Integrating Morphology and Chloroplast Genomics: A New East Asian Species of Aletris (Nartheciaceae) With Insights Into Regional Phylogeny and Evolution
Xiong Li, Yong‐Ling Qiu, Jiang‐Tao Li, Bo Xu, Qi Yu, Wen‐Bin Ju

TL;DR
A new plant species, Aletris medogensis, was discovered in the Himalayas using a combination of physical traits and chloroplast DNA analysis, helping clarify plant evolution in the region.
Contribution
The discovery of a new Aletris species and the integration of morphology with chloroplast genomics to resolve taxonomic and evolutionary questions.
Findings
Aletris medogensis is a new species characterized by unique morphological and chloroplast genomic traits.
Phylogenomic analysis shows A. medogensis is closely related to A. alpestris and has distinct chloroplast genome variations.
Positive selection in genes ccsA, cemA, and rps12 suggests adaptive evolution in the new species.
Abstract
The genus Aletris L. (Nartheciaceae) encompasses approximately 21–24 species distributed in East Asia and North America, yet taxonomic ambiguity persists due to overlapping morphological traits among closely related species. During fieldwork in southeastern Xizang, China, a morphologically distinct candidate species, Aletris medogensis, was discovered. To validate its taxonomic status and explore evolutionary relationships within East Asian Aletris, we integrated detailed morphological observation with comparative chloroplast phylogenomics. The newly proposed species is characterized by creeping stolons, narrow leaves, and glandular‐pubescent inflorescences. Comparative analysis of 14 East Asian Aletris complete chloroplast genomes revealed a conserved quadripartite structure with species‐specific variations, including pseudogenization of ycf1, loss of rrn4.5, and shifts in IR…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 FIGURE 1
FIGURE 1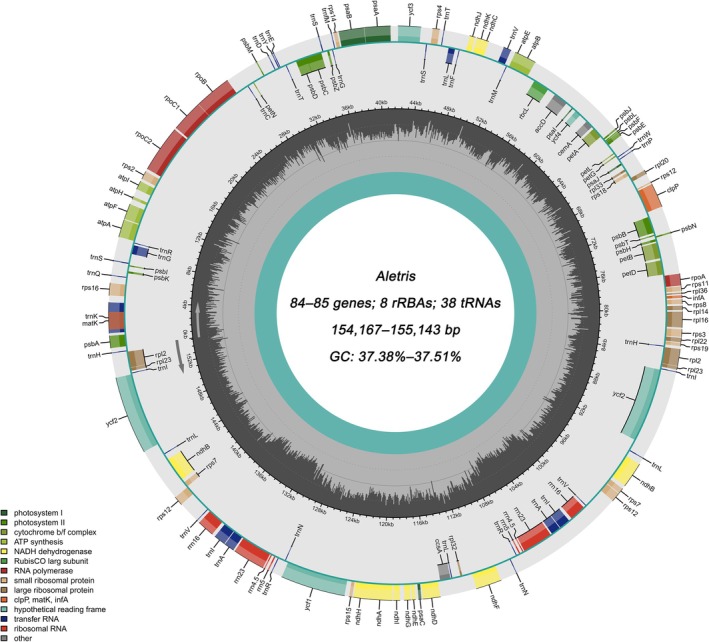 FIGURE 2
FIGURE 2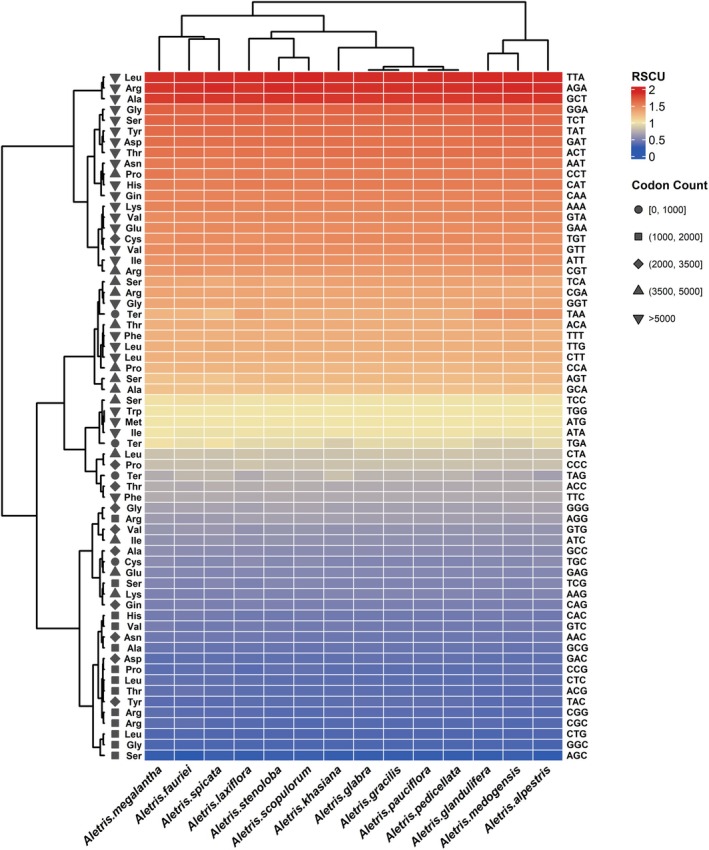 FIGURE 3
FIGURE 3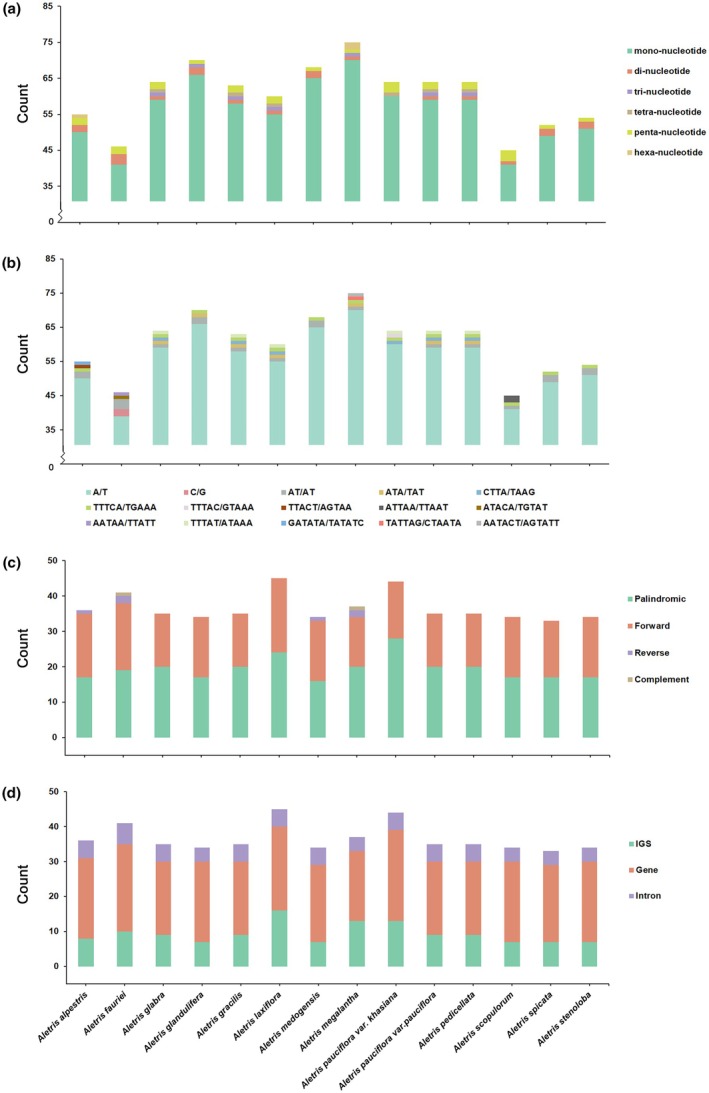 FIGURE 4
FIGURE 4 FIGURE 5
FIGURE 5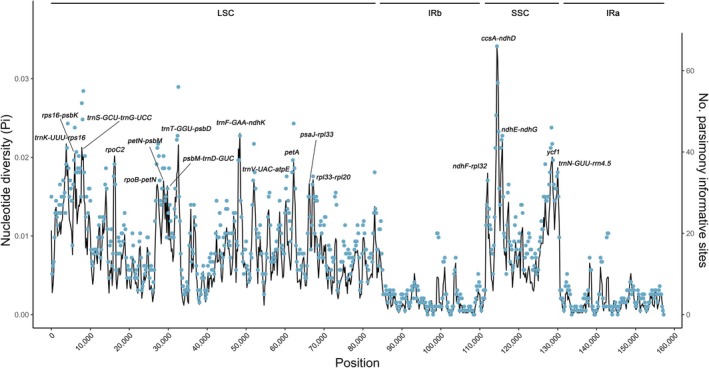 FIGURE 6
FIGURE 6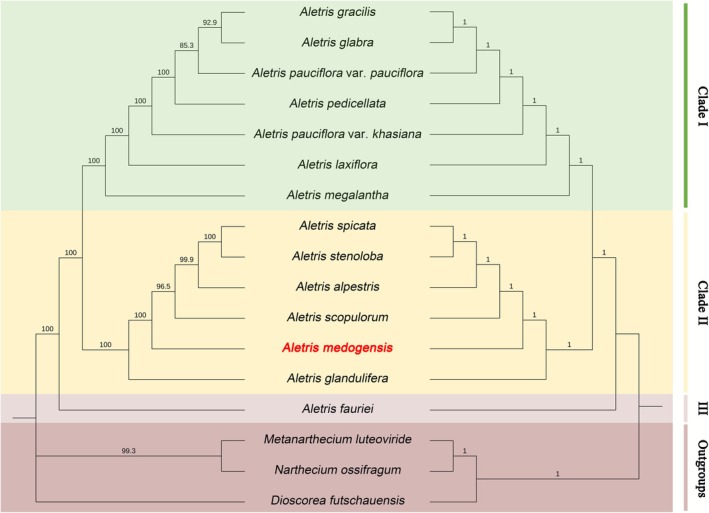 FIGURE 7
FIGURE 7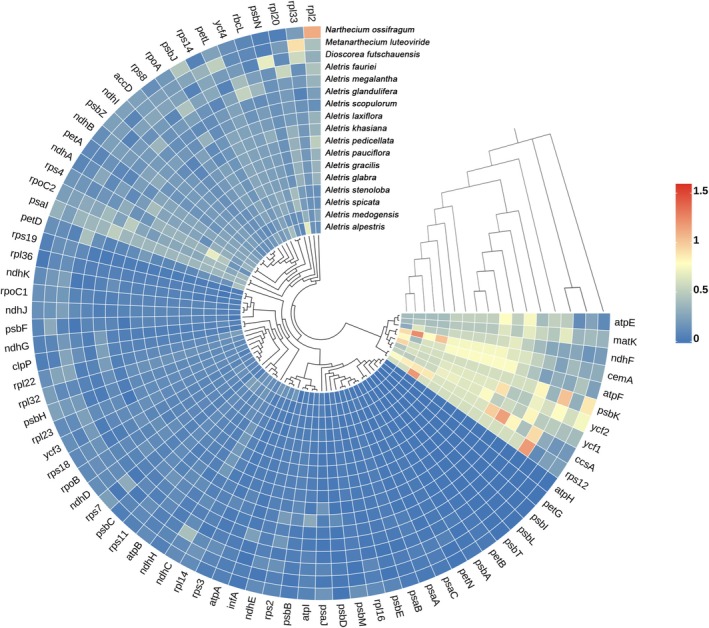 FIGURE 8
FIGURE 8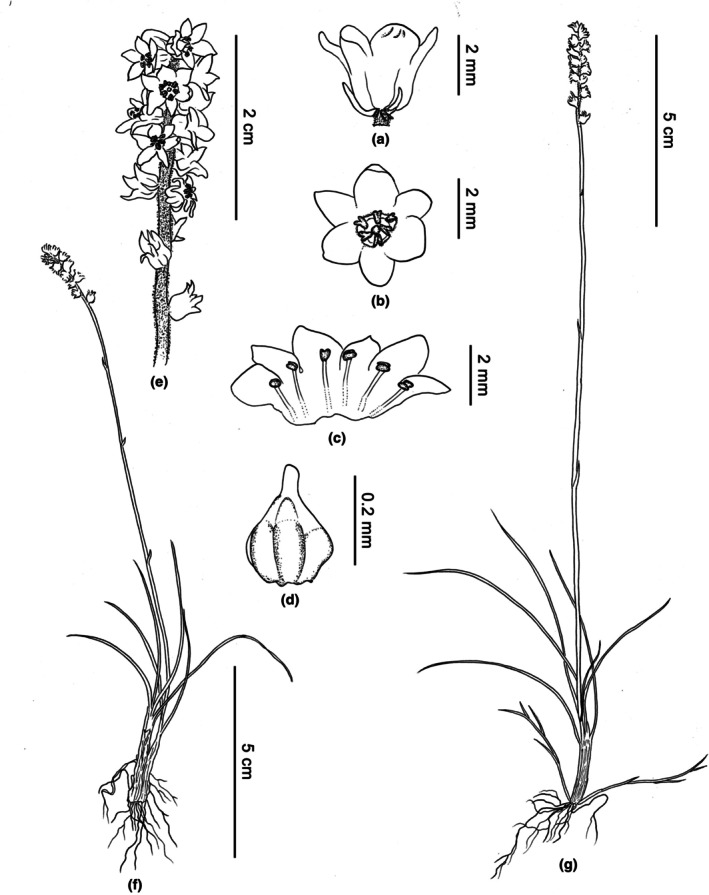 FIGURE 9
FIGURE 9 FIGURE 10
FIGURE 10| Character |
|
|
|
|
|---|---|---|---|---|
| Plant | Tillering with horizontally creeping stolons | None | None | None |
| Leaves | 2.5–9 cm × 0.45–0.9 mm, few and slender, linear | 1.5–8 cm × 2–4 mm, numerous, densely tufted, linear‐lanceolate | 5–25 cm × 2–8 mm, few and laxly tufted, linear‐lanceolate to linear | 5–10 cm × 5–7.5 mm, linear‐lanceolate to linear |
| Scape size | 3.5–16 cm × 0.7–1 mm | 7–20 cm × 0.5–1 mm | 3.5–40 cm × 1.5–2 mm (6.5–8.5 cm × 1.5–2 mm) | 7‐15 cm × 2–2.5 mm |
| Scape hairy | Glabrous, glandular are sparse near the inflorescence | Sparsely puberulent | Densely puberulent | Densely puberulent |
