources if not managed effectively.
The development of next-generation sequencing (NGS) technology enabled the identification of a growing number of viruses in mosquito metagenome studies. These viruses include common pathogens, such as the yellow fever virus (YFV) [14], DENV [15], CHIKV [16], JEV [17], and ZIKV [18]. They also comprise a number of insect-specific viruses (ISVs) and unclassified viruses [19, 20]. Pathogenic insect-borne viruses pose a crucial threat to humans worldwide and place a substantial economic burden on governments. With the intensive growth of global transportation systems and the gradual urbanization of arthropods, arboviruses are emerging and spreading faster and over a wider geographical area [21]. ISVs naturally infect mosquitoes and can replicate in their cells in vitro; notably, they do not replicate in vertebrate cells nor infect humans and other vertebrates [22]. However, several ISVs also play a role in the replication and transmission of arboviruses and the development of gene expression platforms and corresponding vaccines [23, 24]. The Culex ISV Bamaga virus interfered with in vitro replication and in vivo transmission of the pathogenic flavivirus West Nile virus (WNV) [25, 26]. The Nhumirim virus (NHUV) reduced the replication of the WNV, Saint Louis encephalitis virus, and JEV when cocultured in the C6/36 cell line [27, 28]. NHUV also significantly inhibited the replication of the ZIKV and dengue virus 2 in C6/36 cells and reduced the infection rate and transmissibility of ZIKV in Ae. Aegypti [29]. Cell-fusing agent virus and Phasi Charoen-like virus in Ae. aegypti cells affects the growth of the ZIKV, which leads to considerable changes in the cell permissivity to arbovirus infection [30]. The Eilat virus (EILV) can affect the replication of coinfected arboviruses, such as the CHIKV in the Ae. albopictus cell line and Ae. aegypti populations [31]. In addition, the EILV-based chimeras can be used to express arboviruses proteins, such as those from the CHIKV. These chimeric viruses can then be utilized to immunize mice against respective arboviruses [32]. EILV–CHIKV chimeric viruses offer invaluable biosafety advantages for the development of diagnostic reagents [33]. Mapping of mosquito viromes and understanding their composition and roles are crucial for gaining insights into the transmission of arboviruses and vaccine development.
A review of literature revealed the recently rapid increase in the number of published studies on the mosquito metagenome. These studies covered at least 128 mosquito species belonging to 14 genera. Their main focuses were the Culex, Aedes, and Anopheles genera [19]. Another study revealed Flaviviridae, Rhabdoviridae, and Peribunyaviridae as the three most abundant mosquito virus families, with Flaviviridae distributed in mosquitoes on all continents studied and with the highest detection rates in Culex, Aedes, and Anopheles mosquitoes [19, 34]. This work used NGS and integrated bioinformatics methods to analyze the metavirome profiles of Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis, which were collected in Jiangxi Province in 2023. The aim was to compare the metavirome profiles of these species and to identify potential pathogenic viruses as well as ISVs that may influence vector competence and pathogen transmission.
Methods
Mosquito collection
Field collections of Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis were conducted across three representative municipalities in Jiangxi Province (Nanchang, Ganzhou, and Jiujiang) in August 2023. The Electric Handheld Mosquito Trap (catalog no. DX3V) was used for collecting adult mosquitoes. The captured adult mosquitoes (excluded blood-fed female mosquitoes) underwent cryoanesthesia using dry ice or exposure to a laboratory freezer (−20 ℃), after which they were identified at the species level using morphological keys [35]. The species-confirmed female mosquitoes (n = 334) were grouped into nine taxon-specific pools (20–60 individuals per pool) on the basis of the collection site and were then stored at −80 ℃ in cryovials for subsequent analysis. Sampling locations and ecological characteristics are presented in Table 1 and shown in Fig. 1. Table 1. Sampling locations and RNA quality of Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis populationsCitiesCodeSpeciesTotal numberLongitude/latitudeAverage temperature (2023) (℃)Average relative humidity (2023) (%)Average precipitation (2023) (mm)NanchangNCBWAe. albopictus33115° 55′ 39″ E/28° 52′ 35″ NNCSDCx. tritaeniorhynchus52116° 01′ 49″ E/28° 29′ 52″ N19.672.31,848NCZHAn. sinensis42116° 01′ 49″ E/28° 29′ 52″ NJJBWAe. albopictus31115° 54′ 08″ E/29° 43′ 41″ NJiujiangJJSDCx. tritaeniorhynchus40115° 39′ 04″ E/29° 36′ 20″ N18.577.51,251JJZHAn. sinensis25115° 39′ 04″ E/29° 36′ 20″ NGZBWAe. albopictus26114° 50′ 05″ E/25° 42′ 43″ NGanzhouGZSDCx. tritaeniorhynchus40114° 50′ 05″ E/25° 42′ 43″ N20.275.01,542GZZHAn. sinensis45114° 30′ 15″ E/25° 54′ 35″ NFig. 1Sampling locations map of Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis populations
