Nasu-Hakola Disease Presenting as Rapidly Progressive Dementia With Seizures: A TREM2 Mutation Case Without Skeletal Involvement
Zubair Sarkar, Md Mahmood Alam, Ayushi Chaudhari

TL;DR
A rare genetic disorder causing neurological symptoms was diagnosed in a woman without skeletal issues, highlighting the importance of genetic testing in dementia cases.
Contribution
This case highlights TREM2 mutation causing Nasu-Hakola disease without skeletal involvement, expanding clinical recognition.
Findings
A TREM2 gene mutation was identified in a patient with progressive dementia and seizures but no skeletal lesions.
Neuroimaging and genetic testing confirmed Nasu-Hakola disease despite absence of typical bone involvement.
The case emphasizes the importance of genetic testing in atypical early-onset dementia presentations.
Abstract
Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder caused by mutations in the TYROBP or TREM2 genes. It is characterised by the unique combination of neurological and skeletal manifestations. However, isolated neurological variants without bone involvement have been described. A 38-year-old woman presented with a two-year history of progressive cognitive decline, reduced speech output, inappropriate emotional behaviour, recurrent generalised tonic-clonic seizures, gait unsteadiness, and loss of self-care abilities. Neurological examination revealed Parkinsonian features, cerebellar signs, brisk reflexes, and frontal release signs. Laboratory investigations for metabolic, autoimmune, and infectious aetiology were normal. Brain MRI demonstrated diffuse cortical atrophy, periventricular T2/fluid-attenuated inversion recovery (FLAIR) hyperintensities, and bilateral globus…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
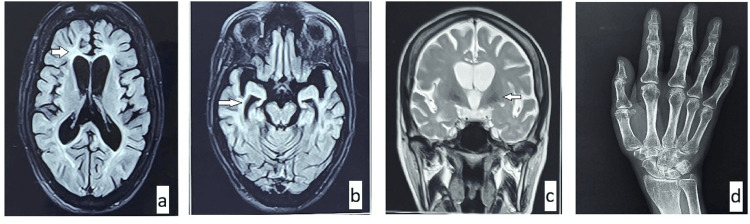 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroinflammation and Neurodegeneration Mechanisms · Muscle Physiology and Disorders · Neurological Disease Mechanisms and Treatments
Introduction
Nasu-Hakola disease (NHD), also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), is an uncommon hereditary disorder that is transmitted in an autosomal recessive manner. Reported cases have been primarily concentrated in Japanese and Finnish cohorts, with only a few cases reported from the Indian subcontinent [1]. The hallmark of this disease is an unusual combination of early-onset dementia with skeletal involvement in the form of bone cysts and pathological fractures [2]. However, this case emphasises the need to consider NHD in cases of early-onset dementia with or without skeletal involvement in appropriate clinical scenarios.
Case presentation
We describe the case of a 38-year-old homemaker who presented with a two-year history of progressive cognitive decline, markedly reduced word output, inappropriate smiling, behavioural disturbances, multiple episodes of seizures (generalised tonic-clonic seizures (GTCS)), difficulty in walking, and inability to care for herself. Her parents were not consanguineous, and there was no family history of similar illness.
The progression was quite rapid, involving multiple cognitive domains within a year of onset. Cognitive assessment with Mini-Mental State Examination (MMSE) [3] and neuropsychological testing could not be done due to advanced inattention and dementia. Upon further examination, she exhibited Parkinsonian features, including axial and limb rigidity, reduced facial expressions, and cerebellar signs, including limb and gait ataxia. Deep tendon reflexes were brisk, and plantar reflexes were bilateral extensor. Frontal release signs, including glabellar tap and palmomental reflex, were present.
The patient was extensively evaluated for reversible causes. All investigations, including biochemical workup, autoimmune markers, and cerebrospinal fluid analysis, were unremarkable. These investigations comprehensively ruled out metabolic, endocrine, nutritional and autoimmune causes of dementia. MRI brain scans revealed diffuse cortical atrophy, along with widespread T2/fluid-attenuated inversion recovery (FLAIR) hyperintensities involving the periventricular white matter and calcification in the bilateral globus pallidus (Figures 1a-1d). X-ray of both hands showed generalised osteopenia without visible cystic lesions (Figure 1e). Clinical exome sequencing identified a homozygous c.371T>G mutation in the TREM2 gene located on exon 2, confirming the diagnosis of NHD.
