DNA methylation analysis of NOTCH1 variants reveals the first episignature for non-syndromic congenital heart defects
Gregor Dombrowsky, Liselot van der Laan, Ananília Silva, Jeroen Breckpot, Enrique Audain, Anna Wilsdon, Michael A. Levy, Niels Vos, Marcel Mannens, Jiao Wang, Anjali Jain, Robert Lesurf, David Winlaw, Connie R. Bezzina, Mary Ann Thomas, Almuth Caliebe, Sabine Klaassen

TL;DR
This study identifies NOTCH1 as a major genetic cause of congenital heart defects and introduces a new DNA methylation signature to improve diagnosis.
Contribution
The first DNA methylation-based episignature for non-syndromic congenital heart defects linked to NOTCH1 variants.
Findings
NOTCH1-haploinsufficiency is the most common monogenic cause in the cohort, accounting for 1% of CHD cases.
A NOTCH1-specific DNA methylation episignature was established with high specificity compared to 99 other signatures.
Methylation profiling can aid in variant interpretation and improve diagnostic management for CHD patients.
Abstract
Congenital heart defects (CHDs) are the most common malformation amongst newborns, with a prevalence of approximately 0.8–2%. The etiology of CHD is highly complex and can be linked to genetic and nongenetic factors. The molecular basis remains partially unclear, and only a minority of patients can be assigned to clear monogenic causes. Here we analyzed a cohort of 3907 CHD cases and population-matched controls using exome sequencing. In addition, we employed epigenetic profiling on a subset of cases that harbored rare NOTCH1 variants. We identified 24 pathogenic or likely pathogenic single nucleotide variants (SNVs) in NOTCH1 in our exome cohort, as well as a further 15 variants of uncertain significance (VUS) likely to have a deleterious effect. Although the cardiac phenotypes showed some heterogeneity, non-syndromic Tetralogy of Fallot (ToF) and related malformations were the most…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
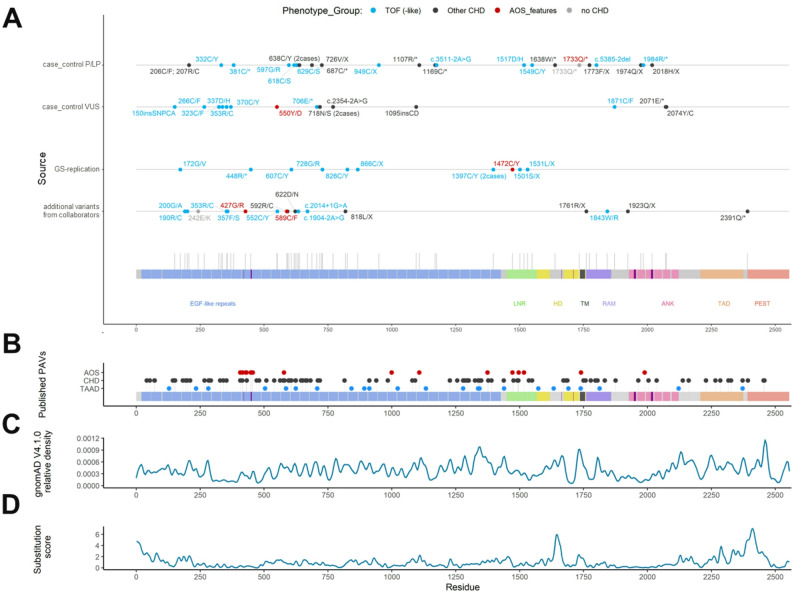 Figure 1
Figure 1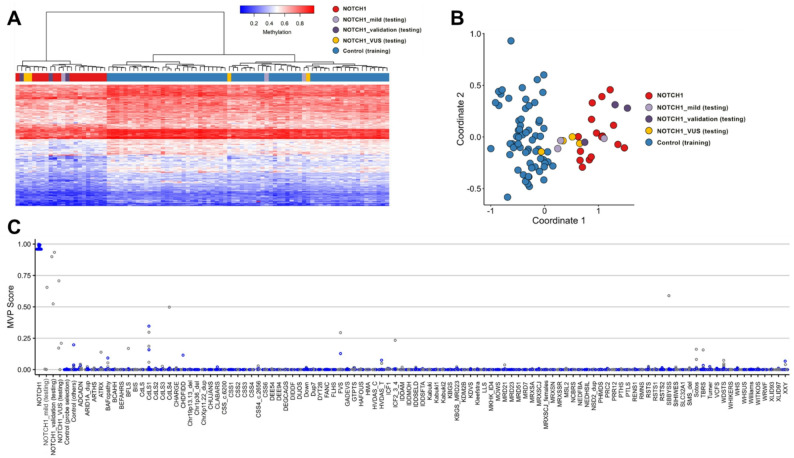 Figure 2
Figure 2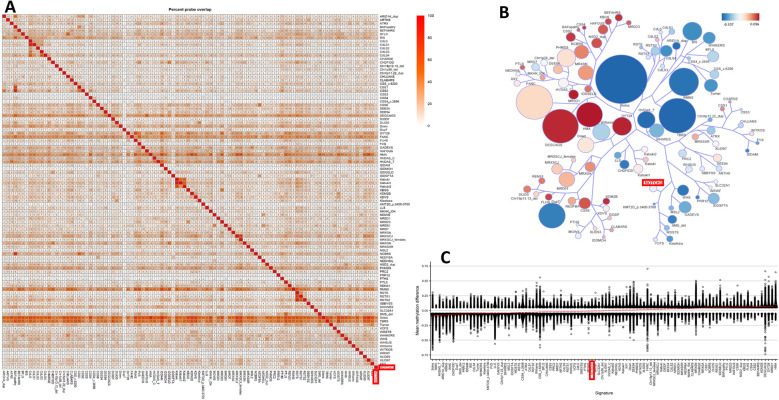 Figure 3
Figure 3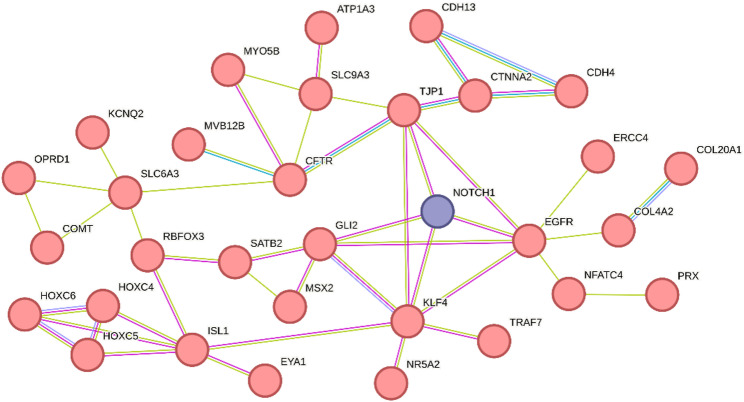 Figure 4
Figure 4- —Carl von Ossietzky Universität Oldenburg (3092)
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenomics and Rare Diseases · Congenital heart defects research · Congenital Heart Disease Studies
Background
Congenital heart defects (CHD) are one of the most common birth abnormalities, affecting approximately 0.8–2% of live births worldwide [1–3]. It is widely accepted as having a multifactorial etiology with complex interactions between genetic and environmental factors during fetal development [4].
NOTCH1-signalling is one of the most important mechanisms during embryogenesis [5]. NOTCH1 encodes a large single-pass membrane receptor that is involved in cell fate determination, differentiation, and the development of the nervous and cardiovascular systems. The latter includes regulation of cardiac precursor development, angiogenesis, vasculogenesis, and epithelial-mesenchymal transition during valve development [5, 6].
Initial reports of non-syndromic CHD in humans associated with NOTCH1-variants focused on malformations of the left ventricular outflow tracts (MIM#109730). However, subsequent publications have significantly broadened associated phenotypes, suggesting that conotruncal malformations of the cardiac outflow tract are a prevailing outcome, with phenotypes such as Tetralogy of Fallot (ToF), truncus arteriosus communis (TAC) and double outlet right ventricle (DORV) [7, 8]. Patients with NOTCH1-variants may have an elevated risk for aneurysms of the ascending aorta [9]. Somatic activating NOTCH1 variants have also been related to tumorigenesis [10, 11]. In addition, NOTCH1-variants are known to cause Adams-Oliver syndrome, a condition mainly characterized by terminal transverse limb defects, aplasia cutis congenita, and various forms of CHD [12].