Nucleic acid extraction and quality control
Frozen mosquito specimens were retrieved from −80 ℃ storage and transferred to sterile grinding tubes. Each tube received 600 µl saline solution and 6–8 zirconium-oxide grinding beads. The tubes were securely capped and homogenized at 5,000 revolutions per minute (rpm) for 45 s using a bead mill homogenizer. Following centrifugation at 12,000g for 10 min at 4 ℃, the supernatants were subjected to RNA extraction using a QIAamp Viral RNA Mini Kit (no. 52904) as per the manufacturer’s protocol. DNase treatment to deplete genomic DNA from the RNA extracts was performed using the Turbo DNA-free kit (Thermo Fisher Scientific) following the manufacturer’s instructions. The purified RNA was quantified using a Qubit 4 Fluorometer (Thermo Fisher Scientific) with RNA HS Assay Kit (Invitrogen, Q32855) and archived at −80 ℃ in nuclease-free microtubes.
Library construction and sequencing
First, viral RNA enrichment was performed. RNA enrichment was performed using the Macro & Micro-test pathogen RNA enrichment platform. Probes that bind to ribosomal RNA (rRNA) were used to capture host rRNA. This was then digested with RNase H, after which DNase I was used to digest the probes and DNA. The remaining RNA was then purified.
Second, fragmentation, reverse transcription, and library construction were performed. Enriched RNA samples were purified and recovered using magnetic beads before fragmentation to ~300 base pairs (bp). Reverse transcription using random primers synthesized the first strand of complementary DNA (cDNA). The complementary strand was synthesized concurrently with end repair and A-tailing to yield the second strand of cDNA. Sequencing adapters were then ligated to both ends of the cDNA fragments. Following adapter ligation, fragment selection was performed via magnetic bead purification (0.6–0.8× bead selection). The target fragment library obtained through magnetic bead purification and selection was then enriched and purified again using magnetic beads to yield a library ready for sequencing.
Finally, the MGISEQ-2000 was used for sequencing. After library quantification, the sequencing libraries were pooled according to the required data volume. The pooled libraries then underwent ligation, digestion, and purification to obtain ligation products. These were then processed through rolling circle amplification to generate DNA nanoballs (DNBs). The DNBs were loaded onto BGI sequencing chips using the MGIDL-200 and tested on the MGISEQ-2000 sequencer (BGI Genomics). On the MGISEQ-2000 platform, PE150 sequencing (150 bp paired-end) was performed using dual-indexing for multiplex library pooling. Each library is projected to yield approximately 10 million reads.
Data analysis
Raw sequencing reads underwent quality control via FastQC (v0.12.1; https://www.bioinformatics.babraham.aCx.uk/projects/fastqc/), followed by adapter trimming and low-quality base removal using Trimmomatic (v0.39; http://www.usadellab.org/cms/?page=trimmomatic). Clean reads were subjected to host-genome depletion against the reference genomes of Ae. albopictus (GCF_035046485.1), Cx. tritaeniorhynchus (GCA_964187845.1), and An. sinensis (GCA_000441895.2) using Kneaddata (v0.12.0; https://huttenhower.sph.harvard.edu/kneaddata). Host-depleted reads were assembled de novo using MEGAHIT (v1.2.9; k-mer range 21–141; https://github.com/voutcn/megahit). The resulting contigs (> 500 bp) were compared against the National Center for Biotechnology Information (NCBI) nt database via BLASTnt (v2.13.0+), with an e-value cutoff of 1 × 10^−5^. Further species identification was carried out on the basis of the annotation results. Viral abundance was calculated on the basis of the number of reads obtained through sequencing. The relative abundance (%) in our study was calculated using the formula: (number of annotated reads for a specific contig/total number of annotated reads) × 100. The α-diversity (Shannon and Simpson) indices were calculated using the “vegan” package in R, which has been widely used for data analysis [36].
