Prenatally Diagnosed 7q11.23 Copy Number Variations: A Retrospective Case Series
Jiong Yan, Ziyang Liu, Song Yi, Nian Liu

TL;DR
This study examines prenatal cases of 7q11.23 copy number variations, showing ultrasound anomalies and high termination rates, with inherited cases having milder outcomes.
Contribution
The study provides new insights into prenatal manifestations and inheritance patterns of 7q11.23 CNVs, emphasizing the importance of genomic testing for counseling.
Findings
7q11.23 deletions were associated with 100% ultrasound anomalies, including cardiovascular defects and growth restriction.
76.5% of ongoing pregnancies with de novo CNVs resulted in termination of pregnancy.
Inherited CNVs were linked to milder outcomes compared to de novo variants.
Abstract
Williams‐Beuren syndrome (WBS; OMIM #194050), caused by 7q11.23 deletions, is well‐characterized postnatally, but prenatal manifestations remain poorly defined. This study aims to delineate the prenatal phenotypes, inheritance patterns, and outcomes of 7q11.23 copy number variations (CNVs). A retrospective study of 20 prenatal cases with 7q11.23 CNVs diagnosed by SNP array or CNV sequencing (CNV‐seq) was conducted. Clinical data, including ultrasound findings, genetic results, and pregnancy outcomes, were analyzed. Classic 7q11.23 deletions (1.42 Mb median size) were associated with ultrasound anomalies in 100% of cases (11/11), predominantly cardiovascular defects (36.4%, 4/11) and growth restriction (18.2%, 2/11). While 7q11.23 duplications (1.42–3.03 Mb) were associated with anomalies in 50% of cases (3/6), including cleft palate and ventriculomegaly. Inheritance pattern analysis…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
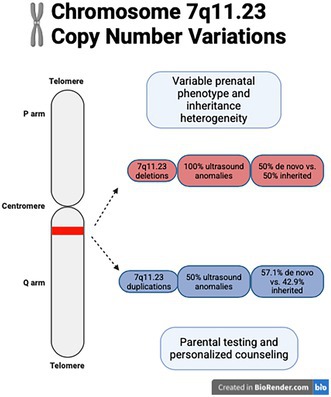 Figure 1
Figure 1| Case | Maternal age (years) | Pregnancy history | GA (weeks) | Indication for genetic test | Ultrasound findings | Sex | Diagnostic method | Genetic results (HGVS) | Size (Mb) | Inheritance | Pregnancy outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 | G2P0 | 23 | Ultrasound findings | Duodenal obstruction | M | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,140,000) × 1 | 1.42 | De novo | TOP |
| 2 | 27 | G2P0 | 24 | Ultrasound findings, adverse pregnancy history | FGR, PLSVC, elevated umbilical artery S/D ratio | M | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,140,000) × 1 | 1.42 | Maternal | TOP |
| 3 | 30 | G2P1 | 29 | Ultrasound findings | Left ventriculomegaly (13 mm), enlarged posterior fossa (11 mm) | M | CNV‐seq | seq[GRCh37]7q11.23 (75,160,000–76,140,000) × 1 | 0.98 | Maternal | Live birth |
| 4 | 29 | G4P1 | 23 | Family history | FGR | M | CNV‐seq | seq[GRCh37]7q11.23 (72,740,000–74,120,000) × 1 | 1.38 | Maternal | TOP |
| 5 | 29 | G1P0 | 20 | Family history | Increased NT (4.8 mm), increased NF (8.1 mm) | F | SNP array | arr[GRCh37]7q11.23q21.11 (76,007,283–80,123,898) × 1 | 4.1 | Maternal | TOP |
| 6 | 35 | G2P0 | 18 | High‐risk NIPT for 7q11.23 microdeletion | Pleural effusion, peritoneal effusion | M | SNP array | arr[GRCh37]7q11.23 (72,664,089–74,143,240) × 1 | 1.48 | De novo | TOP |
| 7 | 38 | G2P1 | 21 | Ultrasound findings, adverse pregnancy history | Increased NT (3.5 mm) | F | SNP array | arr[GRCh37]7q11.23 (72,723,370–74,154,209) × 1 | 1.43 | De novo | TOP |
| 8 | 21 | G1P0 | 31 | Family history, Ultrasound findings | Aortic stenosis, tricuspid regurgitation | M | SNP array | arr[GRCh37]7q11.23 (72,701,098–74,154,209) × 1 | 1.45 | Maternal | TOP |