Understanding the genetic basis of CHD is crucial for improving diagnosis, for outcome or recurrence risk prediction, and for developing targeted therapies. Although next-generation sequencing (NGS) has been successful in identifying genetic variants associated with CHD, the mechanisms by which these pathogenic variants lead to CHD remain largely unknown. In addition, genomic studies are complicated by genetic heterogeneity of CHD and by the abundance of variants of unknown significance (VUS). Individual functional testing of VUSs to confirm or refute their contribution to CHD is complex and time-consuming, and typically not performed in a diagnostic context. Recently, the testing of episignatures (DNAm) has evolved as an easy method to screen such cases, as it uses readily available genomic DNA from peripheral blood samples.
Epigenetics involves the study of heritable changes in gene expression that occur without altering the underlying DNA sequence. Among these mechanisms, DNA methylation is the most thoroughly studied. Numerous rare genetic disorders have been linked to unique DNA methylation profiles, known as episignatures [13]. In recent years, episignatures have emerged as robust and reliable biomarkers, playing a crucial role in diagnosing congenital genetic disorders and reclassifying VUSs [14–18]. Their application in clinical diagnostic laboratories has demonstrated significant utility in providing diagnoses for patients with suspected rare genetic conditions who previously lacked a clear genetic diagnosis [19].
Reports of de novo variants histone modifying genes as well as altered DNA methylation in the context of CHD suggest that these mechanisms might contribute to the etiology of this disease [20–22].
Given that our cohort revealed NOTCH1 as the most common monogenic cause, we aimed to explore whether effects of these variants might also manifest in the DNA-methylation pattern. In light of the high abundance of variants in NOTCH1 in CHD patients, this episignature can have a considerable contribution to the diagnostic management of these patients.
Methods
Discovery cohort description
The work presented herein is primarily based on a cohort of 3907 exome-sequenced patients with CHD and 5157 population-matched controls [23, 24]. 1438 (37%) displayed extracardiac phenotypes (syndromic CHD, S-CHD), while 2469 (63%) non-syndromic heart defects (non-syndromic CHD, NS-CHD). 977 individuals presented with conotruncal defects, including 484 cases with ToF. Cases with various subgroups of CHD were included as long as the patient required intervention within the first year of life. Samples with known structural variations, such as 22q11 syndrome, or chromosomal aneuploidies, such as trisomy 21, were excluded if such a diagnosis was reported. Patient recruitment was conducted through multiple centers across Germany as well as from international centres [23, 24].
Samples were subjected to exome sequencing on DNA from peripheral blood using different versions of the SureSelect Exome chips (Agilent). Enriched libraries were subjected to 75-base paired-end sequencing (Illumina HiSeq). Data curation and quality filtering of the sequencing data were performed in accordance with previous work of our group [23, 24]. Samples were restricted to European ancestry.
Following the quality control steps outlined above, variants were functionally annotated using the Variant Effect Predictor tool (VEP v.104) [25], extended using the plug-ins CADD (version 1.6) and dbNSFP (version 4.1a) and evaluated based on the canonical transcript as defined by Ensembl (https://www.ensembl.org).
Functional domains and sites of post-translational modification in NOTCH1 were retrieved from UniProt (Identifier: P46531) (Table S1). Variants were collapsed into protein truncating variants (PTV), protein-altering variants (PAV) and synonymous variants (SYN).
Analyzed variants were prefiltered to an ultrarare frequency defined as a minor allele count (MAC) of ≤ 2 in gnomAD V4.1.0, in a set of internal unpublished control samples of German origin and the UK-BioBank (UKBB). Variant pathogenicity was assessed following the workflow presented by the American College of Medical Genetics (ACMG) [26, 27]. The severity of PAVs was assessed using in-silico prediction tools CADD, MPC, and REVEL [28–30]. Thresholds were used following the suggestions made by Pejaver et al. [31]
For the classification of splice site variants, SpliceAI was used with a cut-off of ≥ 0.5 [32]. Enrichment testing was carried out using a two-sided Fisher´s exact test (FET) and false-discovery rate (FDR) adjustment for multiple testing (n = 17 tests).
For all cases that underwent methylation profiling, genes related to DNA- or histone-methylation processes were reviewed. Genes were selected based on GO-terms GO:0035514, GO:0009008, GO:0140188, GO:0140940, GO:0140938 and GO:0140939 and filtered for species homo sapiens (see https://geneontology.org/) (Table S2). Variants were screened for pathogenic or likely pathogenic variants following the ACMG guidelines [26, 27].
Assembly of additional cases
The findings from the initial cohort were extended using a genome-sequencing-based dataset of 1044 probands with non-syndromic congenital heart disease (CHD), containing 218 cases with transposition of the great arteries (TGA) and 826 with ToF. These samples were provided as part of a joint cohort from the Heart Centre Biobank Registry at the Hospital for Sick Children (Ontario, Canada), the Kids Heart BioBank at the Heart Centre for Children, The Children’s Hospital at Westmead (Sydney, Australia) and the CONCOR-project (Amsterdam Medical Center; Netherlands) [33, 34].
Sequencing was performed on DNA from blood or saliva of probands using the Illumina HiSeqX using the Illumina TruSeq DNA PCR-Free kit. The reads were trimmed and cleaned by trimmomatic v.0.32 [35], then mapped to human reference genome hg38 using bwa v.0.7.15 [36], followed by realignment and calibration(GATK v.4.1.2.0). HaplotypeCaller was used to generate genotype Variant Call Format (gVCF) files for each sample, combined and joint called (CombineGVCFs and GenotypeGVCFs tools). SNVs and indels were recalibrated separately by variant quality score recalibration (VQSR) tools, and variants that passed VQSR truth sensitivity level 99.5 for SNPs and level 99.0 for indels were retained. The VariantFiltration tool was used to mark out the low Genotype Quality (GQ) SNV and indel sites whose GQ values were lower than 20 and read depths were lower than 10.
Post processing of the data was performed using Bcftools view (v1.9) to subset the joint-called whole genome VCFs for the region of interest (chr9:136,484,054–136,580,643) [37], followed by decomposition and normalisation using vt v0.5, and annotation using VEP (v104.1) and VCFanno v0.3.1 [25, 38, 39].
Filtering and estimation of the deleteriousness of the variant were carried out as described for the initial cohort.
In addition, further variants were retrieved through personal communication with different collaboration partners, as outlined in Table 2.
Review of published NOTCH1-variants
Variants in Clinvar and publications reporting NOTCH1-related cases were collected from PubMed as of June 2024. Search parameters were “NOTCH1 and CHD or congenital heart defects or AOS or Adams-Oliver syndrome”. Publications were manually revised. Variants that were explicitly cited from previous publications or with missing information regarding the position or patient’s phenotype were excluded, as were synonymous variants and variants that were considered benign by the authors (Table S3).
Study cohort - methylation
A total of 26 individuals (12 males and 14 females) with NOTCH1-variants for whom material was available were included in the analysis of DNA methylation. The individuals were divided into one group for the discovery of the episignature (n = 19, of which 3 were negative for the episignature and removed) and additional samples to independently validate (n = 3) and assess VUS variants (n = 4). We used the discovery cohort for probe selection and construction of the classification model for the episignature. All of these individuals had confirmed deleterious variants in NOTCH1.
DNA methylation data
Bisulfite-converted genomic DNA, extracted from peripheral blood, underwent application to the Infinium Methylation EPIC Bead Chip (Illumina, San Diego, CA) array following the manufacturer’s protocol. Subsequently, utilising the minfi R package (version 1.44.0) and the intensity data files (IDATS) containing methylated and unmethylated signal intensities produced post-EPIC array were preprocessed and imported into R (version 4.2.3) [40]. Standard preprocessing methods for Illumina microarrays were employed, involving background correction and normalisation. Quality control procedures included examining density plots and verifying concordance between recorded and predicted sex and age. Finally, probes were filtered by excluding probes overlapping with single-nucleotide variations, cross-reactive probes, probes specific to regions on the X or Y chromosomes, and probes with a detection p-value >0.1. The resulting number of probes after this filtration process was 772,557.