The assembled short sequences were further assembled using MEGAHIT (v1.2.9). Near-full-length viral genomes (> 90% coverage) were identified through BLASTnt alignment against the NCBI database. Maximum-likelihood phylogenetic trees were reconstructed in MEGA (v11.0.13; https://www.megasoftware.net/) using 1000 bootstrap replicates under the best-fit substitution model.
Results
Sampling and sequencing results
Field collections across three municipalities in Jiangxi Province (Nanchang, Ganzhou, and Jiujiang) yielded 334 adult female mosquitoes of Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis (Table 1). These nine pools were then subjected to metagenomic sequencing and generated high-quality data. Each sample pool generated a data volume exceeding 20 Gb, with an average Q30 score of over 97% (Additional file 1: Supplementary Table S1). The detection and quantification of viruses could be influenced by RNA extraction, stability, integrity, and reverse transcription. Therefore, future studies should control for these variables to reach definitive conclusions.
Virus diversity and host analysis
Metavirome assembly of nine mosquito pools identified 86 viruses, which comprised 49 viruses classified within 19 established families and 37 unclassified viruses (Additional file 2: Supplementary Table S2). The Ae. albopictus pools were assembled and annotated to 10 families (n = 37 total viruses), the Cx. tritaeniorhynchus pools to 14 families (n = 37 total viruses), and the An. sinensis pools to 7 families (n = 20 total viruses). Analysis of the α-diversity of these 86 viruses among the three mosquito species revealed no significant differences (Shannon and Simpson diversity P > 0.05; Fig. 2). Of these viruses, 43.02% (37/86) were unclassified viruses and 37.21% (32/86) were ISVs. Notably, the annotated viruses included two human–invertebrate dual-host viruses, namely, Guapiacu virus (GUAPV) [37] and *Wyeomyia virus strain Darien *(WVD) [38].Fig. 2. Shannon and Simpson diversity of 86 viruses in Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis pools
Relative abundance and cluster analysis of viruses
Hierarchical clustering of viral abundance profiles revealed significant compositional divergence of viruses among the nine mosquito pools at the family and species levels (top 30). At the family level (Fig. 3A), unclassified viruses were present in all nine mosquito pools, with the highest relative abundance observed in the NCSD (84.17%), JJSD (70.72%), NCBW (68.38%), and GZSD (40.38%) pools. Phasmaviridae constituted the predominant viral family in the NCZH (50.95%), JJZH (60.25%), GZZH (55.42%), and GZBW (54.82%) pools, whereas Orthomyxoviridae dominated the JJBW pool (61.64%). Notably, Xinmoviridae exhibited an elevated abundance in the NCZH (46.78%) and GZZH (43.92%) pools compared with the other pools. At the species level (top 30) (Fig. 3B), Wuhan mosquito virus 1 (WMV1) dominated all An. sinensis pools but was undetectable in the Ae. albopictus and Cx. tritaeniorhynchus pools. Furthermore, the Xincheng mosquito virus (XCV) and Hubei reo-like virus 12 were only detected in An. sinensis. XCV was most abundant in the GZZH (43.92%) and NCZH (46.78%) pools and WMV1 in the JJZH (60.25%) pool. In the three Cx. tritaeniorhynchus pools, Hubei mosquito virus 2 had the highest relative abundance in the NCSD (84.10%) and JJSD (70.25%) pools but was also detected in one An. sinensis pool (NCZH). Broome luteo-like virus 1 was the most abundant in the GZSD (30.66%) pool. Wuhan mosquito virus 2 (WMV2) was only detected in the three Cx. tritaeniorhynchus pools, and it was least abundant in the NCSD (14 reads) pool. Of the three Ae. albopictus pools, Aedes orthomyxo-like virus 2 (AOMV-2) was only detected in the JJBW (61.41%) pool. Usinis virus was the most abundant in the NCBW (34.89%) pool and present at relatively high levels in the GZBW (38.39%) pool. Barstukas virus was the most abundant in the GZBW (54.82%) pool and was also detected in the JJBW pool. Eight viruses were detected only in the Ae. albopictus pools (High Island virus, Usinis virus, Sichuan mosquito sobemo-like virus, Guangzhou sobemo-like virus, Wenzhou sobemo-like virus 4 [WSLV4], Aedes albopictus anphevirus (AealbAV), Longgang virus, and Guato virus).Fig. 3. The relative abundance of viral families in Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis pools (A family level; B species levels top 30)
