Improved chromosome-level genome assembly of the American cockroach, Periplaneta americana
Rachel L Dockman, Tyler J Simmonds, Kevin J Vogel, Scott M Geib, Elizabeth A Ottesen

TL;DR
Scientists improved the genome assembly of the American cockroach, making it easier to study its biology and evolution.
Contribution
A high-quality, chromosome-level genome assembly of Periplaneta americana was generated, including a previously missing chromosome.
Findings
The genome was assembled into 17 scaffolds with a scaffold N50 of 188.1 Mb.
The assembly includes a chromosome missing in previous references and achieved a BUSCO score of 99.7%.
Annotation identified 16,750 protein-coding genes with a BUSCO score of 97.8%.
Abstract
The American cockroach, Periplaneta americana, is a cosmopolitan insect notorious for thriving among humans undeterred by attempts to eliminate it. The traits that contribute to its ubiquity as an opportunistic pest, such as long lifespan, expansive neurosensory capacity, and nutritional flexibility, also make P. americana an excellent invertebrate model organism as evidenced by its long history in neuroscience and physiological research. Current genetic resources available for P. americana highlight its large, complex genome and richly diverse transcriptional capabilities, but fall short of producing a complete, chromosome-level genome. Here, we present a high-quality de novo genome assembly of a laboratory-raised adult female P. americana using a combination of high fidelity PacBio long reads and Hi-C sequencing. The final 3.23 Gb genome was assembled with chromosomal resolution into…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
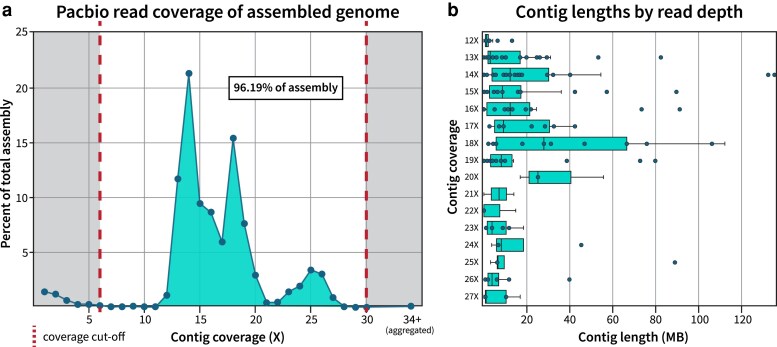 Fig. 1
Fig. 1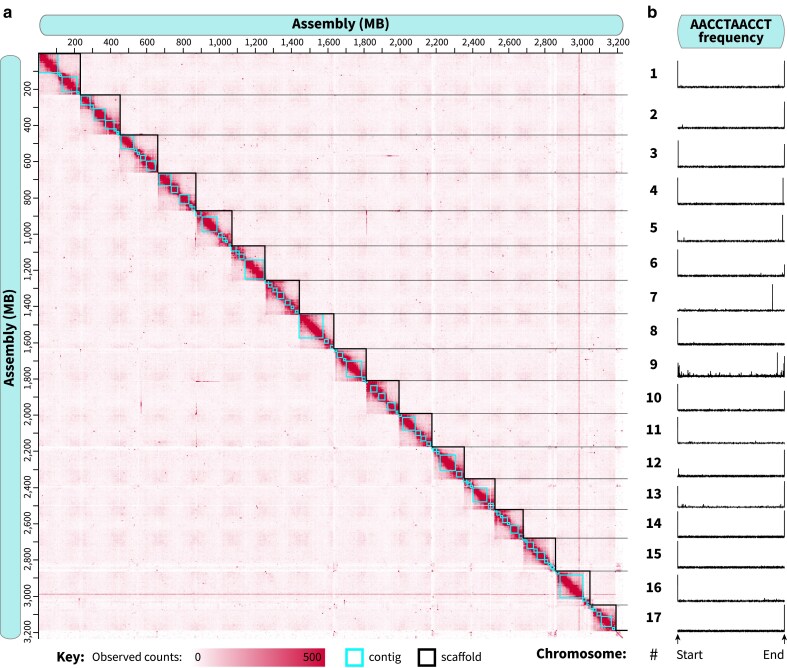 Fig. 2
Fig. 2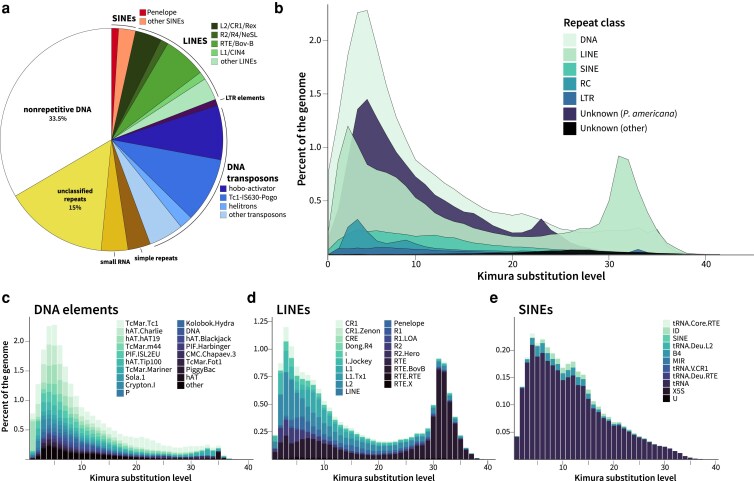 Fig. 3
Fig. 3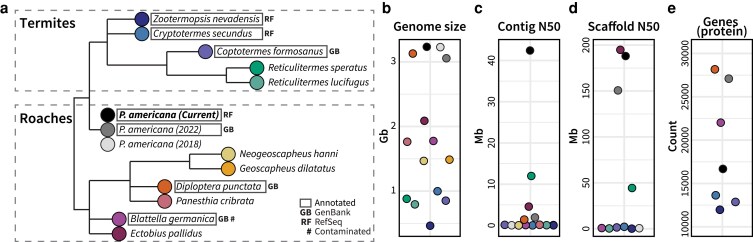 Fig. 4