DNA methylation analyses
DNA methylation analyses were conducted following our previously published methodology [13, 15]. Matched controls were chosen from the EpiSign Knowledge Database (EKD) based on age, sex, batch, and array type using the R package MatchIt (version 4.5.2) [41]. Samples exhibiting batch effects and/or more than 5% probe failure in the EKD were excluded. The training cohort and matched case-control samples underwent examination for data structure and outliers through principal component analyses (PCA). Subsequently, feature selection was performed using matched cases and controls. Differential methylation analysis was carried out utilizing the limma package (version 3.54.2) [42] with linear regression fitting. Methylation beta values served as predictors, and labels were used as the response, adjusting the model for estimated blood cell counts as confounding variables. The empirical Bayes method was applied to control for false discoveries, and adjustments were made using the Benjamini-Hochberg procedure to compute the moderated t-statistics and p-values. To ensure biological relevance, probes with a mean methylation difference below 5% (Δβ < 0.05) between cases and controls were excluded. Each remaining probe was ranked using a composite score that combined effect size (absolute Δβ) with statistical confidence (–log10 FDR-adjusted p-value). From this ranking, the top 800–1000 probes were retained. These were further refined by receiver operating characteristic (ROC) curve analysis (retaining probes with high AUC values) and by removing probes with high inter-probe correlation based on Pearson’s correlation coefficient, yielding a final set of 160–500 informative [13].
Further exploration involved investigating the distinct clustering of cases and controls using heatmaps and multidimensional scaling (MDS) with ggplots2 (version 3.1.3). The optimal clustering was selected based on parameter values. Leave-one-out cross-validation and unsupervised clustering results were employed to assess the reproducibility of the episignature (Figure S1).
Prediction model
The sensitivity and specificity of the NOTCH1-episignature cohort were assessed through a classifier employing all episignature probes. A support vector machine (SVM) model was trained using the R package e1071 (version 1.7–13) with the selected features and matched controls and cases as training data. To enhance specificity, 75% of the samples in the EKD (comprising those with an episignature, unaffected samples, and training controls) were included, while the remaining 25% were designated for testing. This process was iterated four times, ensuring that each sample served as a testing sample once. The average SVM, also known as the methylation variant pathogenicity (MVP) score, was then employed for further analysis. Rare disease episignature classification typically involves a substantial proportion of unaffected or “normal” samples alongside affected cases. In this context, SVM’s provide a superior capacity compared to alternative machine learning models, as it allows more accurate discrimination between disease states and unaffected backgrounds, as well as among different episignature-positive conditions.
Overlap of the NOTCH1 genome-wide dna methylation profile with other episignature positive rare disorders
Functional annotation and comparison of the EpiSign™ classifier v5 cohort were conducted based on previously published articles [43]. The assessment involved determining the percentage of differentially methylated positions (DMPs) shared between the NOTCH1-episignature and the other 99 neurodevelopmental disorder episignatures on the EpiSign™ v5 clinical classifier. Heatmaps were created using the R package pheatmap (version 1.0.12), and circos plots were produced with the R package circlize (version 0.4.15) [44]. To identify relationships across all cohorts with known episignatures, clustering analysis was performed. Utilising the R package TreeAndLeaf (version 1.6.1) [45], a tree and leaf plot was generated to visualise the distances and similarities between the cohorts. For an exploration of the genomic location of the selected DMPs, probes were annotated in relation to CpG islands (CGIs) and genes using the R package annotatr (version 1.20.0) [46] with AnnotationHub (version 3.2.2), as described previously by Levy et al. [43].
In-silico modelling of NOTCH1 variants
For each variant-related region, structural models were generated using AlphaFold3. The modelled structures were subsequently subjected to conformational sampling with PyRosetta [47, 48], using 20 independent FastRelax trajectories under the ref2015 scoring function [49, 50]. Both backbone and side-chain flexibility were allowed, with disulfide bonds constrained according to the Uniprot annotations and AlphaFold3 prediction. Among the resulting models, the structures within the lowest energy were selected for downstream analysis. Underlying models and resulting structures are outlined in Table S4 and Figures S5-S9.
Results
Enrichment of deleterious NOTCH1-variants in a large CHD case-control cohort
In the analyzed cohort of 3907 exome-sequenced patients with CHD and 5157 population-matched controls, deleterious NOTCH1-variants were the most frequent monogenic finding. Filtering regarding ultrarare variants affecting the coding region and canonical splice sites of NOTCH1 yielded 76 variants. Based on this initial variant set, enrichment testing was performed for truncating variants (PTVs), synonymous (SYN) and protein altering variants (PAVs), which were grouped based on in-silico predictions. Furthermore, distinct functional domains were tested for individual enrichment of PAVs. We investigated disulfide bridges, as these are frequent, especially in the extracellular EGF-like domains, and are essential for correct protein folding.
PTVs, as well as PAVs with strong in-silico pathogenicity predictions, were enriched (p_adj_ = 1.09e-04, p_adj_ = 0.047) (Table 1). Furthermore, ultrarare PAVs that disrupted disulfide bridges were enriched (p_adj_ = 0.025) and were almost exclusively found amongst patients with ToF (9/10 cases). In total, this type of variant was present in 1.85% (9/484) of ToF-cases. EGF-like repeats were also significantly more affected in CHD-cases. However, this was attributed mainly to overlap with disulfide-bond affecting variants (the exclusion of these variants results in a loss of significance (ratio 8 to 4, p_raw_ = 0.143)). Amongst the individual EGF-like repeats, no individual repeat was significantly enriched (data not shown). Interestingly, no PAVs in the vicinity of the ligand binding site (residues 420–421, 448–452, 469 each ± 5 residues) were observed. For none of the other tested functional domains, particular enrichment was observed. None of the discovered cases had pathogenic variants in established CHD genes or genes involved in DNA- or histone-methylation.Table 1. Enrichment testing for ultrarare NOTCH1-variants. Testing results of ultrarare variants affecting NOTCH1 in 3907 CHD cases vs. 5157 controls. Testing was performed using FET. P-values were adjusted using false-discovery rate (FDR) with n = 17 tests. Significance was defined as p_adj_ < 0.05. Significant scenarios are printed in bold. PTVs are defined as stop-gain, frameshift, and splice-site variants. PAVs are defined as missense and indels. PP3 corresponds to the severity of in-silico prediction tools evaluating REVEL, CADD and MPC-score as proposed by Pejaver et al. [31] ANK = Ankyrin domain, CI = confidence interval, EGF = Epidermal growth factor, FDR = false discovery rate, HD = heterodimerisation domain, LNR = Lin12/Notch repeats, OR = odds ratio, PAV = Protein altering variant, PEST = PEST domain, PP3 = ACMG criterion for deleterious in-silico predictor, PSEN = Interaction with presenelin 1, PTV = Protein truncating variant, RAM = RBP-Jκ-associated module, TAD = transcriptional activation domain, SYN = synonymous variantScenarioCarrier casesCarrier controlsp__raw_p__FDR_ORCI95%PTVs1716.41e-061.09e-0422.533.5–937.8Disulfide bonds1011.47e-030.02513.231.9–572.8EGF-like repeats1751.82e-030.0304.501.6–15.6PAVs (PP3str)702.76e-030.047Inf1.9 - InfPAVs (PP3mod)1030.0210.3644.411.1–24.9Novel cysteine formation400.0340.586Inf0.9 – InfEGF-like repeats(excl. Disulfide bonds)840.14312.640.7–12.0RAM100.4311Inf0.03 - InfAnkyrin210.58112.640.1–155.7PAVs (PP3sup)320.65811.980.2–23.7SYN660.77211.320.4–4.9TAD33111.320.2–9.9LNR22111.320.1–18.2PEST22111.320.1–18.2PAVs (neutral or benign in-silico)912110.990.4–2.6HD011100–51.4PSEN011100–51.4
Review of published NOTCH1-variants
Through a review of publications that reported NOTCH1 variants, we assembled a list of 204 unique variants reported in 304 cases (Table S3). Of these cases, 238 were reported in the context of CHD, 22 in the context of thoracic aortic aneurysms (TAAD) and 44 in Adams-Oliver syndrome (AOS).