At the family level in the heat map (Fig. 4A), the 20 viral families (including unclassified viruses) can be divided into three major genetic branches. The unclassified viruses in the first genetic branch were closely related to Bromoviridae and Orthomyxoviridae families. At the species level (Fig. 4B), the top 30 viruses can also be divided into three genetic branches. Branch I contained 11 viruses that were relatively abundant in three Ae. albopictus pools. Branch II contained nine viruses that were relatively abundant in three Cx. tritaeniorhynchus pools. Branch III contained ten viruses that were relatively abundant in three An. sinensis pools.Fig. 4. The heat map of viral families in Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis pools (A family level; B species levels top 30)
Complete viral genome assembly and analysis
Further de novo assembly and reconstruction yielded 17 complete viral genomes and nine RNA-dependent RNA polymerase (RdRp) genes (Table 2). The 17 complete viral genomes revealed four viruses from the Ae. albopictus pools: AealbAV (OR715784 and OR729834), Shinobi tetravirus (SHTV, PQ295851, and PQ295852), San Gabriel mononegavirus (PQ295853 and PQ295854), and Longgang virus (PQ305976). Three viruses were found in Cx. tritaeniorhynchus pools: Quang Binh virus (QBV, PQ376979, and PQ376980), Culex pseudovishnui negev-like virus (PQ376981), and Culex tritaeniorhynchus rhabdovirus (CTRV, PQ305975, and PQ305977). Four viruses were found in the An. sinensis pools: CTRV (PQ305978), XCV (PQ376982 and PQ376983), Hubei virga-like virus 23 (PQ376984), and Riboviria sp. (PQ442368). Table 2. Information of concatenated viral sequences from Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis populations in the present studyMosquitoCodeVirusesGene typesGenBank numbersAe. albopictusJJBWAedes albopictus anphevirusComplete genomeOR715784JJBWShinobi tetravirusComplete genomePQ295851JJBWSan Gabriel mononegavirusComplete genomePQ295853JJBWLonggang virusComplete genomePQ305976JJBWBarstukas virusRdRp, complete cdsPQ793244GZBWAedes albopictus anphevirusComplete genomeOR729834GZBWBarstukas virusRdRp, complete cdsPQ793243NCBWAedes albopictus anphevirusRdRp, complete cdsPQ480189NCBWSan Gabriel mononegavirusComplete genomePQ295854NCBWShinobi tetravirusComplete genomePQ295852Cx. tritaeniorhynchusGZSDCulex tritaeniorhynchus rhabdovirusComplete genomePQ305975GZSDQuang Binh virusComplete genomePQ376979GZSDOrthophasmavirus aedisRdRp, complete cdsPQ793245JJSDIsahaya Culex iflavirusRdRp, complete cdsPQ442367JJSDQuang Binh virusComplete genomePQ376980JJSDCulex pseudovishnui negev-like virusComplete genomePQ376981JJSDOrthophasmavirus aedisRdRp, complete cdsPQ793246NCSDCulex tritaeniorhynchus rhabdovirusComplete genomePQ305977An. sinensisNCZHCulex tritaeniorhynchus rhabdovirusComplete genomePQ376978NCZHXincheng mosquito virusComplete genomePQ376982GZZHXincheng mosquito virusComplete genomePQ376983GZZHHubei virga-like virus 23Complete genomePQ376984GZZHRiboviria sp.Complete genomePQ442368GZZHXiangYunbunya-arena-like_virus 14RdRp, complete cdsPQ442370GZZHWuhan mosquito virus 1RdRp, complete cdsPQ480190JJZHXinzhou mosquito virusRdRp, complete cdsPQ442369cds stands for ‘coding sequence’