| 9 | 25 | G1P0 | 19 | High‐risk NIPT for 7q11.23 microdeletion | VSD | F | SNP array | arr[GRCh37]7q11.23 (72,621,346–74,289,047) × 1 | 1.67 | De novo | TOP |
| 10 | 30 | G2P1 | 28 | Ultrasound findings | Rocker‐bottom feet | M | SNP array | arr[GRCh37]7q11.23 (72,650,120–74,154,209) × 1 | 1.5 | De novo | TOP |
| 11 | 25 | G1P0 | 29 | Ultrasound findings, family history | Interruption of the inferior vena cava, micrognathia | M | CNV‐seq | seq[GRCh37]7q11.23 (72,780,000–74,140,000) × 1 | 1.36 | Maternal | Live birth |
| 12 | 27 | G2P0 | 9 | Fetal demise | Fetal demise | M | CNV‐seq | seq[GRCh37]7q11.23 (72,740,000–74,140,000) × 1 | 1.4 | De novo | Fetal demise |
| 13 | 27 | G1P0 | 11 | Fetal demise | Fetal demise | M | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,140,000) × 1 | 1.42 | N/A | Fetal demise |
| 14 | 32 | G1P0 | 8 | Fetal demise | Fetal demise | F | CNV‐seq | seq[GRCh37]7q11.23 (72,200,000–74,140,000) × 3 | 1.94 | De novo | Fetal demise |
| 15 | 27 | G1P0 | 24 | Ultrasound findings | Cleft palate | F | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,140,000) × 3 | 1.42 | De novo | TOP |
| 16 | 20 | G1P0 | 20 | Family history | No abnormalities on ultrasound | F | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,080,000) × 3, seq[GRCh37]2p23.3p23.2 (26,420,000–28,080,000) × 3 | 1.36/1.66 |
Maternal (7q), Maternal (2p) | TOP |
| 17 | 42 | G1P0 | 21 | AMA | No abnormalities on ultrasound | M | CNV‐seq | seq[GRCh37]7q11.23 (72,720,000–74,140,000) × 3 | 1.42 | Paternal | Live birth |
| 18 | 28 | G1P0 | 23 | Ultrasound findings | Polyhydramnios, pulmonary artery crossover | M | SNP array | arr[GRCh37]7q11.23 (72,370,954–75,403,750) × 3 | 3.03 | De novo | TOP |
| 19 | 29 | G2P1 | 29 | Ultrasound findings | Unilateral ventriculomegaly (12 mm) | M | SNP array | arr[GRCh37]7q11.23 (72,608,900–74,184,702) × 3 | 1.58 | De novo | TOP |
| 20 | 28 | G1P0 | 21 | Adverse pregnancy history | No abnormalities on ultrasound | M | CNV‐seq | seq[GRCh37]7q11.23 (72,650,001–74,200,000) × 3, seq[GRCh37]4q12 (55,238,386–55,970,791) × 3 | 1.55/0.73 | Maternal (7q), Paternal (4q) | Live birth |
| Abnormality | Deletions ( | Cases (Deletions) | Duplications ( | Cases (Duplications) |
|---|---|---|---|---|
| Cardiovascular defects | 36.4% (4/11) | 2, 8, 9, 11 | 16.7% (1/6) | 18 |
| Fetal growth restriction | 18.2% (2/11) | 2, 4 | 0% (0/6) | — |
| Increased nuchal translucency | 18.2% (2/11) | 5, 7 | 0% (0/6) | — |
| Ventriculomegaly | 9.1% (1/11) | 3 | 16.7% (1/6) | 19 |
| Other anomalies | 27.3% (3/11) | 1, 6, 10 | 16.7% (1/6) | 15 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsWilliams Syndrome Research · Genomic variations and chromosomal abnormalities · Congenital Ear and Nasal Anomalies
Introduction
1
Williams‐Beuren syndrome (WBS; OMIM #194050) is a multisystem disorder caused by a recurrent 1.5–1.8 Mb heterozygous deletion at chromosome 7q11.23. The syndrome is characterized by distinctive clinical features, including cardiovascular anomalies, intellectual disability, and a recognizable facial phenotype (Pober 2010). While the postnatal manifestations of WBS have been extensively documented, the prenatal presentation of 7q11.23 copy number variations (CNVs)—encompassing both deletions and rare duplications—remains poorly characterized, creating significant challenges for prenatal diagnosis and genetic counseling (Berg et al. 2007; Strømme et al. 2002; Morris and Mervis 2000).