Missense variants were distributed throughout the entire protein without overrepresentation of particular domains. NOTCH1 missense variants affecting disulfide bridges were reported in both CHD and AOS cases with a slight enrichment in AOS (14/238 CHD, 10/44 AOS, p = 0.001; OR 4.7, 95%-CI 1.71–12.4, two-sided FET). One disulfide-altering variant was also found in a TAAD-case. Interestingly, all 14 variants affecting disulfide bridges identified in CHD cases were reported to have ToF or related malformations. A comprehensive overview of all disulfide-impacting variants can be found in Table S5. In addition, PAVs in the vicinity of the ligand binding site were found in 8/44 AOS patients versus 2/238 CHD cases (p = 6.93e-06; OR 25.7, 95%-CI 4.9–256.5.9.5, two-sided FET).
Assembly of additional cases
In an independent genome-sequenced cohort [34], we identified no variants similar to the ones in our case-control cohort among 218 cases of TGA. However, in 826 ToF cases, we identified four ultrarare PTVs, five cases with ultrarare PAVs disrupting disulfide bonds, and two additional ultrarare PAVs with strong in-silico prediction scores (Table 2). Collectively, deleterious variants in NOTCH1 were thus found in 1.5% (10/641) of European ToF cases and 1.3% (11/839) of samples regardless of population (Fig. 1A). In addition, we assembled a further 17 samples from the centres of various co-authors. These either fulfilled the filtering criteria established above, were deemed potentially causal due to evidence from segregation with the disease within the family, or were considered of interest for validation purposes regarding the specificity of the episignature analysis (see below). Collectively, we found 63 ultrarare, deleterious NOTCH1-variants in 67 individuals (Fig. 1; Table 2).Table 2. Overview of all identified NOTCH1-variants. Samples with pathogenic or likely pathogenic variants from the initial case-control cohort are listed, followed by samples with VUSs from the initial cohort. Lastly, variants from the GS-cohort and additional collaboration partners are presented. Variants are classified based on the ACMG-classification scheme. Where available, the segregation of the variant is given with [S] indicating, that a targeted testing was performed using Sanger-sequencing and [ES] indicating that the inheritance pattern was determined based on exome-sequencing data. ACH = Alberta Children´s Hospital, ASD = atrial septal defect, ASDII = secundum atrial septal defect, AUMC = Amsterdam University Medical Centre, AV= Aortic valve, BAV = bicuspid aortic valve, CCHMC = Cincinnati Children’s Hospital Medical Center, CoA = Coarctation of the aorta, DORV = double outlet right ventricle, F = female, HLHS = Hypoplastic left heart syndrome, HSC = Hospital for Sick Children Toronto, LP = likely pathogenic, LVOTO = Left ventricular outflow tract obstruction, M = male, MVP = Mitral valve prolapse, n.a. = not available, n.t. = not tested, P = pathogenic, PA = pulmonary artery atresia, PAH = Pulmonary artery hypertension, pDA = Patent ductus arteriosus, PvA = Pulmonary valve atresia, Pro = Proceed-cohort (replication GS-cohort), PS= Pulmonary stenosis, RV = right ventricle, TAC = Truncus arteriosus communis, TGA = Transposition of the great arteries, ToF = Tetralogy of Fallot, UHL = University Hospitals Leuven, VSD = ventricular septal defect, VUS = Variant of uncertain significancePathogenic or likely pathogenic variants#NOTCH1 genotype(ref: NM_017617.5)Class (ACMG)Previously reportedSegregation statusSampleGenderCardiac phenotypeExtacardiac phenotypeFamilial CHD historyRemarks1c.617G>T p.(Cys206Phe); c.619C>T p.(Arg207Cys)LP;VUSNovel;ClinvarID: 2164530(3x VUS)n.t. C08660MHemitruncusAnisocorian.a.Positive DNAm signature (0.98549) [Discovery]2c.995G>A p.(Cys332Tyr); c.6376G>A p.(Gly2126Arg)LP;VUSCOSM3716156;rs572960572n.t. C07803MToF and variantsSeizure with abnormal EEG, neurodermitis, polyps as infant Mother with TOFPositive DNAm signature (0.98568) [Discovery]3c.1143T>A p.(Cys381Ter)LPNoveln.t. C08987FDORVOsteoporosis, liver insufficiency, kidney insufficiencyEmptyPositive DNAm signature (0.98551) [Discovery]4c.1789G>C p.(Gly597Arg)LPNoveln.t. C08017MToF and variants, dextrocardiaHepatomegalyEmptyAlso carries VUS in TLL1 (NM_012464.5:c.555G>C)5c.1852T>A p.(Cys618Ser)LPNoveln.t. C08732MToF and variantsn.a.Empty6c.1886G>C p.(Cys629Ser)LPNoveln.t. C07885FToF and variants, Aortic aneurysmHypothyroidismn.a.7c.1913G>A p.(Cys638Tyr)LPCOSV53094192n.t.C08084MVSDNeck fistulan.a.Father of C080858c.1913G>A p.(Cys638Tyr)LPCOSV53094192Inherited from affected father [ES] C08085FToF and variantsGeneralized muscular hypotoniaFather with VSD;Cousin with severe developmental delay; maternal uncle with spontaneously closed VSDDaughter of C08084 9c.2061C>A p.(Cys687Ter)LPNoveln.t. C08345FVSDRecurrent infections, mild developmental delayn.a.Positive DNAm signature (0.9855) [Discovery] 10c.2176_2181GTGGAC>TGCAACA p.(Val726fs16)LPNovelDe novo* [S] C07635FHypoplastic right heartn.a.n.a.Positive DNAm signature (0.98549) [Discovery] 11c.2846_2856GTGCCAGTGAC>CCT p.(Cys949Serfs29)LPNovelDe novo* [S] C07558MToF and variantsn.a.n.a.Positive DNAm signature (0.98548) [Discovery]12c.3319C>T p.(Arg1107Ter)Prs41309764, ClinvarID: 12476 (3x P), CM053346,PMID: 16025100n.t. C07810FTricuspid atresiaMild developmental delayn.a.Positive DNAm signature (0.98552) [Discovery]13c.3507C>A p.(Cys1169Ter)LPNoveln.t. C07994MHLHSHydrocephalus internus, mild muscular hypotonia, intracerebral haemorrhage, delayed wound healing, increased scarring, recurring otitis mediaEmptyPositive DNAm signature (0.98548) [Discovery]14c.3511-2A>G p.(?)LPPMID: 26820064De novo [S] C07964FTAC, peripheral PSStrabism, unilateral hearing impairmentEmptyPositive DNAm signature (0.98551) [Discovery]15c.4549G>C p.(Asp1517His)LPCOSM124799, Clinvar: 3048375 (1x LP)n.t. C08171FToF and variantsHypothyroidism, kidney cyst, kidney insufficiency n.a.Positive DNAm signature (0.98549)[VUS-evaluation]16c.4646G>A p.(Cys1549Tyr)LPNovelDe novo [ES] C01022FToF and variantsn.a.n.a.17c.4913G>A p.(Trp1638Ter)LPNoveln.t. C00902MAbnormality of cardiovascular system morphologyHemiatrophy, dentiogenesis imperfecta, inflammation of the large intestine, generalized joint laxityn.a.18c.5197C>T p.(Gln1733Ter); c.452A>G p.(Asn151Ser); c.3880G>A p.(Glu1294Lys)LP;VUS;VUSrs1208976166;rs766362765; COSM5880101,rs1247035429;PMID: 30582441n.t. C02192MToF and variantsCraniosynostosisBrother with aortic anomaly,maternal aunt with heart murmurPositive DNAm signature (0.986) [Discovery]19c.5197C>T p.(Gln1733Ter); c.3880G>A p.(Glu1294Lys)LP;VUS;VUSrs1208976166; COSM5880101,rs1247035429;PMID: 30582441n.t. C02193FControl samplen.a.n.a.20c.5315_5316G>GC p.