| Bract–like leaves | 0.2–0.6 cm long | Less than 1 cm long | 1.5–5.0 cm long | 1.5–5.0 cm long |
| Inflorescence | raceme 1.5–2.5 cm, densely 10–14‐flowered, densely glandular hairs | Raceme 1–4 cm, laxly 4–10‐flowered, rachis puberulent | Raceme 1–5 cm, densely to laxly 4–6‐flowered, rachis pubescent | Raceme 1–5 cm, densely to laxly 18–20‐flowered, rachis pubescent |
| Bract and bracteole | 1.0–2.0 mm long, shorter than flower | 1.5–4.0 mm long, shorter than flower | 3–20 mm long, one of them 1–4 × flower length | 3.5–7.0 mm long, subequaling or slightly exceeding the flower |
| Pedicel | Less than 0.5 mm long | 2–4 mm long | 1–12 mm long | 1–12 mm long |
| Perianth | Glabrous, lobes ovate, parted nearly to 1/2 of its length | Glabrous but often densely papillose, lobes lanceolate, parted nearly to 1/2 of its length | Glabrous, lobes oblong‐ovate to lanceolate, parted nearly to 1/4 of its length | Glabrous, lobes oblong‐ovate to lanceolate, parted nearly to 1/4 of its length |
| Capsule | Apex abruptly narrowed | Apex abruptly narrowed | Apex of valves gradually narrowed | Apex of valves gradually narrowed |
| Species name | Size (bp) | GC content (%) total (LSC/SSC/IR) | No. of genes (PCGs/tRNA/rRNA) | GenBank accession | |||
|---|---|---|---|---|---|---|---|
| Total | Large single‐copy region (LSC) | Small single‐copy region (SSC) | Inverted repeat (IR) | ||||
|
| 154,557 | 83,194 | 18,167 | 26,598 | 37.38 (35.26/31.18/42.82) | 131 (85/38/8) | |
|
| 154,704 | 83,265 | 18,113 | 26,663 | 37.44 (35.33/31.27/42.82) | 131 (85/38/8) | |
|
| 154,440 | 83,501 | 18,183 | 26,378 | 37.5 (35.45/31.31/42.88) | 131 (85/38/8) | |
|
| 154,999 | 83,511 | 18,160 | 26,664 | 37.48 (35.4/31.27/42.84) | 131 (85/38/8) | |
|
| 154,201 | 83,018 | 17,855 | 26,664 | 37.45 (32.44/31.26/42.84) | 131 (85/38/8) | |
|
| 154,993 | 83,505 | 18,160 | 26,664 | 37.48 (34.01/31.28/42.84) | 131 (85/38/8) | |
|
| 154,167 | 82,706 | 18,151 | 26,655 | 37.49 (32.93/31.22/42.84) | 131 (85/38/8) | |
|
| 154,204 | 83,021 | 17,855 | 26,664 | 37.45 (32.44/31.26/42.83) | 131 (85/38/8) | |
|
| 154,309 | 83,076 | 23,519 | 23,857 | 37.42 (32.42/33.44/43.11) | 130 (85/38/7) | |
|
| 154,205 | 83,022 | 17,855 | 26,664 | 37.45 (32.44/31.26/42.84) | 131 (85/38/8) | |
|
| 154,468 | 83,043 | 18,003 | 26,711 | 37.47 (31.31/31.32/42.86) | 131 (85/38/8) | |
|
| 155,143 | 83,583 | 18,248 | 26,656 | 37.39 (32.86/31.17/42.83) | 131 (85/38/8) | |
|
| 154,201 | 83,018 | 17,855 | 26,664 | 37.45 (32.44/31.25/42.84) | 131 (85/38/8) | |
|
| 154,551 | 83,158 | 18,093 | 26,650 | 37.51 (33.69/31.37/42.85) | 131 (85/38/8) | |
|
| 153,777 | 82,852 | 17,777 | 26,574 | 37.38 (35.17/31.19/54.77) | 130 (84/38/8) | |
|
| 155,312 | 84,051 | 18,135 | 26,563 | 37.57 (35.47/31.63/39.59) | 130 (85/37/8) | |
|
| 153,946 | 83,979 | 18,909 | 25,529 | 37.21 (35.04/31.21/42.99) | 124 (86/38/0) | |
| Category | Gene group | Gene name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I |