Phylogenetic reconstruction of RdRp amino acid sequences from the 26 viruses revealed two major clades (I–II; Fig. 5) under the maximum likelihood criterion (1000 bootstrap replicates). Clade I contained three CTRV (Rhabdoviridae; PQ305975, PQ305977, and PQ305978), three AealbAV (Xinmoviridae; OR715784, OR729834, and PQ480189), two XCV (Xinmoviridae; PQ376982 and PQ376983), two QBV (Flaviviridae; PQ376979 and PQ376980), Isahaya Culex iflavirus (Iflaviridae; PQ442367), and two unclassified viruses: San Gabriel mononegavirus (PQ295853 and PQ295854) and SHTV (PQ295851 and PQ295852). Clade II included Xinzhou mosquito virus (Peribunyaviridae; PQ442369), Phasmaviridae members, namely, Orthophasmavirus aedis (PQ793245 and PQ793246), WMV1 (PQ480190), and Barstukas virus (PQ793243 and PQ793244), as well as five unclassified viruses, Culex pseudovishnui negev-like virus (PQ376981), *Hubei Virga-like virus 23 *(PQ376984), Longgang virus (PQ305976), Riboviria sp. (PQ442368), and XiangYun bunya-arena-like virus 14 (PQ442370).Fig. 5. Maximum likelihood phylogeny of RdRp in 26 viruses (1,000 bootstrap replicates)
Discussion
Global surveillance of known mosquito-borne viruses has intensified following recurrent arboviral outbreaks across six continents. Yet, untargeted metaviromics has uncovered an expanding universe of mosquito-associated viruses, which is profoundly reshaping our understanding of virome diversity and evolution [34]. Here, metaviromic sequencing and computational biology were used to characterize the viromes of three principal arboviral vectors, including Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis, in Jiangxi Province, China’s subtropical monsoon zone (high temperatures and humidity; Table 1) [39]. Research has shown that climatic factors in regions with high temperatures and humidity correlate positively with the density of Ae. albopictus populations and increase the risk of spreading diseases such as dengue fever. The natural conditions in Jiangxi Province are conducive to the breeding of mosquitoes such as Ae. albopictus and the transmission of vector-borne diseases. Therefore, it is critical to strengthen vector-borne virus surveillance in this region. The aim was to compare the metavirome profiles of these species and to identify potential pathogenic viruses as well as ISVs that may influence vector competence and disease transmission. These findings contribute to understanding the complex interactions between mosquitoes and their viral communities, with implications for arbovirus surveillance and control strategies in Jiangxi Province.
A total of 86 viruses were identified in the nine mosquito pools examined in this study. The majority of these viruses are unclassified (43.02%). The lack of formal taxonomic classification limits our comprehension of the diversity of unclassified viruses circulating in these vector species [40]. The high proportion of unclassified viruses may be related to the inadequacy of the current database. In addition, the lack of reference sequences for several unknown viruses makes annotation difficult, which can result in viruses remaining unannotated and hinder the discovery of other unknown viruses. These viruses can only be accurately named through the continuous refinement of relevant virus databases. Despite having various ecological niches and geographical ranges, the three mosquito species did not exhibit significantly different α-diversity metrics for the 86 viruses (P > 0.05). Such a similarity may be related to the environmental factors that mosquitoes were exposed to, host selection, and the adaptability of viruses themselves. Research has shown that the viral composition of mosquitoes, individually and in populations, was closely related to host phylogeny, climate, land use type, and other factors [41]. However, examination of the top 30 viruses from each mosquito pool revealed evident differences in the highly abundant viruses of the three species and formed three distinct genetic branches (Fig. 4B). This tripartite segregation implied the strong host phylosymbiosis of several viruses, where virome configuration mirrored mosquito phylogeny, which indicates codivergence over the past million years [40, 42, 43].