(Phe1773fs5)LPNovelDe novo* [S] C07685FHypoplastic right heartRecurring otitis media, hypertrophic osteoarthropathyEmptyPositive DNAm signature (0.98551) [Discovery]21c.5385-2del p.(?)LPCOSM5622683De novo [ES] C01099MToF and variantsn.a.n.a.22c.5922CT>C p.(Gln1974fs6)LPNovelDe novo* [S] C08838FASDIIn.a.n.a.Positive DNAm signature (0.98549) [Discovery]23c.5950C>T p.(Arg1984Ter)PPMID: 26820064n.t. C08330FDORVLiver insufficiencyn.a.Positive DNAm signature (0.9855) [Discovery]24c.6053_6054del p.(His2018fs7)LPNoveln.t. C07725FCoA, BAV, right PAHallux, scoliosis, recurrent severe infections, muscular hypotoniaFather with cardiac phenotype as a child (no details available)Positive DNAm signature (0.98864) [Discovery] Ultrarare variants of uncertain significance#NOTCH1 genotype(ref: NM_017617.5)Class (ACMG)Previously reportedSegregation statusSampleGenderCardiac phenotypeExtacardiac phenotypeFamilial CHD historyRemarks25c.436_450dup p.(150_151insSerAsnProCysAla)VUSClinvarID: 2169548(1x VUS)n.t. C07149MToF and variantsn.a.n.a.Also carries VUS in SMAD6 (NM_005585.5: c.511G>A)Creates novel cysteinePositive DNAm signature (0.98547) [Discovery]26c.797G>T p.(Cys266Phe) c.798C>T p.(Cys266=)VUS;VUSCOSM6981052Maternal [S] C08918FToF and variantsn.a.n.a.Predicted splice donor gain27c.968G>T p.(Cys323Phe)VUSCOSM6903534Paternal [S] C08178MToF and variants n.a.n.a.Disrupts disulfide bridge28c.1009G>C p.(Asp337His)VUSNoveln.t.C08917MToF and variantsn.a.n.a.Moderate in-silico score29c.1057C>T p.(Arg353Cys)VUSrs1300110216, PMID: 30582441n.t.C07205FToF and variantsn.a.n.a.Creates novel cysteine30c.1109G>A p.(Cys370Tyr)VUSrs1564199987, ClinvarID: 576792 (1x VUS)Maternal [ES] C02162FToF and variantsn.a.n.a.Disrupts disulfide bridge31c.1648T>G p.(Tyr550Asp)VUSCOSM753122, ClinvarID: 1033432 (1x VUS) n.t. C06994MTACFacial asymmetry, sprengel anomaly, specific learning disability, high anterior hairline, plagiocephaly, micropenis, sleep disturbance, attention deficit hyperactivity disordern.a.Moderate in-silico score32c.2116G>T p.(Glu706Ter)VUSNovelPaternal [S] C07611MDORVHepatomegalyEmptyTruncating variant33c.2153A>G p.(Asn718Ser)VUSCOSM4411745,ClinvarID: 1029477 (1xLP, 2x VUS),PMID: 31813956n.t. C00994MCoAAutistic behaviour, delayed speech and language development, intellectual disability (mild), motor delayn.a.Predicted splice donor gain, brother of C0100734c.2153A>G p.(Asn718Ser)VUSCOSM4411745,ClinvarID: 1029477 (1xLP, 2x VUS),PMID: 31813956n.t. C01007MVSDAutism, delayed speech and language development, intellectual disability, (mild), poor coordination, moderate global developmental delayn.a.Predicted splice donor gain, brother of C0099435c.2354-2A>G p.(?)VUSCOSV53109619n.t. C01673MHLHSn.a.n.a.Altered splicing potentially preserves reading frame;Mild DNAm signature (0.88909)36c.3280_3285dupp.(1095_1096insCysAsp)VUSNoveln.t.C00068MAbnormal heart morphologyMicrocephaly, congenital cataract, mixed hearing impairment, sleep apnoea, constipationn.a.Creates novel cysteine37c.5612G>T p.(Cys1871Phe)VUSClinvar: 2182606 (2xVUS)n.t.C07622MToF and variantsn.a.n.a.Affects cysteine residue, residue is not known to form a disulfide bond38c.6211del p.(Glu2071fs38); c.2054A>C p.(Asn685Thr)VUS;VUSNovel;rs781473342n.t. C01759MCoA, BAVn.a.n.a.Not predicted to undergo NMD,Positive DNAm signature (0.98551) [Discovery]39c.6221A>G p.(Tyr2074Cys)VUSNovelPaternal [S] C08510MCoA,Aneurysm fossa ovalisEsophagus atresia, polyhydramnionn.a.Creates novel cysteine residuePositive DNAm signature (0.89228)[VUS-evaluation] Variants from GS cohort#NOTCH1 genotype(ref: NM_017617.5)Class(ACMG)Previously reportedSegregation statusSampleGenderCardiac phenotypeExtacardiac phenotypeFamilial CHD historyRemarks40c.515G>T p.(Gly172Val)LPnoveln.t.Pro1FToF and variantsNoPat. grandfather and pat. first cousin with TOF41c.1342C>T p.(Arg448Ter)LPrs869025494, PMID: 30582441n.t.Pro2MToF and variantsShort sightednessPat. first cousin with CoA, MVP and small left ventricle42c.1820G>A p.(Cys607Tyr)LPPMID 30582441n.a.Pro3FToF and variantsNoFather with MVP43c.2182G>C p.(Gly728Arg)LPnoveln.a.Pro4MToF and variantsNoEmptySample is predicted to be of East-asian ancestry44c.2477G>A p.(Cys826Tyr)VUSnovelMaternalPro5MToF and variantsNoEmptyDisrupts disulfide-bridge45c.2595del p.(Cys866Valfs10)LPnoveln.t.Pro6MToF and variantsNoEmpty46c.4190G>A p.(Cys1397Tyr)LPnoveln.t.Pro7FToF and variantsNoUnknown47c.4190G>A p.(Cys1397Tyr)LPnoveln.t.Pro8MToF and variantsNoUnknown48c.4415G>A p.(Cys1472Tyr)LPPMIDs: 30293987, 31813956n.t.Pro9FToF and variantsCutis aplasiaNephew with TOF49c.4501delp.(Ser1501Valfs79)LPnoveln.t.Pro10MToF and variantsNoMat. aunt with TOF, died w/o surgery at 6 m of age,Pat. grandfather has an MI at age of 6050c.4592delp.(Leu1531Argfs49)LPnoveln.t.Pro11FToF and variantsNoUnknown Additional variants from collaboration partners#NOTCH1 genotype(ref: NM_017617.5)Class(ACMG)Previously reportedSegregation statusSampleGenderCardiac phenotypeExtacardiac phenotypeFamilial CHD historyRemarks51c.568C>T p.(Arg190Cys)VUSn.t.Pro12FToF and variantsUnknownUnknownCreates novel cysteine residue52c.599G>C p.(Gly200Ala)VUSnoveln.t.Pro13FToF and variantsUnknownUnknownModerate in-silico prediction for pathogenicity53c.724G>A p.(Glu242Lys)VUSrs564629053Inherited from affectedmotherAUMC1MCardiac evaluation: normalObesity, autism spectrum disorder, intellectual disability, alopecia, delayed pubertyMother with similar phenotypeNegative for NOTCH1-DNAm signature (0.0157)[VUS-evaluation]54c.1057C>T p.(Arg353Cys)VUSPMID: 30582441n.tPro14FToF and variantsNoMother's maternal cousin was born with CHD and died at a few days of ageCreates novel cysteine residue55c.1070T>C p.(Phe357Ser)VUSnovelInherited from affected fatherPro15FToF and variantsMicrocephalyFather - VSD Repair and Implantation of mechanicalprosthetic AVModerate in-silico prediction for pathogenicity56c.1279G>C p.(Gly427Arg)LPnovelDe novoLUV1 (UHL)FPrimary pulm. hypertension, pDAAdams-Oliver Syndrome, alopecia, mild hand/feet anomalies, thrombopenia, granuloma of the upper lipEmptyNegative for NOTCH1-DNAm signature (0.10763)57c.1655G>A p.(Cys552Tyr)LPnovelDe novoLUV2 (UHL)MToF and variantsPlagiocephaly, delayed development (total IQ 83), ASS, long triangular faceEmptyPositive DNAm signature (0.98549)[Validation]58c.1774C>T p.(Arg592Cys)VUSrs1472690723n.a.AUMC2MTGAUnknownUnknownMAC 17 in gnomAD V4.1.0Negative for NOTCH1-DNAm signature (0.45762)59c.1766G>T p.(Cys589Phe)LPnoveln.a.MT1 (ACH)FMild PVS, not clinically significantHeight slightly < p3, Cutis aplasia, 6.5 cm long and 1 cm wide and at the midline top of the scalp, normal digits, possible subtle cutis marmorata, no brachydactylyTwo pregnancies with similar cardiac anomalies (1x ToF and variants, 1x DORV)60c.1864G>A, p.(Asp622Asn)VUSrs367873715, Clinvar 659037n.a.AUMC3MSlight RV dilatation,Idiopathic PAHDiabetesEmptyKnown PAH genes have been excluded,Negative for NOTCH1-DNAm signature (0.53211)[VUS-evaluation]61c.1904-2A>G p.