|
| Subunits of photosystem II |
| |
| Subunits of NADH dehydrogenase |
| |
| Subunits of cytochrome |
| |
| Subunits of ATP synthase |
| |
| Large subunit of rubisco |
| |
| Self‐replication | Proteins of large ribosomal subunit |
|
| Proteins of small ribosomal subunit |
| |
| Subunits of RNA polymerase |
| |
| Ribosomal RNAs |
| |
| Transfer RNAs |
| |
| Other genes | Maturase |
|
| Protease |
| |
| Envelope membrane protein |
| |
| Acetyl‐CoA carboxylase |
| |
| c‐type cytochrome synthesis gene |
| |
| Translation initiation factor |
| |
| Conserved hypothetical chloroplast reading frames |
|
| Start | End | Length | # SNP | # SVS | # PIP | Pi | Gene name |
|---|---|---|---|---|---|---|---|
| 3400 | 4745 | 1346 | 78 | 50 | 22 | 0.016959 |
|
| 5800 | 6578 | 779 | 48 | 32 | 14 | 0.016218 |
|
| 7800 | 8759 | 960 | 78 | 56 | 18 | 0.020378 |
|
| 16,000 | 16,993 | 994 | 42 | 23 | 13 | 0.012838 |
|
| 27,000 | 27,789 | 790 | 54 | 37 | 14 | 0.01674 |
|
| 28,800 | 29,361 | 562 | 38 | 26 | 12 | 0.017218 |
|
| 29,800 | 30,389 | 590 | 31 | 11 | 20 | 0.016653 |
|
| 32,200 | 33,185 | 986 | 64 | 45 | 13 | 0.016593 |
|
| 48,000 | 48,892 | 893 | 51 | 32 | 18 | 0.016814 |
|
| 52,000 | 52,586 | 587 | 42 | 28 | 12 | 0.017529 |
|
| 62,200 | 62,744 | 545 | 39 | 30 | 8 | 0.015932 |
|
| 66,200 | 66,722 | 523 | 26 | 14 | 11 | 0.016847 |
|
| 67,000 | 67,779 | 780 | 39 | 23 | 14 | 0.01477 |
|
| 112,000 | 112,596 | 597 | 33 | 15 | 17 | 0.018093 |
|
| 114,200 | 116,332 | 2133 | 154 | 104 | 49 | 0.020851 |
|
| 117,400 | 117,996 | 597 | 32 | 18 | 14 | 0.014381 |
|
| 128,200 | 129,199 | 1000 | 68 | 42 | 26 | 0.018297 |
|
| 130,000 | 130,599 | 600 | 35 | 17 | 18 | 0.018681 |
|
- —Special Investigation and Monitoring of the Yarlung Zangbo Grand Canyon National Nature Reserve in Xizang
- —Science and Technology Major Project of Xizang
- —Second Tibetan Plateau Scientific Expedition and Research
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Diversity and Evolution · Plant and Fungal Species Descriptions · Bryophyte Studies and Records
Background
1
The genus Aletris L. was established by Linnaeus (1753), with Aletris farinosa L. from southeastern North America designated as the type species. It was traditionally placed in the family Liliaceae Juss. (Ambrose 1980; Wu et al. 2003), but molecular phylogenetic studies have since shown closer relationships to the genera Lophiola, Metanarthecium, Narthecium, and Nietneria, leading to their reclassification into Nartheciaceae by Caddick et al. (2002). Aletris is the largest genus in this family, with approximately 21–24 species found in East Asia and North America (Zhao et al. 2012). In China, 15 species and one variety have been recorded, nine of which are endemic, primarily distributed in the central and southwestern regions.