Solemoviridae, Xinmoviridae, Phasmaviridae, Flaviviridae, Rhabdoviridae, Peribunyaviridae, Orthomyxoviridae, and unclassified viruses accounted for the highest number of reads in this study. The Solemoviridae family is a group of positive-sense single-stranded RNA viruses that infect plants, with genome lengths ranging from four to six kilobases (kb). These viruses are transmitted through mechanical damage, vegetative propagation, insect vectors, or nonbiological pathways such as soil [44].This study identified viruses belonging to the Solemoviridae family that were present in all three Ae. albopictus pools and in two Cx. tritaeniorhynchus pools. The Sichuan mosquito sobemo-like virus and Guangzhou sobemo-like virus were relatively highly abundant in all three Ae. albopictus pools. However, none of these viruses were present in the three Cx. tritaeniorhynchus pools. Multiple sobemo-like viruses were present in Ae. albopictus. Several studies, including this one, have detected WSLV4 in this mosquito species [45, 46]. In addition, Guangzhou sobemo-like virus was detected in Ae. albopictus from Hainan Island and Cameroon, which further supports its widespread presence in this mosquito species [47, 48]. The other two viruses in the Solemoviridae family, Yongsan sobemo-like virus 1 (YSLV1; JJSD pool, 0.46%) and Bat sobemovirus (BSV; NCSD, 13.61%), achieved high read counts in the two Cx. tritaeniorhynchus pools. However, they were not detected in any of the three Ae. albopictus pools. YSLV1 was shown to be prevalent in Aedes vexans nipponii in Korea, likely owing to its high adaptability and capacity for stable replication in mosquito cells [49]. BSV is a virus found in bats, but reports of its presence in mosquitoes are lacking. To date, research primarily focuses on the diversity and distribution of the virus within bat populations [50]. The present study revealed a higher abundance of BSV viruses in the NCSD pool, which deserves further investigation.
The family Xinmoviridae comprises negative-sense RNA viruses (9–14 kb genomes) infecting diverse arthropods, though their host range remains poorly defined [51]. Metaviromic profiling revealed a high relative abundance of Xinmoviridae in all An. sinensis and Ae. albopictus pools, with AealbAV dominating Ae. albopictus and XCV mainly reported in An. sinensis. These two viruses were highly similar and clustered together on a single branch (Fig. 5). Crucially, neither virus was detected in Cx. tritaeniorhynchus pools. AealbAV is a mosquito-specific virus widespread in Ae. albopictus populations globally. It has been detected in laboratory and field-collected mosquitoes and was distributed across the mosquito life cycle [52]. AealbAV can moderately reduce DENV replication in vitro, and it is a promising tool for future biocontrol measures [53]. Our data confirm AealbAV’s pan-regional dominance in Ae. albopictus in Jiangxi. Complete genomes assembled from two pools (GenBank: OR715784 and OR729834) showed minimal divergence (similarity: 99.98%). XCV is a mosquito-specific virus classified within the genus Anophevirus and has been first characterized in An. sinensis from China [54]. XCV exhibited ubiquitous colonization in Anopheles mosquito populations with no documented vertebrate pathogenicity. As a mosquito-specific virus, it may modulate vector competence of those mosquitoes [55, 56]. This study revealed the high abundance of XCV in all three An. sinensis pools (GZZH 43.92%, JJZH 1.28%, and NCZH 46.78%), which suggests that XCV may occupy an important ecological niche within these populations.
Phasmaviridae comprises enveloped viruses with trisegmented negative-sense RNA genomes (9.7–15.8 kb). These viruses are primarily maintained and transmitted by insects [57]. In this study, Phasmaviridae reads dominated all three An. sinensis pools (GZZH 55.42%, JJZH 60.25%, and NCZH 50.95%). High read numbers were also recorded in the GZBW (54.28%), GZSD (28.57%), and JJSD (10.64%) sample pools.* Barstukas virus* was the main virus species annotated in the GZBW pool. It has also been detected in Ae. albopictus mosquitoes on Hainan Island and is recognized as an environmentally sourced virus [48]. This virus showed a high degree of similarity with other viruses (WMV1 and Orthophasmavirus aedis) in the Phasmaviridae family (Fig. 5). WMV2 was the predominant virus species identified in Cx. tritaeniorhynchus sample pools. WMV1 was the predominant virus species in the three An. sinensis pools. Both viruses are ISVs and were first detected in 2015 in Cx. tritaeniorhynchus sample pools from Hubei Province, China [54, 58]. WMV2 was also detected in Cx. tritaeniorhynchus pools from Yunnan and Shandong Provinces in China and from Japan [59–61]. In addition, WMV2 is widely found in other mosquitoes, such as Armigeres subalbatus, An. sinensis, Cx. quinquefasciatus, and Ae. aegypti [59, 62]. WMV1 is widely found in Cx. tritaeniorhynchus and Anopheles mosquito populations but has not been reported in other mosquito species [19, 55]. This research supports this assertion.