Fig. 4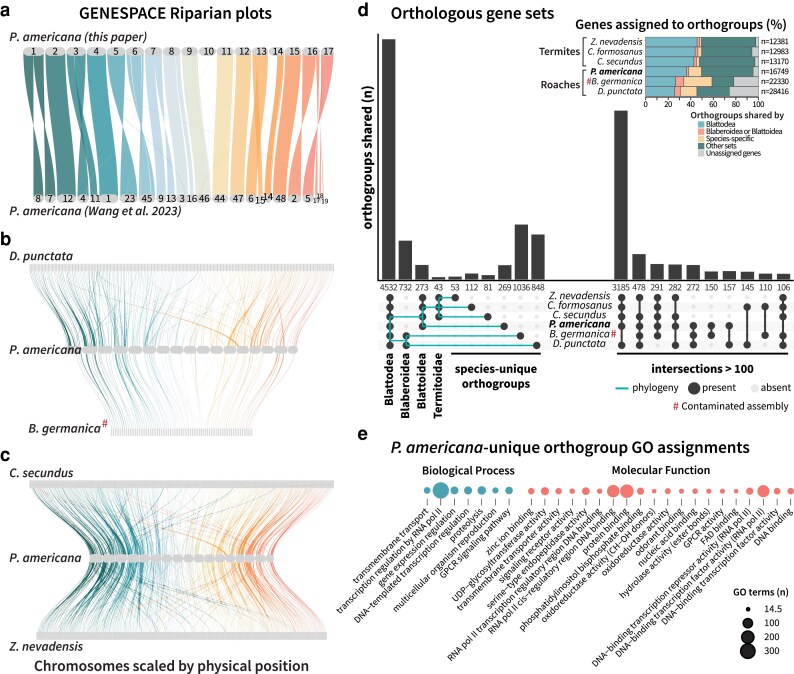 Fig. 5
Fig. 5| Assembly name | ASM293952v1 | ASM2559430v2 | PAMFEO1_priV1 | |
|---|---|---|---|---|
| Accession | GCA_002939525.1 | GCA_025594305.2 | GCF_040183065.1 | |
| Total size (Gb) | 3.4 | 3.1 | 3.2 | |
| Genome | Ungapped (Gb) | 3.2 | 3.1 | 3.2 |
| GC (%) | 35.5 | 35.42 | 35.5 | |
| Count (#) | 18,601 | 48 | 91 | |
| Scaffolds | N50 (Mb) | 0.3325 | 150.7 | 188.1 |
| L50 (#) | 2951 | 9 | 8 | |
| Count (#) | 122,589 | 9217 | 259 | |
| Contigs | N50 (Mb) | 0.0508 | 1.9 | 42.4 |
| L50 (#) | 17,827 | 416 | 22 | |
| Repeats (%) | 57.80 | 62.90 | 65.86 | |
| Genome BUSCO (% complete) | 97.60 | 94.60 | 99.70 | |
| Protein BUSCO (% complete) | 91.20 | 90.50 | 97.80 | |
| Protein-coding genes (#) | 21,336 | 29,939 | 16,750 |
| Feature | Class | Count |
|---|---|---|
| Genes | Protein-coding | 16,750 |
| Non-coding | 8,596 | |
| Other | 1 | |
| Genes with variants | 8,362 | |
| Pseudogenes | Transcribed | 11 |
| Non-transcribed | 271 | |
| Transcripts | mRNA | 37,240 |
| Miscellaneous RNA | 2,220 | |
| tRNA | 4,974 | |
| lncRNA | 4,188 | |
| snoRNA | 31 | |
| snRNA | 192 | |
| rRNA | 326 | |
| Single-exon transcripts | 421 |
- —National Institutes of Health10.13039/100000002
- —USDA-ARS10.13039/100007917
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect and Arachnid Ecology and Behavior · Insects and Parasite Interactions · Insect Utilization and Effects
Introduction
The American cockroach (Periplaneta americana) is a notorious pest found across the world, living and thriving alongside humans in widely variable environmental conditions. Despite their unsavory reputation, cockroaches are of considerable interest to researchers across disciplines. Cockroaches combine the large size, complex physiology, and tractable nature of mice with the simpler and cost-effective care requirements that are characteristic of model insects.