Current literature on prenatal WBS primarily consists of isolated case reports or small case series, with fetal growth restriction (FGR) and congenital cardiovascular defects emerging as the most frequently reported findings (Borrell et al. 2018; Yuan et al. 2020; Lv et al. 2023). However, emerging evidence suggests a broader spectrum of phenotypic variability, including less common associations such as aortic coarctation and persistent left superior vena cava (PLSVC) (Yuan et al. 2020; Lv et al. 2023; Wang et al. 2023). Furthermore, the clinical implications of atypical deletions or duplications outside the classic WBS critical region (WBSCR), as well as their correlation with fetal structural abnormalities, remain insufficiently explored (Alesi et al. 2021).
In this study, we present a cohort of 20 consecutive prenatal cases diagnosed with 7q11.23 CNVs. We comprehensively analyzed the indications for genetic testing, prenatal ultrasound findings, inheritance patterns of the CNVs, and pregnancy outcomes. Our objective was to provide a detailed review of prenatal cases involving 7q11.23 CNVs, with the aim of improving the understanding of this genomic disorder in the context of prenatal diagnosis and facilitating more informed clinical decision‐making.
Materials and Methods
2
Study Design and Participants
2.1
This retrospective cohort study was conducted at the Prenatal Diagnosis Center of the Maternal and Child Health Hospital of Hubei Province (Wuhan, China). From April 2017 to December 2024, 18,459 prenatal samples were analyzed via SNP array/copy number variation sequencing (CNV‐seq), with 3261 (17.7%) diagnosed as chromosomal abnormalities. The study cohort included fetuses with confirmed 7q11.23 deletions or duplications identified through CNV‐seq or SNP array analysis, irrespective of ultrasound findings. Maternal demographics, gestational age at diagnosis, primary indications for genetic testing, ultrasound findings, comprehensive genetic results, inheritance patterns, and pregnancy outcomes were extracted from medical records. The study was approved by the Institutional Ethics Committee of the Maternal and Child Health Hospital of Hubei Province (Approval No. 230741011). Written informed consent was obtained from all participants.
Genetic Testing Methods
2.2
SNP Array
2.2.1
Genomic DNA was extracted from uncultured prenatal samples (amniocytes, chorionic villi) and parental peripheral blood using the QIAamp DNA mini kit (QIAGEN, Germany). SNP array analysis was performed using the CytoScan 750 K Array following manufacturer protocols. Data were analyzed using Affymetrix Chromosome Analysis Suite (ChAS) v3.3, with genomic coordinates mapped to GRCh37/hg19 (Affymetrix Inc 2013).
Copy Number Variation Sequencing (CNVseq)
2.2.2
Genomic DNA (10 ng) was fragmented, ligated with adapters, and amplified to construct sequencing libraries as previously described. Libraries were sequenced on the NextSeq CN500 platform (Illumina, USA), yielding approximately 5 million 45‐bp single‐end reads. Reads were aligned to GRCh37/hg19, and CNVs were identified using in‐house bioinformatics pipelines (Wang et al. 2018; Dong et al. 2016).
Results
3
Demographics and Genetic Findings
3.1
This study included 20 prenatal cases with 7q11.23 CNVs, comprising 13 deletions and 7 duplications, diagnosed between 2017 and 2024. The incidence of 7q11.23 CNVs was 0.11% (20/18,459) overall and 0.61% (20/3261) among chromosomal abnormalities. The mean maternal age was 28.3 years (range: 17–42 years), with a median gestational age at diagnosis of 21 weeks (range: 8–31 weeks). Genetic testing revealed deletions ranging from 0.98 to 4.1 Mb (median: 1.42 Mb) and duplications spanning 1.42–3.03 Mb (median: 1.58 Mb) (Table 1).
Seventeen cases (85%) involved the classic WBSCR (chr7:72,700,000–74,100,000). Among the remaining cases, two exhibited larger deletions or duplications extending towards the centromere (Case 5, 4.4 Mb deletion; Case 18: 3.03 Mb duplication), while one case carried a smaller 0.98 Mb deletion (Case 3). Three cases (Cases 12–14) were diagnosed in the first trimester (8–11 weeks) following ultrasound‐detected fetal demise. The remaining 17 cases underwent invasive prenatal testing due to ultrasound anomalies (n = 9), family history (n = 4), or high‐risk noninvasive prenatal testing (NIPT) results (n = 2).