(?)VUSnoveln.a.DW1(CCHMC)FToF and variantsChoanal atresiaFather with smallhole in heartNot predicted to disrupt reading frame62c.2014+1G>A p.(?)Prs515726232, Clinvar 139664, PMID: 25931334Inherited from affected fatherLUV5 (UHL)MDORV, TGANoFather with ASDII, two siblings (fetuses) with HLHSPositive DNAm signature (0.98551)[Validation]63c.2433_2452dup p.(Leu818Profs65)PnovelInherited from affected motherJB1(UHL)MCoA, asymmetric tricuspid AV,parachute-like MVNoMother with BAV, congenital strabism and ptosis of the right eyelid,Mat. grandfather with aneurysm of the abdominal aorta, he´s not carrier of the variant.Mat. grandmother had AV-replacement (no genetic testing)64c.5281delCp.(Arg1761Glyfs37)Prs515726231, PMID: 25931334Inherited from affected fatherLUV4(UHL)FMild CoA, asymmetric tricuspid AVNoFather with CoA, paternal half-brother with ToF, PA, VSD, type DORV,several other family members with LVOTO, or non-affected carriersPositive DNAm signature (0.9855)[Validation]65c.5527T>C p.(Trp1843Arg)VUSnoveln.t.Pro16MToF and variantsNoUnknownModerate in-silico prediction for pathogenicity66c.5767del p.(Gln1923fs67)PnovelInherited from affected fatherSM1 (HSC)MHLHS/DORVNoFather with PA & VSD, pat. uncle with ToF and variants,pat. grandmother with unspecified valve defect.67c.7171C>T p.(Gln2391Ter)VUSnoveln.t.SM2 (HSC)MHLHSDevelopmental delay, speech disorder, hemiparesis right-sidedEmptyNot predicted to undergo nonsense-mediated decay
Fig. 1. Overview of variants found in NOTCH1.** A**: Representation of NOTCH1 variants found in the analyzed samples. Each dot represents an identified variant colour-coded by the corresponding phenotype group. “TOF (-like)” includes ToF, DORV and TAC (blue). “AOS_features” marks cases that displayed extracardiac anomalies possibly consistent with an AOS phenotype (red). Other CHD (darkgray) reprents all non-TOF-like phenotypes. Cases without CHD are shown in light gray. Functional domains are shown based on Uniprot (for details refer to Table S1). B: Overview of previously reported missense-variants. (see Table S3 for details). Variants are split and color-coded based on the corresponding reported phenotype. Black: (ns-)CHD, Red: Adams-Oliver-Syndrome, Blue: thoracic aortic aneuyrism. C: Density plot of missense variants present in gnomAD V4.1.0. D: Amino acid conservation as retrieved from Aminode.[53] Depicted is the substitution score per amino acid. High values indicate a low conservation of the residue
Of note, regions surrounding and preceding the extracellular ligand binding site, as well as the intercellular ankyrin-repeats, display high conservation and low variant abundance in the population. Interestingly, we did not observe variants near the ligand binding site (Fig. 1A), while this appears to be a region frequently affected in AOS (Fig. 1B).
Discovery and validation of the NOTCH1-episignature
The collected cases displayed a heterogeneous phenotypic outcome as well as the type and classification of variants affecting NOTCH1 which called for an accurate and accessible evaluation of these variants. We thus explored DNA methylation testing on a subset of available samples. The resultant probe set generated from a discovery cohort of 19 samples effectively distinguished between cases and controls (Fig. 2, Table S6). However, three samples from the discovery group (cases #35, #56, #58) didn’t align with the episignature and were excluded from the training set (Fig. 2, light purple). Two of these samples consistently grouped with controls in the heatmap and MDS plots, displaying MVP scores close to 0; the other sample had a higher MVP score (0.88) but was not consistently grouped with the discovery cases and couldn’t be included in the discovery cohort. To validate the *NOTCH1-*episignature, we used three additional samples with confirmed pathogenic variants in NOTCH1 (cases #57, #62, #64), all of which aligned with the episignature (Fig. 2A, dark purple). Furthermore, each of these cases yielded a high MVP score, affirming their resemblance to our *NOTCH1-*episignature (Fig. 2B, dark purple).
Fig. 2. Discovery and validation of the NOTCH1-episignature**. A**: The Euclidean hierarchical clustering heatmap depicts each column representing one NOTCH1 discovery case (highlighted in red), along with mild signatures (light purple), VUS (yellow), and validation samples (dark purple). Each row corresponds to a specific probe selected for this episignature. Notably, a distinct separation is observed between the cases (in red) and controls (in blue). However, it’s worth mentioning that the negative cases tend to cluster together with the controls, except for one. B: The multidimensional scaling (MDS) plot illustrates the separation between NOTCH1 cases and controls, including the negative sample identified in (A). C: In the SVM classifier model, the selected NOTCH1-episignature probes were used to train the model. 75% of controls and 75% of samples from other neurodevelopmental disorders (depicted in blue) were utilised for training, while the remaining 25% of controls and 25% of other disorder samples (grey) were reserved for testing
After having successfully established an episignature, we investigated whether we could use it to reclassify VUSs. To this end, we evaluated four VUS samples (cases #15, #39, #53, #60). Our analysis revealed that two samples (cases #15, #39) aligned with the *NOTCH1-*episignature, while the other two did not (Fig. 2A and B, yellow).
Comparison of the NOTCH1 global DNAm profile with other neurodevelopmental disorders included in the EpiSign V5 classifier.
To explore the concurrence between the DNAm profiles defining the NOTCH1 cohort and those previously identified in 99 other disorders using the EpiSign™ v5 classifier [13, 19, 51, 52], a functional analysis focusing on the overall DNAm alterations observed in the NOTCH1 cohort was conducted. Regarding the genomic location of the DMPs, most were located within Inter_CGI regions (39%), shores (23%), CDS (40%) and intergenic regions (26%) (Figure S2). Using clustering analyses on the top 500 most significant DMPs for each cohort, the NOTCH1 cohort exhibited the highest proportion of DMP overlap with Sotos syndrome (34%) and Tatton-Brown-Rahman syndrome (TBRS) (25%) in comparison with 99 other episignatures (Fig. 3A). Furthermore, cluster analysis using tree and leaf plots unveiled similarities between NOTCH1 and other disorders, notably Lysine Methyltransferase 2D (KMT2D_p.3400–3700) and Smith-Magenis syndrome (SMS_del; 17p11.2) groups (Fig. 3B). Finally, the mean differences in β-values between NOTCH1 and other known episignature disorders revealed more hypomethylation changes in the NOTCH1 cohort (Fig. 3C).