There is a rich body of work compiled throughout the last century describing basic cockroach biology (Crampton 1925; Cornwell 1968; Guthrie and Tindall 1968; The American cockroach 1982). Early research leveraging P. americana as a model system has enhanced the overall understanding of nervous system function, connectivity, and regeneration (Roeder 1948; Yamasaki and Narahashi 1958; Jacklet and Cohen 1967; Sattelle 1992; Jankowska et al. 2021). Unique traits that enable P. americana to survive extreme environmental stressors have also been discovered, such as its co-evolutionary relationship with the endosymbiotic Blattabacterium, a bacterium which provides essential amino acids to its host (Cochran et al. 1979; Vicente et al. 2018). Studies on the cockroach immune system have linked its expansive repertoire of immune-associated proteins to common allergens (Arumugam et al. 2017; Wang et al. 2023) and avenues of pesticide resistance (Zhang et al. 2016; Zhang et al. 2018; Sun et al. 2024). The extensive antimicrobial and regenerative capabilities of P. americana have also earned it respect in both ancient and modern Chinese tradition as an important medicinal insect (Tsuneo et al. 1988; Zhang et al. 2023). Modern sequencing technologies and genetic techniques have further supported the use of P. americana as a model organism. The American cockroach is highly susceptible to RNA inhibition (RNAi) via multiple administration methods, making it an especially useful organism for investigations deciphering gene involvement in insect physiological development and pesticide resistance (French et al. 2015; Huang et al. 2018; Li et al. 2021). P. americana also shows potential as an emerging insect model for host–microbiome interactions. The gut microbiome of omnivorous cockroaches reflects that of humans and omnivorous mammalian model systems (Zurek and Keddie 1996; Tinker and Ottesen 2016; Ayayee et al. 2018; Dockman and Ottesen 2024), and germ-free nymphs can be easily produced via ootheca sterilization, a highly desirable trait for defined-community research interests (Mikaelyan et al. 2016; Tegtmeier et al. 2016; Dukes et al. 2021; Vera-Ponce de León et al. 2021).Altogether, these traits support the research potential of this insect and argue toward the necessity of a complete, high-quality American cockroach genome. While there are 2 previous genome assemblies publicly available, the assembly presented by Li et al. (2018) lacks publicly available protein annotations and is limited by the short-read technology available at the time and the assembly prepared by Wang et al. (2023), while a significant improvement, contains more scaffolds than supported by karyotype analysis (male/XO: 33 diploid; female/XX: 34 diploid) (John and Lewis 1960).
Here, we present the first chromosome-level assembled genome of P. americana. We used long-read high fidelity (HiFi) PacBio sequencing in addition to chromatin contact mapping (Hi-C) to produce a genome scaffolded into a 17-chromosome assembly, consistent with previous karyotype findings (John and Lewis 1960). This high-quality genome assembly is an important tool for facilitating future genetic research in the American cockroach.
Materials and methods
Insect origin and selection
An adult female P. americana individual was selected from a stock colony maintained at the University of Georgia by the Ottesen laboratory; this colony has been maintained the laboratory for 10 years and originated in another long-term laboratory colony of unknown origin. The specimen was flash frozen in liquid nitrogen and shipped on dry ice to the United States Department of Agriculture—Agricultural Research Service (USDA-ARS)—Pacific Basin Agricultural Research Center (PBARC) in Hilo, Hawaii.
Sample preparation and sequencing methods
For PacBio sequencing, high molecular weight (HMW) DNA extraction was performed from insect leg tissue using the Qiagen MagAttract HMW DNA Kit (Qiagen, Hilden Germany). DNA was sheared with the Diagenode Megaruptor 2 (Denville, New Jersey, USA) then prepared for PacBio sequencing using the SMRTBell Express Template kit 2.0 (Pacific Biosciences, Menlo Park, California, USA). The library was size-selected prior to sequencing on a Sequel II System (Pacific Biosciences, Menlo Park, California, USA) using Binding Kit v2.0, Sequencing kit v2.0, and SMRT Cell 8M. To target HiFi reads, the library was sequenced using a 30-h movie time on 3 SMRTcells. Raw subreads were converted to HiFi data by processing with circular consensus sequencing (CCS) to call a single high-quality consensus sequence for each molecule, using a 99.5% consensus accuracy cutoff.
For Hi-C sequencing, cross-linked leg tissue from the same insect was processed according to the Arima Hi-C low input protocol using an Arima Hi-C kit (Arima Genomics, San Diego, California, USA). After proximity ligation, DNA was sheared with a Diagenode Bioruptor then size-selected for 200–600 bp DNA fragments. The Swift Accel NGS 2S Plus kit (Integrated DNA Technologies, Coralville, Iowa, USA) was used to prepare an Illumina library from the size-selected DNA, which was then sequenced with an Illumina NovaSeq 6000 (Illumina, San Diego, California, USA).