Phenotypic Spectrum
3.2
Ultrasound anomalies were observed in 100% (11/11) of cases with available prenatal imaging (Cases 1–11, Table 2). Cardiovascular defects were the most common anomalies, present in 36.4% (4/11) of cases, including PLSVC (Cases 2), aortic stenosis (Case 8), ventricular septal defect (VSD, Case 9), and interruption of the inferior vena cava (Case 11). Other recurrent phenotypes included fetal growth restriction (FGR 18.2%; 2/11; Cases 2, 4) and increased nuchal translucency (NT) (18.2%; 2/11; Cases 5, 7). Unique structural anomalies such as duodenal obstruction (Case 1), unilateral ventriculomegaly (Case 3), and rocker‐bottom feet (Case 10) were each observed in one case.
Among duplication cases (Cases 15–20), ultrasound anomalies were observed in 50% (3/6), including cleft palate (Case 15), pulmonary artery crossover (Case 18), and unilateral ventriculomegaly (Case 19).
Inheritance Patterns
3.3
Inheritance pattern analysis was available for 19 cases. Deletions arose de novo in 50% (6/12) and were inherited in 50% (6/12) (Cases 2, 3, 4, 5, 8, 11). Among the six maternal transmissions, three mothers (Cases 4, 5, 8) presented with mild ID. Two mothers (Cases 2, 4) had adverse pregnancy histories: Case 2's mother had a prior termination of pregnancy (TOP) due to FGR, and Case 4's mother had a child with developmental delay and ocular problem. Case 11 presented with micrognathia, while Case 3's mother was phenotypically normal.
Duplications arose de novo in 57.1% (4/7) and were inherited in 42.9% (3/7) (Cases 16, 17, 20). Case 16's mother harbored 7q11.23 duplication and a 2p23.3‐p23.2 duplication, presenting with short stature (147 cm), poor speech communication, and strabismus. Case 20's mother had prior TOP due to the absence of cerebellar vermis. Case 17's father was phenotypically normal.
Pregnancy Outcomes
3.4
TOP was elected in 76.5% (13/17) of ongoing pregnancies, primarily for de novo CNVs or significant ultrasound anomalies (e.g., aortic stenosis, FGR). Families opted to continue the pregnancy in 23.5% (4/17) of cases, resulting in live‐born newborns, all involving inherited CNVs (Cases 3, 11, 17, 20).
Discussion
4
This study presents the largest single‐center prenatal cohort to date characterizing the phenotypic spectrum, inheritance patterns, and outcomes of 7q11.23 CNVs. Our findings expand current understanding of the intrauterine manifestations associated with these genomic alterations and have important implications for prenatal diagnosis and genetic counseling.
The universal presence of ultrasound anomalies (100%) in classic WBSCR deletions confirms the high penetrance of prenatal phenotypes in WBS (Kozel et al. 2021). The predominance of cardiovascular defects (36.4%) in our cohort, including aortic stenosis and PLSVC, reinforces the crucial role of ELN (OMIM #130160) haploinsufficiency in vascular development. These observations align with the well‐established mechanism whereby ELN deficiency disrupts elastin fiber deposition, leading to arterial wall stiffening and subsequent stenosis (Urbán et al. 2000; Hoareau et al. 2023). The novel identification of prenatal markers, such as interrupted inferior vena cava (Case 11), may reflect broader developmental consequences of elastin pathway dysregulation (Collins II 2013).
The chromatin‐remodeling gene BAZ1B (OMIM #605681) emerges as another critical contributor to the prenatal phenotype. Our observation of duodenal obstruction (Case 1) and craniofacial dysmorphism (Case 11) provides clinical evidence supporting BAZ1B's role in neural crest cell migration and differentiation (Lalli et al. 2016; Zanella et al. 2019). These findings suggest that BAZ1B haploinsufficiency may disrupt transcriptional networks governing both enteric nervous system development and craniofacial morphogenesis, offering a potential explanation for these structural anomalies.
Our cohort included cases with atypical deletions extending beyond the classic WBSCR. Case 5, for instance, carried a 4.4 Mb deletion extending into the 7q21.11 region and exhibited increased NT (4.8 mm) and nuchal fold thickness (8.1 mm), indicative of severe fetal anomalies. Larger deletions often encompass additional genes, contributing to more severe and variable phenotypes, including pronounced neurodevelopmental and structural abnormalities (Weiss et al. 2008). Conversely, Case 3 carried a 0.98 Mb deletion and presented with moderate ventriculomegaly and an enlarged posterior fossa, despite the mother being phenotypically normal. This observation aligns with previous studies suggesting that smaller deletions may retain functional copies of some genes, resulting in attenuated clinical manifestations (Berg et al. 2007). The phenotypic variability observed in atypical WBS underscores the importance of precise molecular characterization of CNVs to improve clinical outcome prediction and facilitate informed genetic counseling (Manning and Hudgins 2010).