Fig. 3. Assessment of the amount of DMPs shared between the NOTCH1 cohort and other syndromes with known episignatures. A: Methylation probe overlap. The percentage of DMPs shared between disorders is shown on the colour scale, ranging from white (0%) to red (100%). Each square in the graph represents the percentage of common probes between two syndromes, with the percentage of DMPs from the syndrome on the lower bar that also exist in the DMPs of the syndrome on the right-hand sidebar. B: A tree-and-leaf diagram is used, where each node represents a cohort, and syndromes with more similarity in methylation levels are located closer on the tree. Node size is related to the ratio of the number of DMPs to the total number of probes, while node colour demonstrates the overall mean methylation difference in the corresponding cohort. C: Comparison of the global mean methylation differences between syndromes with known episignatures
Enrichment analysis of probes of the episignature
Of the 210 probes contained within the identified episignature, 120 overlap with protein-coding genes (Table S7). Seven of these (EYA1,* ISL1*,* MSX2*,* NFATC4*,* PRDM16*,* RAI1* and TRAF7) represent genes previously published in the context of cardiac defects [53–59]. Upon performing a STRING analysis of NOTCH1 with all 120 genes, we noted several interactions, particularly for one large network containing 32 of the 121 genes (Fig. 4, Full String network: Figure S3). Interestingly, GO-term enrichment analysis of the 32 genes of this network revealed “Regulation of secondary heart field cardioblast proliferation” (GO:0003266) as the highest-ranking term, involving NOTCH1, ISL1 and EYA1 (Table S8).
Fig. 4STRING interaction network of genes which are collocated with probes in the NOTCH1-episignature. Nodes represent overlapping genes. Edges represent data indicating an interaction, comprising “Textmining” (green lines), “Experiments” (pink lines) and “Databases” (blue lines). The minimal required interaction score cutoff was defined at >0.4. The full interaction network can be found in Figure S3 [84].
Discussion
Here we report on a large exome-sequenced cohort, in which we identified ultrarare variants affecting NOTCH1 as the most common monogenic cause of CHD. In addition, we established a distinct episignature in patients with NOTCH1-associated non-syndromic CHD. Given the high abundance of variants in NOTCH1 in CHD patients, this episignature can possibly contribute to the diagnostic options for these patients.
Our results indicate that deleterious NOTCH1 variants might account for up to 1% of all CHD cases (38/3907 in our cohort), 2.2% amongst conotruncal defects (22/977) and ToF in particular (17/484; 3.5%). As illustrated by the literature reviewed as part of this work, NOTCH1 is a well-established contributor to CHD and ToF appears to be one of the predominant cardiac manifestations besides left-sided malformations.
Importantly, for none of our cases with ultra-rare NOTCH1 variants, was an alternative genetic explanation identified. We also excluded the presence of pathogenic variants in DNA- and histone-methylation related genes, which could possibly interfere with DNA-methylation independently of NOTCH1. This further strengthens our conclusion that the episignature correlates to NOTCH1-variants.
An interesting finding was the identification of ultra-rare NOTCH1 variants that specifically impair disulfide bridges in the extracellular region of NOTCH1 in patients with conotruncal defects. This observation is substantiated by previous reports [8, 60, 61]. Disulfide bridges contribute to the correct three-dimensional protein structure and are highly conserved. We hypothesize that alterations of these residues alter the conformation of the extracellular regions, thereby hindering ligand binding and activation of NOTCH1-signalling.
Consequently, specific attention should be paid to cysteine-altering variants when analysing NOTCH1 variants. Conversely, we also observed four CHD samples with variants that created novel cysteines, while none were found in controls. Novel cysteines might similarly result in an altered protein conformation. Whether this is a relevant disease mechanism remains to be elucidated in further studies.
Of note, an experimentally-derived structure of the full NOTCH1-protein is not available, thus limiting interpretability of structural effects. Nevertheless, we applied in-silico modelling for variants that might potentially alter disulfide-bonding patterns. Not all variants yielded a clear pattern. In some instances, however, modelling information concurred with the methylation signal, providing suggestions for the molecular basis of pathogenicity.
Given the considerable size and clinical importance of the NOTCH1-gene, variant assessment is a frequently recurrent task in the diagnostic setting. Indeed, we observed a considerable number of variants with uncertain effects in our cohort.
The mapping of Mendelian disorders with disease-specific DNAm episignature biomarkers and the identification of global disruptions in DNAm profiles are increasingly prevalent [13, 15]. Aberrant DNAm within gene promoters can disturb gene expression and result in abnormal phenotypes [62, 63]. As such episignatures are also extensively employed for the evaluation and reclassification of VUS [15, 63], we investigated whether a differential DNAm episignature is associated with NOTCH1-related CHD. We delineate a distinct DNAm episignature and demonstrate that it is sensitive and robust through cross-validation analysis. Additionally, we illustrate the specificity of this signature relative to controls and other episignature disorders.
Episignatures are consistently detectable in peripheral blood across more than 200 genes studied to date, the vast majority of which are associated with conditions lacking any hematologic phenotype. While systematic cross-tissue validation remains an important future research direction, current evidence supports the reliability and clinical utility of peripheral blood–derived episignatures, even though this is not the primary tissue affected in NOTCH1-related CHD.
Three cases with ultra-rare NOTCH1 variants (#35, #56, #58), suspected to be pathogenic, clustered with controls and demonstrated an absence of the *NOTCH1-*episignature. Case #35, carrying a variant in a canonical splice acceptor site, displayed an intermediate overlap with the episignature. Exon skipping through disruption of this splice site could potentially preserve the reading frame and result in a shortened, but intact protein. Such an altered protein might still have residual activity, resulting in a hypomorphic effect and generate the intermediate signature overlap. However, due to lack of patient material, we could not validate this using RNA sequencing. Case #56 carried a de novo NOTCH1 missense variant (p.(Gly427Arg)). This patient displayed symptoms consistent with Adams-Oliver syndrome, with a comparably minor cardiovascular involvement (patent ductus arteriosus, pulmonary hypertension). Given the phenotype and the poor prediction score for the NOTCH1 signature of this sample, it could indicate that the identified signature might be more informative for severe NOTCH1-related cardiac phenotypes, rather than AOS. Lastly, case #58 diagnosed with TGA and carrying the p.(Arg592Cys) NOTCH1 variant, also demonstrated lack of overlap with the NOTCH1-episignature. However, this variant was reported in 17 samples in the newest gnomAD freeze (v.4.1.0) and has been listed as benign and as a VUS in ClinVar. Retrospectively, this variant would no longer be considered as (likely) pathogenic. Accordingly, in-silico modelling suggests that this variant is unlikely to result in the formation of a new stable disulfide-bridge.
Epigenetic signatures can be very useful to (re)classify VUS. We therefore investigated four individuals with a NOTCH1 VUS. Two cases (#15 and #39) clustered with the *NOTCH1-*episignature, indicating that those variants might contribute to disease etiology. Case #15 represents a ToF case with hypothyroidism and renal phenotypes, segregation was not possible due to lack of parental DNA.
For case #39 we could establish paternal inheritance for the VUS. However, to our knowledge, the father does not have CHD or NOTCH1-related phenotypes. As incomplete penetrance is frequent in CHD families, analysing the segregation of the episignature in this family would provide valuable insights, as it allows better understanding whether the episignature is a result of altered NOTCH1 activity, or rather indicates a modifying mechanism that enforces a CHD phenotype expression in carriers of NOTCH1-variants. Unfortunately, the paternal DNA was not available for methylation testing. Based on in-silico modelling, this variant might lead to a disruption of the hydrophobic environment, increases flexibility of the ANK region, and thereby destabilises MAML1-interaction. Despite the paternal inheritance, both modelling and episignature testing concordantly suggest an effect of this variant. Two cases (#53, #60) did not have overlap with the episignature. Case #53 carrying the p.(Glu242Lys) variant has no cardiac phenotype, but does exhibit various syndromic features. The variant is maternally inherited and the phenotype is also present in the mother, suggesting a possible segregation with the phenotype. The other case (#60), that did not have an overlap with the episignature, carried the p.(Asp622Asn) variant, and comes from a family with pulmonary arterial hypertension. Given the above, although suggestive, we conclude that a negative episignature cannot yet be used as definitive evidence for the absence of pathogenicity. Further analyses, on larger numbers of cases with NOTCH1 VUS and their epigenetic signatures, are needed to establish this.