RNA-seq
Transcriptomic data of P. americana digestive tissues that had been processed for a separate study were used to assess this genome assembly (BioProject: PRJNA1105088; Sequence Read Archive (SRA) experiments: SRX27556002-SRX27556025). Briefly, midgut, hindgut, and fat body tissues were collected from 8 mixed-sex adult insects and individually stored in RNAlater until extraction using the EZNA Total RNA Kit II (Omega-Biotek). Gut samples were split lengthwise and vortexed to remove lumen contents, and half of each gut sample (or 15 mg of fat body tissue) was homogenized directly in 1 mL RNA-Solv reagent with an Ultra-Turrax rotor-stator homogenizer (Janke & Kunkel, Germany). Following kit instructions, homogenized samples were phase-separated with 200 μL chloroform (VWR), column-purified, and eluted into 50 μL nuclease-free water. Total RNA was incubated with 1 μL TURBO DNase enzyme (Invitrogen) and 5 μL TURBO DNase reaction buffer for 20 min at 37 °C to destroy contaminating DNA, then purified using the column-based EZNA MicroElute RNA Clean-Up Kit (Omega-Biotek) into 30 μL nuclease-free water. RNA quality was assessed with an Agilent Bioanalyzer and samples were stored at −80 °C until library preparation, which was performed using the NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB), the NEBNext® Ultra II Directional RNA Library Prep Kit for Illumina (NEB), and Illumina-compatible index primers (NEBNext® Multiplex Oligos for Illumina). In all, 24 separate libraries (3 per insect) with 300 bp inserts were sent for 150 bp paired-end Illumina NovaSeq sequencing at Novogene (Sacramento, CA, USA). Resulting reads were processed with Joint Genome Institute (JGI) programs BBduk and BBSplit (https://jgi.doe.gov/data-and-tools/software-tools/bbtools/) to remove sequencing adapters and screen for Blattabacterium contamination prior to ribosomal RNA removal with SortMeRNA (Kopylova et al. 2012). Cleaned RNA reads were aligned to the repeat-masked genome as an additional contaminant filtering approach.
Genome assembly and polishing
The first genome assembly was generated and deduplicated using hifiasm v0.19.6-r595 Hi-C integration to obtain primary, alternate, and haplotype-phased assemblies (Cheng et al. 2021). Three flow cells of PacBio HiFi data were obtained from long-read sequencing and concatenated into a single fastq file for assembly and used with Hi-C data obtained from the same source insect. The primary assembly was selected for polishing and contigs were filtered to retain those with coverage reported from hifiasm as between 6× and 30×.
Contamination filtering was performed as described by Lu and Salzberg (2018) . Contigs were separated into individual files, then fragmented with SeqKit v2.5.1 into 100 bp pseudo-reads with 50 bp overlaps (Shen et al. 2016). These pseudo-reads were fed through Kraken v2.1.3 with default parameters to align to the default eukaryotic and prokaryotic databases, and contigs identified as Blattabacterium or with 70% identity assigned to Homo sapiens were discarded (Wood et al. 2019). Remaining contigs were masked using RepeatMasker v4.1.5 (https://www.repeatmasker.org/RepeatMasker/) with Dfam library v3.7 to screen for and remove contigs composed primarily of repetitive DNA, with remaining masked contigs used for RNA-seq alignment with HISAT2 (Kim et al. 2019; Storer et al. 2021). For final repeat annotation, a more detailed repeat-masking procedure was followed as described below.
The Arima Genomics mapping pipeline (https://github.com/ArimaGenomics/mapping_pipeline) was used to map Hi-C data to the filtered assembly. The pipeline described utilizes the programs BWA-MEM for separate alignment of the paired Illumina Hi-C reads, the Picard (https://broadinstitute.github.io/picard/) “MarkDuplicates” command for PCR duplicate removal, and SAMtools for file sorting and handling (Li 2013; Danecek et al. 2021). The resulting Hi-C alignment files and the hifiasm assembly were fed into Yet Another Hi-C Scaffolding tool (YaHS) without breaking contigs, and the resulting contact map was visualized with Juicebox for manual curation (Durand et al. 2016; Zhou et al. 2023). The corrected genome was exported and screened for possible telomeres using tidk (https://github.com/tolkit/telomeric-identifier) and repeat regions identified de novo (AACCTAACCT) were graphed (Brown et al. 2023). An additional round of polishing was performed in Juicebox to correct repeat-heavy telomeric loci, and completeness of the final scaffolded genome was assessed with the Insecta set (version odb10) of Benchmarking Universal Single-Copy Orthologs (BUSCOs) (Manni et al. 2021).
Mitochondrion identification
The program MitoHiFi v2 was used to identify a consensus mitochondrial genome sequence from the original hifiasm assembly (Uliano-Silva et al. 2023). The NCBI reference sequence NC_016956.1 was selected as the reference P. americana mitochondrion for identification (Xiao et al. 2012).
In-depth repeat modeling and masking
For in-depth repeat identification, RepeatModeler v2.0.4 was used to create an ab initio repeat library specific to P. americana, which was then separated into libraries of known and unknown repeat families (Flynn et al. 2020). Using the script repclassifier.sh (https://github.com/darencard/GenomeAnnotation/blob/master/repclassifier), unknown repeats were iteratively re-annotated for 7 rounds, when the percent of repeats classified as “known” rather than “unknown” plateaued. Four rounds of repeat masking using RepeatMasker v4.1.5 were then performed as described in (https://darencard.net/blog/2022-07-09-genome-repeat-annotation/), during which simple repeats were identified and masked first, followed by insect-specific Dfam repeats, known P. americana repeats, and lastly unknown P. americana repeats. The output from the 4 RepeatMasker rounds were combined to generate an overall masked genome, annotation files, and table describing the repeats, and repeat landscapes were summarized with the script parseRM.pl (https://github.com/4ureliek/Parsing-RepeatMasker-Outputs).