Aletris species typically grow on mountain slopes, roadsides, and shrublands. They prefer moist, shaded environments but can tolerate drought and adapt to various conditions (Yang et al. 2024; Akahori et al. 1971). These perennial herbs possess fibrous roots and basal rosettes of grass‐like leaves (lanceolate to linear). A simple, erect scape bears a terminal raceme of bisexual flowers. Each flower has a bract and a smaller bracteole on the pedicel, a 6‐lobed perianth (white to golden‐orange; cylindrical to campanulate) with rough abaxial surfaces. Six short stamens insert on the perianth tube, and the 3‐lobed stigma yields a beaked, loculicidal capsule enclosed by the persistent perianth (Liang and Turland 2000; Nong et al. 2024). Many Aletris species are of ecological and medicinal importance, traditionally used to treat coughs, hemoptysis, dysmenorrhea, and pulmonary abscesses (Li et al. 2014; Challinor et al. 2015).
During a 2024 field investigation in southeastern Xizang, we encountered an unusual Aletris species growing on moss‐covered rocks along streams (Figure 1). It resembled A. pauciflora var. pauciflora (Klotzsch) Hand.‐Mazz. (Brotherus and Handel‐Mazzetti 1936), A. pauciflora var. khasiana F. T. Wang & Tang (Wang and Tang 1978), and A. alpestris Diels (Adolf 1905) in having rosette‐like clustered leaves, perianth glandular, with lobes shorter than or equal to the perianth, and the bract and bracteole borne at the apex of the pedicel. However, it differed in its rhizomes horizontally creeping from the base of the plant, narrower linear leaves, inflorescences with glandular hairs, and shorter pedicels, bracts, and bracteoles. We propose it as a new species, Aletris medogensis W.B.Ju, Y.L.Qiu & Bo Xu, based on morphological traits and molecular phylogenetic analysis derived from the chloroplast (cp) genome.
Habit of Aletris medogensis on moss‐covered rocks along streams (photographed by Wen‐Bin Ju).
In recent years, comparative chloroplast genome analysis has emerged as an essential tool in plant phylogenetics and species identification due to its highly conserved structure, maternal inheritance, and relatively slow evolutionary rate (Zhao et al. 2019; Dopp et al. 2021; Lu et al. 2016). Repeat sequences, inverted repeat (IR) boundaries shift, and nucleotide diversity sites within the chloroplast genome can reveal hypervariable regions and develop molecular markers to evaluate genomic evolutionary history and facilitate species delimitation (Keller et al. 2017; Sabir et al. 2014). Furthermore, the chloroplast genome is not merely a neutral phylogenetic marker; it plays a fundamental role in photosynthesis, stress response, and other essential metabolic pathways. Analyzing selective pressures on chloroplast genes can therefore reveal signatures of adaptive evolution directly linked to environmental factors, such as light intensity, temperature, and water availability, which are particularly relevant for species inhabiting specific niches like the montane ecosystems of the Himalayas (Dong et al. 2018; Rui et al. 2025).