MRI brain and X-ray findings.(a) and (b) FLAIR images showing periventricular white matter hyperintensity and diffuse cortical atrophy. (c) T2 image showing bilateral globus pallidus calcification. (d) X-ray of the right hand demonstrating periarticular osteopenia.FLAIR: fluid-attenuated inversion recovery.
Discussion
NHD was first recognised in the 1970s when it was independently described by Nasu and Hakola [2]. This autosomal recessive inherited disorder is characterised by progressive dementia and repeated pathological fractures [2]. Pathogen mutations in either TRYOBP or TREM2 genes underlie NHD. The TREM2-TRYOBP protein complex is involved in the regulation of differentiation and function of osteoclasts [4]. Within the central nervous system, the same complex is primarily expressed in microglia and plays a role in regulating their survival and phagocytic activity [5]. The dysfunction of this signalling pathway leads to impaired clearance of apoptotic neurons and increased proinflammatory responses, causing brain injury and neurodegeneration [6]. NHD is therefore increasingly recognised as a prototypical disorder arising from primary microglial dysfunction [7].
Hakola described the clinical course of NHD in four stages: an initial asymptomatic or latent phase, followed by a skeletal phase marked by pathological fractures and bone cysts, and subsequent early and late neuropsychiatric phases characterised by progressive cognitive and behavioural decline [8]. Neurological symptoms usually start in the third or fourth decade, starting with behaviour change and memory disturbances, like features observed in cases of frontotemporal dementia. Other symptoms include gait disturbances, emotional dysregulation, apathy, and seizures. These symptoms are generally progressive and lead to severe impairment of memory and function. However, presentations vary considerably, and the disease has been shown to affect only the central nervous system, without bone involvement. Patients with TREM2 mutations have been specifically reported with pure neurological involvement [9]. The differential diagnosis included causes of early onset and rapidly progressive dementia, including those with autoimmune aetiology and familial forms of frontotemporal dementia and Alzheimer’s disease.
The absence of family history, early onset in the fourth decade, and a prominent association with recurrent seizures, along with characteristic neuroimaging features such as diffuse cortical atrophy, periventricular hyperintensities, and bilateral basal ganglia calcifications, led us to consider NHD in the differential diagnosis. After ruling out all the reversible causes with relevant investigations, a clinical exome sequencing was ordered, which confirmed the TREM2 mutation and, consequently, the diagnosis of NHD.
Conclusions
This case emphasises the need to consider NHD as a differential diagnosis in cases of presenile dementia, especially when associated with atypical symptoms like generalised seizures and Parkinsonism. Furthermore, the absence of skeletal manifestations shall not preclude the diagnosis of NHD. Early recognition through genetic testing facilitates accurate diagnosis, appropriate counselling, and avoidance of unnecessary investigations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy): first report from India Neurol India Chepuru R Shaik AJ Tandra SR Gaddamanugu P Alladi S Kaul S 5385416620182954718810.4103/0028-3886.227319 · doi ↗ · pubmed ↗
- 2Nasu-Hakola disease: the first case reported by Nasu and review: the 50th Anniversary of Japanese Society of Neuropathology Neuropathology Kaneko M Sano K Nakayama J Amano N 4634703020102050045010.1111/j.1440-1789.2010.01127.x · doi ↗ · pubmed ↗
- 3“Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician J Psychiatr Res Folstein MF Folstein SE Mc Hugh PR 189198121975 https://nesdo.onderzoek.io/wp-content/uploads/2016/08/MMSE_Folstein-1975.pdf 120220410.1016/0022-3956(75)90026-6 · doi ↗ · pubmed ↗
- 4DAP 12/TREM 2 deficiency results in impaired osteoclast differentiation and osteoporotic features J Exp Med Paloneva J Mandelin J Kiialainen A 66967519820031292568110.1084/jem.20030027 PMC 2194176 · doi ↗ · pubmed ↗
- 5TRE Ms in the immune system and beyond Nat Rev Immunol Colonna M 445453320031277620410.1038/nri 1106 · doi ↗ · pubmed ↗
- 6Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2J Exp Med Takahashi K Rochford CD Neumann H 64765720120051572824110.1084/jem.20041611 PMC 2213053 · doi ↗ · pubmed ↗
- 7Developmental regulation of TREM 2 and DAP 12 expression in the murine CNS: implications for Nasu-Hakola disease Neurochem Res Thrash JC Torbett BE Carson MJ 38453420091840437810.1007/s 11064-008-9657-1PMC 2655126 · doi ↗ · pubmed ↗
- 8CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts Neurology Paloneva J Autti T Raininko R 155215585620011140211410.1212/wnl.56.11.1552 · doi ↗ · pubmed ↗