Genome annotation
The finished assembly was submitted to NCBI for structural and functional gene annotation via the automated Eukaryotic Genome Annotation Pipeline v10.3. Evidence fed into the GNOMON gene prediction tool included existing RNA-seq data and transcriptome assemblies for P. americana, NCBI RefSeq protein sets from Acromyrmex echinatior, Hyalella azteca, Acyrthosiphon pisum, Caenorhabditis elegans, Tribolium castaneum, Drosophila melanogaster, and Apis mellifera, as well as Insecta and P. americana GenBank protein sets. Details of the annotation release (GCF_040183065.1-RS_2024_10) are available at https://www.ncbi.nlm.nih.gov/refseq/annotation_euk/Periplaneta_americana/GCF_040183065.1-RS_2024_10/.
Blattodea comparison
Protein-coding genes identified in this assembly were compared with 6 Blattodea species that have publicly available protein annotations in GenBank or RefSeq. Translated CDS fasta and corresponding GFF files were downloaded from NCBI for the following organisms: a previous P. americana genome (GenBank: GCA_025594305.2) (Wang et al. 2023), Zootermopsis nevadensis (RefSeq: GCF_000696155.1) (Terrapon et al. 2014), Blattella germanica (GenBank: GCA_003018175.1) and Cryptotermes secundus (RefSeq: GCF_002891405.2) (Harrison et al. 2018), Coptotermes formosanus (GenBank: GCA_013340265.1), and Diploptera punctata (GenBank: GCA_030220185.1) (Fouks et al. 2023). The program GENESPACE v1.3.1, which implements OrthoFinder (v2.5.5) as part of its pipeline, was run in R Studio to identify orthologous gene sets based on the longest isoform peptide sequences and to generate riparian plots for synteny visualization (Emms and Kelly 2015, 2019; Lovell et al. 2022). The distribution of orthologous gene sets between and across genomes were visualized with the R package UpSetR v1.4.0 (Conway et al. 2017). To evaluate the general functional capacity of orthogroups containing gene products from (i) only P. americana and (ii) all evaluated Blattodea species, gene ontology (GO) IDs associated with P. americana gene products that had been assigned to these 2 groups were used to represent orthogroup activities. GO terms were extracted from the GO IDs in R with the package clusterProfiler v4.6.2 (Yu et al. 2012; Wu et al. 2021).
Results and discussion
Assembly
As American cockroaches have an XX/X0 sex chromosome system (Jankásek et al. 2021), a single female insect was selected for genome sequencing. Over 6.6 million long-read HiFi reads covering 90.6 Gb of DNA were obtained from PacBio SMRTBell sequencing, with an additional 217.3 Gb of Hi-C proximity ligated 150 bp reads obtained through paired-end Illumina sequencing. The primary genome assembly produced by hifiasm was 3.36 Gb long and comprised 1818 contigs with lengths ranging from 8 Kb to 135 Mb (mean = 1.85 Mb); the lengths and coverage values of individual contigs are presented in Supplementary File 1. The overall genome had an estimated sequencing coverage of ∼90X based on HiFi and HiC reads and possessed a contig N50 of 40.2 Mb. From this initial assembly, we identified several mitochondrially derived contigs that were used to determine the 15,578 bp circular consensus mitogenome (Supplementary Fig. 1). We evaluated the quality of the initial assembly based on contig-level read coverage and sequencing depth reported from hifiasm (Fig. 1) and determined that coverage peaks occurred at 14X, 18X, and 25X, with 96.19% of the initial assembly contained in contigs with coverage values between 6X and 30X. Based on this distribution, we removed 1,548 contigs with abnormally low (5X or lower) or high (31X or higher) coverage from the assembly (Supplementary File 1). Compared to the contigs which passed our filtering parameters, the 1,487 contigs with insufficient sequencing depth were highly fragmented (median length of 84.3 Kb vs 2.4 Mb; Supplementary Fig. 2, Fig. 1b), and their removal had little impact on overall genome assembly statistics (Supplementary Table 2). Since repetitive genomic material may result in abnormally high coverage, we additionally screened high coverage contigs with Kraken2 and found that all 61 contigs matched the cockroach endosymbiont Blattabacterium, validating their removal from the assembly (Supplementary File 1). Subsequent contamination filtering of the filtered assembly resulted in the removal of 27 additional contigs, of which 19 were derived from Blattabacterium, 3 from Homo sapiens, and 5 were very small contigs with at least 40% of their DNA masked as repetitive content (Supplementary File 1). No other contaminants were identified within the retained contigs, with most contigs marked as at least 90% unclassified by Kraken2. After the removal of unsupported and contaminating contigs, the pre-scaffolded assembly contained 243 contigs with an N50 of 42.4 Mb, overall producing a high-quality contig-level genome with no detectable contamination (Supplementary Table 2).
Quality profile of initial contig and final scaffold hifiasm assembly. a) Pacbio reads were mapped to the primary genome assembly to determine overall coverage of individual contigs, and the percent each coverage level contributed to the total genome size was plotted. b) The length of contigs per coverage level were plotted, with individual contigs represented as dots. Retained contigs with coverage between 6X-11X (n = 73) and 28X-30X (n = 5) were excluded from plotting in (b) due to short lengths.