In the genus Aletris, although traditional morphological classification has amassed considerable knowledge, taxonomic controversies persist among some closely related species due to overlapping morphological traits and limited molecular data (Zhao et al. 2012; Liang and Turland 2000; Nong et al. 2024). In this study, we first described the morphological traits of Aletris medogensis in detail and presented its complete chloroplast (cp) genome for the first time. Then, comparative genomics and phylogenomic analyses were conducted by integrating the previously published cp genome data of East Asian Aletris species and related genera in Nartheciaceae, with the following objectives: (1) assess the validity of the newly proposed species status for Aletris medogensis; (2) characterize the global structural features and investigate variations in repeat elements among cp genomes; (3) identify highly variable regions suitable for species identification and phylogenetic studies; (4) reconstruct a robust phylogeny of East Asian Aletris species and its relatives within Nartheciaceae; and (5) investigate adaptive evolution patterns of cp genes in Aletris. This study confirms the distinct taxonomic status of A. medogensis , reveals the genetic differentiation of East Asian Aletris cp genomes, and sheds light on the evolutionary history and adaptive mechanisms within Nartheciaceae. This integrative framework offers new perspectives for taxonomic studies and biodiversity conservation in complex plant groups.
Materials and Methods
2
Sampling and Morphological Comparison
2.1
Specimens of Aletris medogensis were collected from Hanmi to Xiaoyandong, opposite Beibeng Township, Medog County, Nyingchi City, Xizang, China. The voucher specimen was deposited in the Herbarium of the Chengdu Institute of Biology (CDBI), Chinese Academy of Sciences (https://cib.cas.cn/zzjg/zcbm/bbg/, Bo Xu, [email protected]), under the accession number CDBI0298322 (Figure S1). Specimens of related species were examined from herbaria PE, HNWP, WUK, SZ, and SM for comparative analysis. Morphological data, including leaves/bract‐like leaves length and width, scape size, inflorescence length, and floral parts (Table 1), were precisely quantified using ImageJ v1.48 (Schneider et al. 2012).
Sequencing, Assembly, Annotation, and Comparison
2.2
The total genomic DNA (gDNA) was extracted from the dried leaves of Aletris medogensis using a modified CTAB method (Porebski et al. 1997). The gDNA was enzymatically fragmented into 200–500 bp fragments, followed by end‐repair, poly‐A tail addition, and adapter ligation. After PCR amplification, a small‐fragment DNA library was constructed and sequenced on the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) in PE150 mode. To obtain high‐quality sequences, fastp v0.23.2 (Chen et al. 2018) was used to filter out all low‐quality reads, including those with adapters, more than 20% of bases having Phred quality < 5, and reads with > 10% N content. The chloroplast (cp) genome was assembled using GetOrganelle v1.7.7.7.1 (Jin et al. 2020) and annotated using PGA (Qu et al. 2019), with Aletris pauciflora var. pauciflora as a reference. A Python script (Appendix S1) was used to examine gene length and count, as well as the start and stop codons. In addition, Geneious Prime 8 software (https://www.geneious.com) was employed to determine the pseudogenes. The final circular map was drawn using OGDRAW v1.3.1 (Greiner et al. 2019). In addition, the genomic data of other closely related species within Nartheciaceae, including 13 Aletris, 1 Metanarthecium, 1 Nartheciaceae, and 1 Dioscorea accession (Table 2), were also downloaded and curated. Specifically, genomes with problematic annotations were systematically re‐annotated using our standardized PGA protocol to ensure data uniformity.
To compare the contraction and expansion of inverted repeat regions among the 14 cp genomes of Aletris and its closely related genera, we identified and visualized boundaries of large single copy (LSC), small single copy (SSC), and IRa/IRb regions in the 17 cp genomes using IRscope (Amiryousefi et al. 2018). Furthermore, to explore whether there were additional alterations, such as gene rearrangements or inversions, a synteny analysis of the Aletris cp genomes was performed utilizing the mVISTA program (Mayor et al. 2000).
Repeat Sequence Analysis
2.3
Simple sequence repeats (SSRs) in Aletris medogensis and the 13 published Aletris accessions (Table 2) were identified using MISA software (Beier et al. 2017), with threshold settings of 10 repeat units for mononucleotides, 6 for dinucleotides, 5 for trinucleotides, 4 for tetranucleotides, and 3 for penta‐ and hexanucleotides. Then, long sequence repeats (LSRs), including forward (F), reverse (R), palindrome (P), and complementary (C) repeats were detected within the LSC, IRa/b, and SSC regions of the Aletris chloroplast genomes using the REPuter program (Kurtz et al. 2001) with a maximum number of computed repeats of 5000 and minimal repeat size of 30 bp.
Molecular Marker Identification
2.4
The initial alignment of the 13 Aletris complete chloroplast genomes was conducted using MAFFT v7 (Katoh and Standley 2013). Subsequent identification of hypervariable loci with potential phylogenetic utility was detected based on a sliding‐window analysis method to quantify nucleotide diversity (Pi) in Aletris, with a window length of 600 bp and a step size of 200 bp. A Python script (Appendix S2) was developed for identifying hypervariable regions. Adjacent windows with a top 5% Pi value and several single nucleotide polymorphism sites (SNPs) > 25 were concatenated into contiguous hypervariable regions. The final characterization of these regions included quantitative assessments of SNPs, singleton variable sites (SVSs), parsimony informative sites (PIPs), and mean Pi values of contiguous hypervariable regions.