While postnatal studies reported WBS as predominantly de novo, our cohort included maternal carriers with mild intellectual disability (Cases 4, 5, 8) who transmitted deletions of variable severity, as well as a phenotypically normal mother (Case 3) who passed a 0.98 Mb deletion. These findings highlight the necessity of parental testing, even in the absence of overt parental phenotypes (Bayés et al. 2003).
The high TOP rate for de novo CNVs (76.5%) reflects parental concerns regarding neurodevelopmental risks, despite the limited prenatal predictability of WBS‐associated ID (Morris and Mervis 2000). Conversely, live births in cases with inherited CNVs (Cases 3, 11, 17, 20) suggest that familial variants may confer milder prognoses, emphasizing the need for nuanced risk communication during genetic counseling.
This study is limited by its retrospective design and small sample size. Future studies should expand cohorts to refine phenotype–genotype correlations, particularly for duplications and atypical deletions. Evidence‐based guidelines should be developed for managing pregnancies with inherited CNVs, integrating molecular diagnostics and multidisciplinary counseling approaches.
Conclusion
5
This study advances the prenatal understanding of 7q11.23 CNVs by delineating their heterogeneous phenotypes, inheritance patterns, and counseling challenges. The integration of molecular diagnostics into prenatal workflows is critical for accurate diagnosis and risk stratification. These insights underscore the importance of parental testing and personalized counseling to improve family outcomes. Future studies should prioritize long‐term follow‐up and functional validation to further elucidate the developmental consequences of these genomic alterations and optimize clinical management strategies.
Author Contributions
Jiong Yan: conceptualization, supervision, writing – review and editing. Ziyang Liu: formal analysis, writing – original draft. Song Yi: investigation. Nian Liu: methodology, validation, writing – review and editing.
Funding
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Affymetrix Inc . 2013. Cyto Scan 750K Array User Guide. Affymetrix Inc.
- 2Alesi, V. , S. Loddo , V. Orlando , et al. 2021. “Atypical 7q 11.23 Deletions Excluding ELN Gene Result in Williams‐Beuren Syndrome Craniofacial Features and Neurocognitive Profile.” American Journal of Medical Genetics, Part A 185, no. 1: 242–249. 10.1002/ajmg.a.61937.33098373 · doi ↗ · pubmed ↗
- 3Bayés, M. , L. F. Magano , N. Rivera , R. Flores , and L. A. Pérez Jurado . 2003. “Mutational Mechanisms of Williams‐Beuren Syndrome Deletions.” American Journal of Human Genetics 73, no. 1: 131–151. 10.1086/376565.12796854 PMC 1180575 · doi ↗ · pubmed ↗
- 4Berg, J. S. , N. Brunetti‐Pierri , S. U. Peters , et al. 2007. “Speech Delay and Autism Spectrum Behaviors in 7q 11.23 Duplications.” Genetics in Medicine 9, no. 7: 427–441. 10.1097/GIM.0b 013e 3180986192.17666889 · doi ↗ · pubmed ↗
- 5Borrell, A. , M. Grande , M. Pauta , L. Rodriguez‐Revenga , and F. Figueras . 2018. “Chromosomal Microarray Analysis in Fetuses With Growth Restriction and Normal Karyotype: A Systematic Review and Meta‐Analysis.” Fetal Diagnosis and Therapy 44, no. 1: 1–9. 10.1159/000479506.28889126 · doi ↗ · pubmed ↗
- 6Collins, R. T., II . 2013. “Cardiovascular Disease in Williams Syndrome.” Circulation 127, no. 21: 1865–1874. 10.1161/CIRCULATIONAHA.112.000064.23716381 · doi ↗ · pubmed ↗
- 7Dong, Z. , J. Zhang , P. Hu , et al. 2016. “Low‐Pass Whole‐Genome Sequencing in Clinical Cytogenetics: A Validated Approach.” Genetics in Medicine 18, no. 9: 940–948. 10.1038/gim.2015.199.26820068 · doi ↗ · pubmed ↗
- 8Hoareau, M. , N. El Kholti , R. Debret , and E. Lambert . 2023. “Characterization of the Zebrafish Elastin a (elnasa 12235) Mutant: A New Model of Elastinopathy Leading to Heart Valve Defects.” Cells 12, no. 10: 1436. 10.3390/cells 12101436.37408270 PMC 10217568 · doi ↗ · pubmed ↗