Scaffolding with YaHS placed 95.67% of the genome into 17 scaffolds (Supplementary Fig. 3), with manual curation via Juicebox increasing this percentage to 98.93% (Fig. 2a). We determined the telomeric sequence to be AACCTAACCT with tidk and identified long stretches of telomeric repeats in all chromosome-sized scaffolds, with 10 scaffolds flanked by telomeres on both ends and 6 scaffolds flanked on one end (Fig. 2b). Only scaffold #7 contained telomeric repeats embedded within a large contig rather than at the beginning or end of the scaffold; while this may indicate an assembly error, we did not have evidence to support adjusting its placement. The 74 remaining contigs were unable to be matched to just 1 scaffold, likely due to a high density of centromeric repeat regions, and were therefore left unplaced. Generally, these unplaced scaffolds contained contigs which were poorly covered by HiC and RNA sequencing of cockroach digestive tissues (Supplementary Fig. 4). BUSCO analysis of the putative chromosomes found 99.7% of Insecta BUSCOs were present and complete in this genome assembly, of which only 1.5% were duplicated (Supplementary Fig. 5, Table 1).
Chromatin contact sequencing produced an assembly with near-chromosomal resolution. Following scaffolding with YaHS, scaffold boundaries and contig placement were adjusted in Juicebox to optimize chromatin contacts for the 17 chromosome-level scaffolds. Final chromosomal boundaries are shown in the heatmap in (a), and the occurrence of telomeric sequences within each chromosome are displayed in (b).
In summary, we have produced a high-quality genome assembly for P. americana using a combination of long-read and chromosomal contact sequencing data. This 3.23 Gb genome is almost entirely contained within 17 chromosome-scale scaffolds, aligning with previous karyotype and flow cytometry findings for P. americana that identified 17 haploid (male/female: 33/34 diploid) chromosomes and a predicted genome size of 3.338 Gb (John and Lewis 1960; Hanrahan and Johnston 2011; Jankásek et al. 2021). This assembly represents an improvement over previous P. americana genome assemblies available in NCBI databases (Table 1), which resemble our assembly in length and GC content but have lower contiguity and BUSCO scores (Li et al. 2018; Wang et al. 2023). Therefore, we argue that this genome assembly qualifies as both comprehensive and chromosomally resolved.
Repetitive DNA elements
Overall, 50.86% of the genome was identified as repetitive content classified as DNA elements, simple repeat regions, or retroelements, with an additional 15% of the genome determined to be P. americana-specific repeats that remain otherwise unclassified (Fig. 3a, Supplementary Table 1). The DNA transposon and retroelement subgroups contributed similarly to overall repeat content, comprising 24.3% and 20.05% respectively of the genome, but differ in their overall Kimura divergence landscapes (Fig. 3b). The DNA elements found in this genome primarily belong to the Tc1-Mariner and hobo-Activator-Tam3 subfamilies with higher relative abundances at a Kimura substitution level of 5% (Fig. 3c). In contrast, the primary retrotransposon class of long interspersed nuclear elements (LINEs) contained 2 peaks in Kimura substitution, with CR1, L1, L2, and CRE subgroups showing elevated substitution levels around 5% while RTE-clade retrotransposons represent a more ancient repeat lineage, with substitution levels peaking between 31% and 32% (Fig. 3d). Short interspersed nuclear elements (SINEs) and retroelements containing long-terminal repeats (LTRs) were less commonly identified, only making up 3.47% and 0.94% of the genome respectively. Most SINEs contained internal promoters derived from tRNA and showed more gradual patterns of divergence, with a relatively stable plateau between 5% and 15% divergence before tapering (Fig. 3e). Repeat content varies widely even between insects from the same family, but generally Blattodea species show similar repeat distributions with especially low LTR content and expanded LINEs, with larger genomes correlated to higher repeat content (Sproul et al. 2023).
Repeat summary of P. americana. RepeatMasker and RepeatModeler were used to identify (a) the abundance of repetitive element families present in this assembly and (b) the relative abundance of each repeat class vs Kimura substitution level. The repeat landscapes for the classes c) DNA elements, d) LINEs, and e) SINEs were further visualized at the repeat subtype level.
Orthology analysis between Blattodea species
The order Blattodea encompasses both termites and cockroaches, with over 4,700 species identified in NCBI's taxonomy repository. Despite these many representatives, only 12 species have sequenced genomes (Terrapon et al. 2014; Harrison et al. 2018; Li et al. 2018; Shigenobu et al. 2022; Fouks et al. 2023; Martelossi et al. 2023; Wang et al. 2023; Ewart et al. 2024; Hunter 2025), of which half have publicly available annotations uploaded to NCBI. These species include 2 cockroaches from the superfamily Blaberoidea (Blattella germanica, Diploptera punctata), a previous American cockroach genome assembly (superfamily: Blattoidea), and 3 termite species (Blattoidea; Termitidae: Zootermopsis nevadensis, Cryptotermes secundus, Coptotermes formosanus) (Fig. 4a). In general, cockroaches possessed larger genomes than termites, with P. americana's 3.2 Gb genome more than double the size of the available termite genomes (Fig. 4b). Among these Blattodea, this assembly had the highest contig N50 (Fig. 4c) and a high scaffold N50 that was second only to the cockroach Ectobius pallidus (Hunter 2025) (Fig. 4d). Compared to the 6 Blattodea genomes with publicly available annotations, P. americana encoded more protein-coding genes than termites, but fewer were reported in comparison to the 3 cockroach genomes (Fig. 4e). It is unclear whether this difference between annotations in our assembly and the other cockroach assemblies is biological or a result of annotation technique; this assembly was annotated by NCBI's Gnomon pipeline which produced 16,750 protein-coding genes (Table 2), while previous P. americana genomes reported 21,336 (annotations not available) and 29,939 (GenBank) protein-coding genes (Li et al. 2018; Wang et al. 2023). The other 2 cockroach genomes, B. germanica and D. punctata, had user-submitted annotations which may have overestimated their gene content, although the termite genome with GenBank annotations (C. formosanus) contained a similar number of genes to the RefSeq-annotated termites. Notably, the B. germanica assembly is flagged as “contaminated” by NCBI, which may have also contributed to the high gene count in its GenBank annotations.