Phylogenetic Relationship Reconstruction
2.5
The complete chloroplast genome sequences of A. medogensis and 16 published species were retrieved from GenBank (https://www.ncbi.nlm.nih.gov/genbank/; Table 2) and aligned with MAFFT. After removing poorly aligned regions using Gblocks v0.91b (Castresana 2000), the best‐fit nucleotide substitution model (TVM+I+G) was determined by jModelTest v2.1.10 (Darriba et al. 2012). Maximum likelihood (ML) and Bayesian inference (BI) trees were then constructed using IQ‐tree v2.1.4 (Nguyen et al. 2015) and MrBayes v3.2.7a (Ronquist et al. 2012), respectively, based on the best‐fit model. Dioscorea futschauensis was set as the outgroup. Finally, the resulting trees were visualized and further landscaped in FigTree v1.4.4 (https://github.com/rambaut/figtree/releases/tag/v1.4.4).
Analysis of Selective Pressure
2.6
Given the central role of the chloroplast in photosynthesis and its interaction with environmental stressors, signatures of positive selection in its genes can provide insights into molecular adaptations at the intersection of metabolism and ecology. To this end, we calculated the nonsynonymous (d N) and synonymous (d S) substitution rates (ω) for each PCG. In general, ω > 1 indicates that the gene is under positive selection, which may drive species adaptation; ω = 1 implies neutral evolution, where no selection is acting; and ω < 1 reflects negative or purifying selection, with lower values signifying stronger selective constraints (Wang et al. 2010).
We first retrieved the PCGs shared by 17 Nartheciaceae species via CPStools v2.0.2 (Huang et al. 2024) and translated them into amino acid sequences. Homologous amino acid sequences in 17 cp genomes were then aligned using ParaAT v2.0 (Zhang et al. 2012), and the corresponding CDS alignments (codon‐based) were generated with the Epal2nal.pl script from ParaAT. Finally, KaKs_Calculator v3.0 (Wang et al. 2010) software was employed to calculate ω values for all PCGs in each species pair.
Results
3
Morphological Analyses
3.1
Morphological observations of both fresh samples from the field and dried herbarium specimens revealed that the new species exhibited morphological affinities with Aletris alpestris, A. pauciflora var. pauciflora, and A. pauciflora var. khasiana. The new species shares several characteristics with A. alpestris , including features of the plant, inflorescence structure, involucral bracts, perigone segments, corolla tube, and ovary. It also shows similarities to A. pauciflora var. pauciflora and A. pauciflora var. khasiana in leaf shape, stamens, and lobes. However, the new species is morphologically distinct from three taxa, characterized by its narrower leaves, obtuse apex, basal division, the inflorescence axis being densely covered with glandular trichomes, and two bracts that are both shorter than the flowers and unequal in length. Detailed morphological comparisons are provided in Table 1.
Characteristics of the Aletris Chloroplast Genomes
3.2
This study generated and deposited a new chloroplast genome of Aletris medogensis in GenBank (accession no. PV472299), along with 13 published genomes (two of which were re‐annotated) for comparison (Table 2). The results indicated that the whole cp genomes of Aletris ranged from 154,167 ( A. scopulorum ) to 155,143 bp ( A. glandulifera ), exhibiting a typical quadripartite structure consisting of two IR regions (IRa and IRb) of 23,857–26,711 bp, an LSC region of 82,706–83,583 bp, and an SSC region of 17,855–23,519 bp (Table 2). The GC contents of the Aletris cp genomes were similar (37.38%–37.51%), with the IRs showing the highest GC levels (42.82%–43.11%), followed by the LSC (31.31%–35.45%) and SSC regions (31.17%–33.44%; Table 2).
The cp genomes of Aletris are highly similar in terms of gene content, with most encoding 131 genes, including 85 protein‐coding genes (PCGs), 38 tRNA genes, and 8 rRNA genes (all located in the IRs; Table 2; Figure 2). Two species had few missing genes and/or pseudogenes (Table 3). Specifically, a rrn4.5 gene is missing in Aletris pauciflora var. khasiana and an additional ycf1 gene is a pseudogene in A. fauriei (Table 2). Among the 85 PCGs, 73 were unique, and six (ndhB, rpl2, rpl23, rps7, rps12, and ycf2) were duplicated due to their location in the IRs. Likewise, 22 of the tRNA genes are unique, while eight tRNA genes (trnA‐UGC, trnH‐GUG, trnI‐CAU, trnI‐GAU, trnL‐CAA, trnN‐GUU, trnR‐ACG, and trnV‐GAC) and all four rRNA genes (rrn23, rrn16, rrn5, and rrn4.5) were duplicated. Additionally, a total of 18 genes were found to contain introns across the 14 cp genomes, with nine PCGs (atpF, ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, and rps16) and six tRNA genes (trnA‐UGC, trnG‐UCC, trnI‐GAU, trnK‐UUU, trnL‐UAA, trnV‐UAC) containing one intron, while only three PCGs (rps12, ycf3, and clpP) contained two introns (Table 3).
The chloroplast genome map of East Asian Aletris species. The outer circle shows distribution of genes (different colors represent different roles). The arrows indicate the transcription directions of the genes inside and outside of the circle. The gray inner circle represents the GC content.
Codon Usage of Aletris cp Genomes
3.3
The RSCU analysis revealed a similar codon usage pattern among the 14 Aletris species, with each containing all 64 codons that encode 20 amino acids (Figure 3; Table S1). In these genomes, leucine exhibited the highest number (29,369) of codons, while cysteine was the least abundant, with 3288 codons (Figure 3; Table S2). The highest mean RSCU value was identified as UUA (1.88), and the lowest was AGC (mean RSCU = 0.28), which encode leucine and serine, respectively (Figure 3; Table S2). Furthermore, 31 codons were found with a mean RSCU of > 1, of which 29 were A/U‐ending codons; 33 codons were found with a mean RSCU of ≤ 1, of which 30 were G/C‐ending codons. This suggests strong codon usage bias favoring A/U‐ending codons over G/C‐ending codons in Aletris chloroplast genomes (Figure 3; Table S2).
Heatmap of RSCU values among Aletris species. The color gradient from blue to red represents the range of RSCU values and different graphics on the left side represent different quantity ranges.