This study identified six distinct viruses from the Flaviviridae family in the nine mosquito pools [Aedes flavivirus (AEFV), dengue virus 3 (DENV-3), GUAPV, QBV, Culex tritaeniorhynchus flavi-like virus, and Kamiti River virus (KRV)]. In total, five viruses were annotated to the NCBW pool (except for Culex tritaeniorhynchus flavi-like virus), but the number of reads for the other four viruses was low, except for AEFV (4.88%). AEFV is an ISVs first isolated from* Ae. albopictus* and Ae. flavopictus in Japan [63]. In several cases, these ISVs can interfere with pathogenic flavivirus infections, which suggests potential applications in disease control [63]. DENV-3 is one of the four DENV serotypes [64] that triggered several large-scale epidemics worldwide [65–67]. This work revealed that the DENV-3 was present in the NCBW and GZBW pools, which suggests that the virus is being transmitted by Ae. albopictus in Jiangxi Province and thus needs further research. QBV is an ISV that was first identified in Culex mosquitoes collected in Quang Binh, Vietnam, in 2002 [68]. A high rate of QBV was detected in Cx. tritaeniorhynchus populations, whereas a relatively low rate was observed in An. sinensis [69–72]. It was annotated to all three Cx. tritaeniorhynchus pools and the NCZH pool in this study. QBV isolated from Guizhou Province formed a unique branch with the Vietnamese strain but was closely related to QBV isolates from other regions of China. This genetic diversity may be related to the virus’s adaptability and transmissibility [73]. Other viruses, such as GUAPV, Culex tritaeniorhynchus flavi-like virus, and KRV, have not been fully clarified in terms of their transmission mechanisms in mosquitoes. However, these viruses should be further studied to assess their pathogenic potential and their potential public health risks is also worth exploring further [71, 74, 75].
Rhabdoviridae comprises enveloped viruses with negative-sense single-stranded (ss)RNA genomes (10–16 kb), and it demonstrates exceptional host breadth [76]. The Rhabdoviridae virus annotated to two Ae. albopictus pools (JJBW and NCBW) was Piry virus (PIRYV). This virus belongs to the Vesiculovirus genus, which is described as a potential human pathogen; it was first isolated from Philander opossum captured in the state of Pará in northern Brazil [77]. This virus has been detected in cell cultures of Ae. aegypti and Ae. albopictus, which further confirms its ability to grow within these mosquitoes [78]. Although PIRYV has not shown direct pathogenicity to humans, its continued transmission in mosquitoes and potential infectivity in mammals remain a cause for concern. The virus identified in the Cx. tritaeniorhynchus (NCSD and GZSD) and An. sinensis (NCZH) pools was the CTRV, which was also isolated from Cx. tritaeniorhynchus subspecies [79]. This virus is widespread in the natural populations and cell lines of Cx. tritaeniorhynchus. It is primarily transmitted by mosquitoes and does not cause diseases in vertebrates [80, 81]. The detection of CTRV in this study further indicates its presence in the natural populations of Cx. tritaeniorhynchus. CTRV infection may alter the physiological state of mosquitoes, which affects the transmission efficiency of JEV [80]. Furthermore, the presence of CTRV in a sample pool of An. sinensis in Nanchang implies its potential to adapt to various environments and hosts.
Peribunyaviridae encompasses several genera of viruses distributed worldwide [82]. In this study, Culex pseudovishnui bunya-like virus (CPBV) was annotated in high abundance in a Cx. tritaeniorhynchus pool (JJSD 17.77%), and WVD virus was annotated to the An. sinensis pool (JJZH 0.53%). CPBV is an ISV that was first identified in Cx. pseudovishnui [61]; its distribution, biology, and potential public health significance in mosquito species are unknown. WVD, a pathogenic virus that was first isolated in humans, is highly homologous to the Wyeomyia mosquito virus [38, 83]. In this study, a relatively high abundance of WVD virus was annotated to the JJZH pool, which provides a new opportunity for an in-depth investigation of the ecological properties of this virus in mosquitoes and its transmission cycle and potential public health significance.