Available Blattodea genomes from NCBI. Phylogeny of sequenced Blattodea genomes as they appear in the NCBI taxonomy browser is presented in (a), in addition to their b) genome size, c) contig N50, d) scaffold N50, and (e) number of protein-coding genes (if available). Points on the genome statistic plots (b–e) correspond to the colors in (a) and species in (a) with protein annotations available on NCBI are boxed. Assemblies flagged as contaminated are denoted with #.
Since only this assembly and 2 termite genomes (Z. nevadensis and C. secundus) possessed RefSeq annotations, we included the available user-submitted GenBank annotations (C. formosanus, B. germanica, and D. punctata) for ortholog analysis of proteins identified in these Blattodea species. We initially included the previous P. americana genome assembly for OrthoFinder and GENESPACE to compare the annotations between these assemblies. Analyzing the longest isoforms per gene product, OrthoFinder identified 16,711 orthologous gene families among these 7 annotated genomes, of which 2,311 were shared across all 7 Blattodea and 2,220 were shared by all assemblies excluding the previous P. americana assembly (Supplementary Fig. 6). Following the GENESPACE pipeline to generate riparian plots (Fig. 5a), we discovered that, while most chromosomes in our assembly were entirely captured by 1–2 scaffolds from the previous assembly, chromosomes 10 and part of 14 were missing from the other P. americana annotations. The existence of these regions was supported by synteny analysis between our P. americana genome and annotations from the other cockroaches (Fig. 5b) and termites (Fig. 5c), and included proteins encompassing a wide range of functions with immune (Dscam, toll-like receptors, leucine-rich repeat proteins), neurologic (GABA transport, neurotrophic factors), endocrine (vitellogenin synthesis, sterol binding proteins), and nutritional (xanthine dehydrogenase, salivary peptide) importance. Therefore, we chose to rerun OrthoFinder with only our assembly representing P. americana.
Ortholog analysis within Blattodea. Protein GFF3 files available on NCBI for Blattodea species were compared using OrthoFinder and GENESPACE. Riparian plots were generated to assess synteny between the P. americana genome presented here and a) a previous P. americana assembly (GCA_025594305.2), b) the cockroaches Diploptera puntata (GCA_030220185.1) and Blattella germanica (GCA_003018175.1), and c) the termites Cryptotermes secundus (GCF_002891405.2) and Zootermopsis nevadensis (GCF_000696155.1). d) Orthologous gene clusters shared by or unique to the analyzed genomes were visualized via UpSet plot, organized by phylogeny on the left and the next 10 largest sets on the right and summarized by d) the percentage of genes assigned to shared or unique orthogroups. Genes belonging to the 269 orthogroups identified as P. americana-unique were analyzed by their associated GO terms, and e) biological processes and molecular functions with more than 14 occurrences (weighted if multiple terms were assigned to 1 gene) were visualized. Assemblies flagged as contaminated are denoted with #.
Upon rerunning OrthoFinder, we obtained 14,844 orthologous gene families, of which 4,532 orthogroups contained genes from all included Blattodea species (Fig. 5d, Supplementary File 2). Gene families conserved at the order level comprised large fractions of each insect's total gene count, ranging from 5,548 genes (44.8%) in Z. nevadensis to 7,321 genes (25.8%) in D. punctata (Fig. 5d inset). These core gene families encompass a wide range of functions necessary to life and common in insects, with GO terms relating to gene expression and genome maintenance especially prevalent in this subset (Supplementary Fig. 7). The full list of GO terms assigned to these shared gene families can be found in Supplementary File 3.