SSRs and Long Repeat Sequences of Aletris Cp Genomes
3.4
The number of SSRs in the 14 Aletris species varied from 45 in A. scopulorum to 75 in A. megalantha, in which mono‐nucleotide SSRs were the most abundant, followed by tetra‐ and di‐nucleotide SSRs (Figure 4a; Table S3). Among the motifs in the SSRs, A/T were the most frequently occurring motifs, followed by AT/AT and TTTCA/TGAAA motifs (Figure 4b; Table S4). In addition, most of the SSRs were located in the LSC (35–59) and SSC (6–12) regions, and very few were located in the IRa/IRb (1/1–4/4; Table S3). REPuter identified 34–45 repeat sequences with a length > 30 bp, of which palindromic (17–28) and forward (14–21) repeat sequences were most abundant, while complement (0–2) and reverse (0–1) repeat sequences were very rare and almost nonexistent in many species (Figure 4c; Table S5). Most of the repeat sequences (21–26) were located in gene regions of the 14 cp genomes, followed by intergenic regions (7–16, IGS) and introns (4–6; Figure 4d; Table S5). In addition, most of the repeat sequences were less than 50 bp, and a few were larger than 100 bp (Table S6). Notably, A. pauciflora var. khasiana had a palindrome repeat sequence with a length of 2820 bp (Table S6).
Patterns of simple sequence repeats [SSRs (a, b)] and long sequence repeats [LSRs (c, d)] for the 14 Aletris chloroplast genomes. (a) Number of motifs and their abundance of SSRs in each species. (b) Type of motifs and their abundance of SSRs in each species. (c) Type and abundance of LSRs in each species. (d) Number of LSRs at different locations in each genome.
Variance Analysis of the IR Boundaries
3.5
We compared the IR boundaries among 17 Nartheciaceae cp genomes, including Dioscorea futschauensis, Metanarthecium luteoviride, Narthecium ossifragum, and the 14 Aletris accessions mentioned above (Table 2), observing minor variations in gene content, structure, and order at the quadripartite borders (Figure 5; Figure S2). The JLA (IRa‐LSC) and JSA (IRa‐SSC) boundaries were highly conserved in all 14 Aletris cp genomes, with the former positioned between psbA and trnH, and the latter cut through the ycf1 gene. The distance from the JLA boundary to psbA was consistently 138 bp, whereas that to trnH varied from 56 to 249 bp (Figure 5). The JLB (IRb‐LSC) boundaries cut through rps19 in most Aletris species, with 104 bp of rps19 extending into the IRb, while the JLA boundary of A. fauriei lay 77 bp away from rps19 due to the contraction of IRb. Similarly, the ndhF gene extended 2–16 bp into the JSB (IRb‐SSC) boundaries in most Aletris species except A. fauriei, where ndhF extended 153 bp into IRb because of IR expansion (Figure 5). Differences in the expansion/contraction of the IRs were also found in other Nartheciaceae species; only 2 and 28 bp of rps19 extended into the IRb boundary in Dioscorea futschauensis and Narthecium ossifragum, respectively. In addition, an inversion event involving the ndhF and ycf1 genes in the Dioscorea futschauensis cp genome occurred, with the former now extending 6 bp into the JSA, and the latter was cut through by the JSB boundary.
Comparison of LSC, SSC, and IR region boundaries in 17 Nartheciaceae chloroplast genomes.
Candidate Molecular Markers Identification
3.6
Using sliding window analysis, 18 hypervariable regions were identified as potential East Asia‐specific molecular markers for Aletris taxonomy, primarily occurring in the LSC and SSC regions and ranging from 523 to 2133 bp (Figure 6; Table 4). Among them, 11 intergenic spacers and 2 gene regions are located in the LSC region, 2 intergenic spacers and 1 gene are located in the SSC region, and the remaining intergenic spacer of trnN‐GUU and rrn4.5 (trnN‐GUU‐rrn4.5) is positioned in the IRa region (Figure 6). Notably, the ccsA‐ndhD intergenic spacer was the longest (2133 bp) and contained the greatest number of parsimony informative sites (PIP, 49) as well as the highest Pi value (0.020851; Table 4). In addition, the ycf1, petA, and rpoC2 genes also displayed sufficient variability and could be utilized as molecular markers for species identification and genetic studies within Aletris (Figure 6).
Nucleotide diversity (Pi, black line, vertical left axis) and number of single nucleotide polymorphism sites (blue dots, vertical right axis) of 14 Aletris cp genomes based on sliding window analysis. The window length is 600 bp and the step size is 200 bp. The horizontal axis indicates the position of the midpoint of a window. The 18 regions with high diversity (top 5%) are indicated above the peaks.
Phylogenetic Relationship of Aletris
3.7
We found that the relationships among all 14 East Asian Aletris species were completely congruent between ML and BI analyses, forming a strongly supported monophyletic group (BS = 100%, PP = 1.0) that was sister to the outgroup taxa: Dioscorea futschauensis, Metanarthecium luteoviride, and Narthecium ossifragum (BS = 100%, PP = 1.0; Figure 7). While the BI analysis fully resolved the outgroup relationships, with Metanarthecium luteoviride and Narthecium ossifragum forming a sister group that was then sister to Dioscorea futschauensis (PP = 1.0), the ML analysis left them as an unresolved polytomy (Figure 7).
Phylogenetic tree inferred from Maximum Likelihood (ML, on the left) and Bayesian Inference (BI, on the right) methods for East Asian Aletris species and their closely related species based on whole chloroplast genomes. The numbers above branches indicate ML bootstrap support (BS) and Bayesian posterior probabilities (PP) respectively.
Within Aletris species, three evolutionary clades emerged with strong support (BS = 100%, PP = 1): A. fauriei occupies an isolated position as the earliest‐diverging lineage (Clade III), sister to the remaining species, which can be further subdivided into two additional clades (Figure 7). The new species, A. medogensis , clustered as a sister group to a subclade consisting of A. spicata , A. stenoloba, A. alpestris , and A. scopulorum , and together as a sister to A. glandulifera , forming Clade II. Clade I united A. gracilis , A. glabra , A. pauciflora , A. pedicellata , A. pauciflora var. khasiana, A. laxiflora , and A. megalantha. Critically, A. medogensis exhibited stronger phylogenetic affinity to A. alpestris in Clade II than to its morphological relatives A. pauciflora var. pauciflora and A. pauciflora var. khasiana in Clade I (Figure 7), revealing significant morphology–molecular discordance.