Orthomyxoviridae viruses are a group of viruses that have important implications for medicine and public health. For example, influenza viruses (A and B) from this family are the most likely to cause seasonal epidemics and occasional pandemics in humans [84]. This study annotated the AOMV-2 and Wuhan mosquito virus 5 (WMV5) from Orthomyxoviridae family to the Ae. albopictus (JJBW 61.41%) and An. sinensis (JJZH 3.50%) pools, respectively. AOMV-2 was detected with high prevalence in laboratory and field strains of Ae. albopictus populations [85–87], and a strong association with host cell nuclei was found [88]. In this study, AOMV-2 was also detected in high abundance in the Ae. albopictus population in Jiujiang, which warranted further investigation to explore the intrinsic and extrinsic factors contributing to its high relative abundance. WMV5 was first detected in Cx. tritaeniorhynchus in Hubei Province, China, in 2015 [54]. However, it is not an ISV and was found to be distinct from WMV1 and WMV2. Further research is needed to investigate the transmission patterns of WMV5 in An. sinensis populations in natural environments.
Unclassified viruses constituted 43.02% (37/86) of the total virome, and they exhibited a relatively high abundance in four mosquito pools (Fig. 3). High Island virus and Usinis virus were the most frequently detected viruses in Ae. albopictus; Hubei mosquito virus 2 was the most frequently detected virus in Cx. tritaeniorhynchus; and Hubei reo-like virus 12 was the most frequently detected virus in An. sinensis. These viruses were also previously documented in diverse mosquito vectors globally [23, 81, 88, 89]. The abundance and diversity of unclassified viruses represent the bulk of the planet’s undescribed genetic diversity and are key to understanding viral evolution, host ranges, ecological distributions, and origins [58, 90]. Their discovery has provided a new perspective on the origin of viruses and their role in the evolutionary history of hosts. Further research into the biological characteristics, pathogenicity, and effects on vector competence of these unclassified viruses would be worthwhile.
Conclusions
This study used NGS technology to perform metavirome sequencing on Ae. albopictus, Cx. tritaeniorhynchus, and An. sinensis, which were collected in Jiangxi Province, China. A total of 86 viruses were annotated, and they included 49 known viruses belonging to 19 viral families and 37 unclassified viruses. The latter accounted for the highest proportion. No significant difference was observed in the diversity of the 86 viruses among the three mosquito species. However, the most frequently detected viruses of the three species showed distinct differences. The most frequently detected viruses of the Ae. albopictus population include the Sichuan mosquito sobemo-like virus and Guangzhou sobemo-like virus (Solemoviridae), AealbAV (Xinmoviridae), Barstukas virus (Phasmaviridae), AEFV (Flaviviridae), PIRYV (Rhabdoviridae), and the most abundant High Island virus and Usinis virus. The most frequently detected viruses of the Cx. tritaeniorhynchus population include the YSLV1 and BSV (Solemoviridae), WMV2 (Phasmaviridae), QBV (Flaviviridae), CTRV (Rhabdoviridae), CPBV, and Hubei mosquito virus 2 (Peribunyaviridae). The most frequently detected viruses of the An. sinensis population comprise the XCV (Xinmoviridae), WMV1 (Phasmaviridae), WMV5 (Orthomyxoviridae), and the Hubei reo-like virus 12. This study utilized bioinformatics analysis to further assemble and analyze several of these viruses. It explored their potential biological significance and evolutionary relationships through a combination of de novo genome assembly and phylogenetic reconstruction. This provides foundational data for further research into the interactions between these viruses and mosquitoes. Although this study allowed for the extensive screening of viral diversity within mosquito sample pools, notable limitations that require refinement were still noted. First, short-read sequencing technology struggles to accurately assemble complex viral genomes, which has a particular effect on variant analysis. Second, the lack of standardization in bioinformatics analysis workflows and incomplete databases restricted the annotation and functional prediction of highly variable or novel viruses. Furthermore, this method only detects the presence of viruses, rather than proves mosquito vector capacity, vector competence, or viral biological properties directly. Subsequent traditional virus isolation and experimental validation are required to clarify the ecological and epidemiological significance of the findings.
Supplementary Information
Supplementary material 1. Additional file 1: Table S1. Data output summary of Ae. albopictus*, Cx. tritaeniorhynchus and An. sinensis pools. Additional file 2: Table S2. Virus composition and read numbers in Ae. albopictus, **Cx. tritaeniorhynchus *and An. sinensis pools
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, Mc Glinn D, et al. Vegan: Community Ecology Package; 2019. 2020. Available from: https://cran.r-project.org/web/ packages/vegan/index.html.