The percentage of genes assigned to orthogroups varied between Blaberoidea and Blattoidea (termites and P. americana) (Fig. 5d inset). The termite genomes and our P. americana assembly had 94.1%–97.5% of genes assigned to orthogroups, while only 74.1 and 78.1% of genes from D. punctata and B. germanica, respectively, were assigned an orthogroup (Fig. 5d inset, Supplementary File 2). Unassigned genes are unique both between analyzed genomes and within a single genome, so it is unclear whether these genes stem from a larger number of single-copy genes within the cockroaches or represent erroneous gene models without real biological function. Most likely, the Gnomon pipeline used for RefSeq annotations produced more robust and conservative gene models than other pipelines, explaining the high number of P. americana, Z. nevadensis, and C. secundus genes sorted into orthogroups. Despite these questions, differences between cockroaches and termites in their species-specific gene families highlight increased gene expansion events among the cockroaches. Our P. americana assembly, which encoded over 3,000 more genes than the termite assemblies, had 11.3% (n = 1901) of its genes assigned to 269 P. americana-unique gene families, a substantial increase from the 1.5%–3.3% range found in the 3 termite assemblies (Fig. 5d inset). The cockroaches B. germanica and D. punctata showed similar gene duplication patterns to P. americana, with 24.9% and 14.6% of their genes respectively assigned to species-specific gene families. While these Blaberoidea assemblies appear to be over-annotated and should be interpreted cautiously, especially given the contamination in B. germanica flagged by NCBI, the similar patterns shared between them and P. americana suggests that there is a cockroach-related association with gene duplication events. Since ortholog analysis depends on the data supplied, it is difficult to determine whether this difference stems from biological cockroach-termite delineation or is a consequence of few available cockroach representatives. Nonetheless, these results, in combination with the large size of this cockroach genome (Fig. 4b) and the late spike in LINE retrotransposon divergence (Fig. 3d), suggests that acquisition and expansion of new genes contributed to cockroach divergence.
We analyzed common GO terms associated with the 269 orthologous gene families present in our P. americana assembly but not the other Blattodea species (Fig. 5e). Among the most abundant GO terms assigned to genes within these orthogroups were terms related to gene expression and genomic maintenance, a pattern also observed within the Blattodea-shared gene families (Supplementary Fig. 7). Since over a third of the genes assigned to P. americana-unique orthogroups lacked associated GO terms (n = 724), we explored common products and functions that were encoded within these expanded gene families based on the gene descriptions from NCBI (Supplementary Fig. 8). These gene families were enriched in immune and digestive functions, such as lipopolysaccharide recognition, protease activity, odorant binding, and lipase activity (Supplementary Fig. 8). Some abundant functions were found to have expanded from within a single gene family, such as the starvation-associated gene takeout, and chymotrypsin products. In contrast, other functions were both expanded within gene families and found in multiple distinct orthogroups, such as trypsin and lipopolysaccharide-binding proteins. Possessing diverse orthologs within these protein families likely enhances P. americana's capacity to fend off a wider variety of infectious agents, or to efficiently digest protein sources that would be inaccessible to termite relatives. The growth of these gene families may have facilitated cockroach divergence from the protective eusociality found in termite colonies toward a more independent and self-sufficient lifestyle. Additional sequencing of Blattodea genomes may clarify relationships between these gene families and Blattodea evolution. Overall, synteny and ortholog comparison between these insects reveal possible mechanisms of divergence between termites and cockroaches and highlight the potential applications of a chromosomally resolved P. americana genome.
Summary
We sequenced the genome of the American cockroach using high fidelity PacBio long reads in conjunction with Hi-C Illumina short reads. This 3.23 Gb assembly is highly contiguous, with a contig N50 of 42 Mb and a scaffold N50 of 188 Mb, and 98.93% of the assembly is contained within 17 putative chromosomes. The quality of this assembly is further exemplified by its genomic and protein BUSCO scores, which are 99.7% and 97.8% complete, respectively. This high-quality assembly, generated with cutting edge sequencing technology, is a substantial improvement over existing P. americana genomes, and we report an entire chromosome that was missing from a previously published assembly. This genome is expected to facilitate future study of cockroach physiology and Blattodea evolution.
Supplementary Material
jkaf247_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Arumugam G, Sreeramulu B, Paulchamy R, Thangavel S, Sundaram J. 2017. Purification and functional characterization of lectin with phenoloxidase activity from the hemolymph of cockroach, Periplaneta americana. Arch Insect Biochem Physiol. 95:e 21390. 10.1002/arch.21390.28557066 · doi ↗ · pubmed ↗
- 2Ayayee PA, Ondrejech A, Keeney G, Muñoz-Garcia A. 2018. The role of gut microbiota in the regulation of standard metabolic rate in female Periplaneta americana. Peer J. 6:e 4717. 10.7717/peerj.4717.29844953 PMC 5971104 · doi ↗ · pubmed ↗
- 3The American cockroach. 1982. Bell WJ, Adiyodi KG, editors. Chapman and Hall.
- 4Brown M, González De la Rosa PM, Mark B. 2023. A telomere identification toolkit. Zenodo. 10.5281/zenodo.10091385 · doi ↗
- 5Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18:170–175. 10.1038/s 41592-020-01056-5.33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 6Cochran DG, Mullins DE, Mullins KJ. 1979. Cytological changes in the fat body of the American cockroach, Periplaneta americana, in relation to dietary nitrogen levels. Ann Entomol Soc Am. 72:197–205. 10.1093/aesa/72.2.197. · doi ↗
- 7Conway JR, Lex A, Gehlenborg N. 2017. Up Set R: an R package for the visualization of intersecting sets and their properties. Bioinformatics. 33:2938–2940. 10.1093/bioinformatics/btx 364.28645171 PMC 5870712 · doi ↗ · pubmed ↗
- 8Cornwell PB . 1968. The cockroach: a laboratory insect and an industrial pest. Huthinson.