While systematic ancestry-focused studies on episignature biomarkers have not been performed, experimental design for feature selection, and available evidence from large scale studies and testing programs supports the robustness of episignatures across ancestral and ethnic backgrounds [19]. Nevertheless, future studies should preferably focus on non-European samples to confirm independence of the episignature from ancestry effects. While some of the cases also presented with extracardiac phenotypes, we did not observe generalizable similarities, especially none for which the episignature might have a predictive value. The current data suggests a limited sensitivity for mild or syndromic cases. Testing additional samples with AOS-features might thus help to assess predictive value outside of nsCHD-cases.
Looking more closely at the differentially methylated positions (DMPs) of the identified *NOTCH1 *signature, we found minimal overlap between the NOTCH1 signature's DMPs and other established signatures (Fig. 3A), underscoring the highly specific nature of the NOTCH1 episignature. Sotos- (34%) and TBR-syndrome (25%) had the largest overlaps. Sotos syndrome is caused by heterozygous mutations in the NSD1 gene and is characterized by overgrowth, facial abnormalities, brain anomalies, seizures, and impaired intellectual development [64]. However, some Sotos patients also present with CHD, which could explain the overlap in some of the DMPs [65]. TBR syndrome is caused by dominant variants in the DNMT3A gene and is characterized by impaired intellectual development, face abnormalities, tall stature, seizures, scoliosis and large head circumference. There is only limited literature about patients with TBR syndrome presenting with CHD [66].
In addition, the closest established episignatures resembling NOTCH1 were found to be KMT2D-related as well as Smith-Magenis syndrome, which is associated with impairment of RAI1 (Fig. 3B). Both genes are associated with CHD [67, 68]. Furthermore, one probe of the NOTCH1-episignature overlaps with the UTR of RAI1.
One important question is how pathogenic NOTCH1-variants could generate a specific DNAm-signature. NOTCH-signalling has been reported to interact with histone-methyltransferases such as KDM5A and SETD1A [69, 70]. Moreover, crosstalk between histone-modification and regulation of DNA-methylation patterns is well documented [71]. Given the above, we hypothesize that pathogenic NOTCH1 variants lead to altered NOTCH1-signalling which in turn affects histone-methyltransferases thereby impacting DNA-methylation patterns. Alternatively, it is known that the NOTCH1 intracellular domain (ICN) localizes to endothelial cell mitochondria, where it enhances mitochondrial metabolism [72]. In addition, a NOTCH1 variant observed in a non-syndromic ToF patient was demonstrated to decrease ICN mitochondrial localization and pyruvate dehydrogenase activity in heart tissues [72]. Pyruvate dehydrogenase is integral for mitochondrial bioenergetics. This is of interest as recent findings suggest that mitochondrial dysfunction can, in turn, cause alterations in metabolic processes tightly intertwined with DNA methylation, such as the methionine cycle [73]. NOTCH-signalling is one of the earliest and most significant events in (cardiac) development and remains active throughout life. It is therefore conceivable that alterations of this pathway have long lasting implications, amongst others on DNA-methylation. Additional functional testing e.g. applying ChIP-seq and RNA-seq could help shed light on underlying interactions. Due to unavailability of material, these tests could not be integrated in the scope of the presented data.
To look further into the connection between NOTCH1, methylation and CHD we sought to determine whether genes underlying the DMPs of the NOTCH1episignature are associated with cardiac development. We identified 120 genes overlapping DMPs, several of these (7/120) are indeed known to be involved in cardiogenesis and have been associated with CHD.
A surprisingly large STRING-interaction network containing 32 genes was found and revealed overrepresentation of the GO-term regulation of secondary heart field cardioblast proliferation. In particular, ISL1 and EYA1 emerged as interesting contributors to this process. ISL1 encodes a transcription factor of the LIM/homeodomain family regulating cell proliferation and survival [74]. It is described as a marker of early progenitor cell populations that contribute to the outflow tract, right ventricle, a subset of left ventricular cells and a large number of atrial cells [75, 76]. Moreover, pathogenic variants in ISL1 have been reported in patients with CHD (DORV, VSD) [77, 78]. and NOTCH1-signalling has been shown to positively regulate ISL1-expression in cardiac progenitor cells [79, 80].
EYA1 is a member of the eyes absent (EYA) family of proteins, which acts as a protein phosphatase and transcriptional coactivator [81]. In humans, variants in EYA1 are associated with the branchiootorenal syndrome type 1 (OMIM 113650), a condition involving malformations of the ears and kidneys, as well as craniofacial abnormalities. Cardiac abnormalities are typically not part of the spectrum, but double-null Eya1-mice display impaired cardiovascular development with an interrupted or right-sided aortic arch, amongst others [82]. Interestingly, the Eya1-Notch1 axis has been shown to play a role in various developmental processes [83]. Dephosphorylation of the intracellular NOTCH1 (ICN) through EYA1 is thought to stabilise the protein, thus contributing to an enhanced NOTCH1-signalling [81].
Concluding, genes underlying the DMPs of the NOTCH1-episignature have direct and indirect links to (cardiac) development. It is therefore possible that aberrant alterations in methylation of these genes, as a consequence of pathogenic NOTCH1 variants, could lead to CHD. Future research into these interactions is needed to elucidate the detailed mechanisms.
In general, given the high prevalence of NOTCH1 variants among patients with CHD and having identified a specific NOTCH1 DNAm signature, we argue that DNAm analysis can contribute substantially to a more accurate variant assessment, ultimately resulting in improved case management. Moreover, this signature broadens the potential applications of epigenetic testing as DNA from peripheral blood is usually available for individuals undergoing clinical genetic testing. Furthermore, this work can lead to follow-up studies, such as refining sub-signatures amongst the individual subtypes, extending the signature with regards to syndromic NOTCH1-related phenotypes, elucidating the underlying mechanism of this episignature and potentially extending this approach to other genes related to non-syndromic CHD.
Conclusions
In this work we report on one of the largest cohorts with NOTCH1-associated CHD cases. Our analysis identified variants disrupting disulfide-bonds as a novel and frequent mechanism for conotruncal malformations. Overall, deleterious variants in NOTCH1 are found in 1% of CHD cases and over 2% of conotruncal malformations, making it the most common monogenic cause in this type of disorder. In addition, we established a NOTCH1-specific DNAm-signature, representing the first such signature in non-syndromic CHD-cases. We also show that genes underlying this signature have direct and indirect links to (cardiac) development and CHD. Overall, this novel signature considerably broadens the applicability of epigenetic testing and facilitates assessment of NOTCH1 variant pathogenicity.
Supplementary Information
Additional file 1: Supplementary figures S1 to S9 including cross validation plots, annotations of differentially methylated probes, full STRING interaction network for genes overlapping the presented signature, a NOTCH1-protein plot representing the DNAm prediction scores for tested samples and figures depicting the results of in-silico modelling for cysteine-altering variants.
Additional file 2: Supplementary tables S1 to S8 containing domains and modification site annotations, GO-enrichment results, published NOTCH1-variants, details on the in-silico modelling, an overview of disulfide-bond residues in NOTCH1 as well as prediction scores and probe information for the NOTCH1-episignature.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Hoffman JIE, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890-900. 10.1016/S 0735-1097(02)01886-7.10.1016/s 0735-1097(02)01886-712084585 · doi ↗ · pubmed ↗
- 2Pierpont ME, Brueckner M, Chung WK, et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation. 2018;138(21):e 653-e 711. 10.1161/CIR.0000000000000606, https://pubmed.ncbi.nlm.nih.gov/30571578/. 10.1161/CIR.0000000000000606 PMC 655576930571578 · doi ↗ · pubmed ↗
- 3Lehman A, Wuyts W, Patel MS. Adams-Oliver Syndrome In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. Gene Reviews®. Seattle (WA): University of Washington, Seattle; April 14, 2016. https://pubmed.ncbi.nlm.nih.gov/27077170/.
- 4Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. 2020. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v 4-01-2020.pdf.
- 5Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, Mac Arthur DG et al. Regional missense constraint improves variant deleteriousness prediction [Internet]. Genomics; 2017 Jun [Cited 2023 May 26]. Available from: http://biorxiv.org/lookup/doi/10.1101/148353
- 6Ho DE, Imai K, King G, Stuart EA. Match It: Nonparametric Preprocessing for Parametric Causal Inference. J Stat Softw . 2011 [Cited 2024 Jun 20];42(8). Available from: http://www.jstatsoft.org/v 42/i 08/