Selective Pressure of cp Genes in Nartheciaceae
3.8
A total of 77 PCGs shared among 17 Nartheciaceae cp genomes were analyzed for selective pressure, and most of them were subjected to purifying selection (K a/K s < 1; Figure 8; Table S7). By calculating the K a/K s ratios for homologous genes within species pairs, only ccsA in Aletris scopulorum, cemA in A. medogensis and A. stenoloba, rps12 in A. spicata and A. fauriei, psbK in Dioscorea futschauensis, and rpl2 in Narthecium ossifragum showed signatures of positive selection (K a/K s > 1). These findings suggest that some mutations were positively favored by selection in specific species. Furthermore, eight genes (atpE, matK, ndhF, cemA, atpF, ccsA, rps12, and ycf1) evolved faster in most Aletris species than in the other three Nartheciaceae species branches (Figure 8; Table S7), potentially reflecting species‐specific adaptations.
Heatmap of the mean K a/K s values of 77 PCGs shared by 17 Nartheciaceae species.
Discussion
4
The discovery of Aletris medogensis as a new species (Figure 1), supported by both morphological distinctiveness and chloroplast genome analysis, highlights the importance of integrating traditional taxonomy with modern molecular tools. This study not only expands known endemism in the eastern Himalayas but also provides critical insights into the evolutionary dynamics and genomic features of Nartheciaceae.
Chloroplast Genome Evolution in Aletris
4.1
Consistent with other angiosperms (Sugiura 1992; Daniell et al. 2016), the comparative analysis of the 14 East Asian Aletris cp genomes revealed a conserved quadripartite structure with minor variations in size (154,167–155,143 bp) and gene content (Table 2). Various elements influencing chloroplast structure and function, such as the contraction and expansion of the IR regions, gene insertions, deletions, duplications, inversions, and alterations in introns, have been reported in many species (Tsudzuki et al. 1992; Lin et al. 2012; Li et al. 2020). Notably, the pseudogenization of the ycf1 gene in A. fauriei and the loss of rrn4.5 in A. pauciflora var. khasiana are of functional interest. The former is significant given its role in plastid protein import and stress responses, while the latter might reduce ribosomal redundancy in stable riparian niches, potentially enhancing DNA repair capacity in high‐UV montane habitats (Dong et al. 2015; Wei et al. 2024). Additionally, the IR boundary dynamics also observed here—particularly the expansion of ndhF into IRb and the contraction of rps19 from IRb in A. fauriei (Figure 5)—reflect species‐specific genomic rearrangements. Such IR contractions/expansions can alter gene dosage and disrupt operon integrity, potentially affecting photosynthetic efficiency (Kim and Lee 2005; Zoschke and Bock 2018).
The strong codon usage bias favoring A/U‐ending codons (Figure 3) aligns with angiosperm‐wide trends but shows intensified bias in Himalayan species (Liu and Xue 2005; Smarda et al. 2014). This likely optimizes translational efficiency under low‐temperature stress, as A/U‐rich transcripts fold less stably, facilitating ribosome binding in cold environments (Chiba et al. 2013; Krasileva et al. 2017). Additionally, the excess of SSRs and LSRs in intergenic regions (Figure 4d) suggests their role in regulating genome plasticity; the 2820 bp palindromic repeat in A. pauciflora var. khasiana (Table S6) could promote recombination‐driven innovation, potentially accelerating adaptation to microhabitat heterogeneity (Wicker et al. 2007; Oliver et al. 2013).
Phylogenetic Implications for Aletris Taxonomy
4.2
Although A. medogensis possesses a unique combination of morphological characters not found in its morphologically similar relatives (e.g., A. alpestris , A. pauciflora ), morphological characteristics alone may be insufficient to resolve species boundaries, where subtle differences in floral structures and vegetative traits often overlap among taxa (Table 1). Our chloroplast phylogenomic analysis resolves long‐standing controversies in East Asian Aletris systematics. First, Metanarthecium luteoviride formed a clade with Narthecium ossifragum (PP = 1, Figure 7), providing robust genomic evidence to exclude it from Aletris—a conclusion consistent with Zhao et al. (2012) but contradicting fragment‐based studies (Merckx et al. 2008; Fuse et al. 2012). Second, our data confirm that A. pauciflora var. khasiana and A. pauciflora var. pauciflora are phylogenetically distinct (Figure 7), validating Zhao et al.'s (2012) proposal to recognize the former as a separate species. Critically, the whole cp genome tree provided robust molecular evidence to corroborate the distinctiveness of Aletris medogensis by demonstrating that it was phylogenetically distant from the two varieties of A. pauciflora (which were nested within Clade I), and that, despite belonging to the same major clade (Clade II), it was not closely related to A. alpestris , which formed a separate subclade (Figure 7). This discordance between morphological affinity and phylogenetic placement underscores the necessity of molecular data to clarify evolutionary relationships in taxonomically challenging groups (Redwan et al. 2015).
While our integrative evidence from morphology and chloroplast phylogenomics robustly supports the recognition of A. medogensis as a distinct species, phylogenetic inferences based solely on plastid data may not always reflect the true species tree because of evolutionary processes such as incomplete lineage sorting and introgression, due to its uniparental inheritance. Therefore, the employment of nuclear markers, or a combination of nuclear and plastid data, is strongly advocated in future studies to further validate and refine the phylogenetic relationships proposed here.
Hypervariable Regions and Molecular Markers
4.3
The hypervariable regions identified here (Figure 6; Table 4), including ycf1 (Pi = 0.0183), petA (Pi = 0.0183), and rpoC2 (Pi = 0.0183) gene regions, offer superior resolution for species delimitation compared to traditional markers (e.g., matK/trnL‐F) (Zhao et al. 2012). For instance, the A. spicata –A. stenoloba complex could not be resolved in previous studies due to identical sequences (Zhao et al. 2012), whereas our SNPs of the ycf1 gene clearly distinguish them, implying the plastid gene ycf1 could serve as a key barcode for Aletris. In addition, these hypervariable regions will serve as valuable molecular markers for future phylogenetic and population genetic studies (Wei et al. 2024). The prevalence of SSRs (45–75 per genome) and LSRs (34–45 per genome) underscores the potential role of repetitive elements in shaping cp genome evolution (Tables S3–S5) (Xu et al. 2023). The exceptional 2820 bp palindromic repeat in A. pauciflora var. khasiana (Table S6) may serve as a hotspot for recombination, contributing to genomic plasticity (Feng et al. 2022).
Selective Pressures and Adaptive Evolution
4.4
Positively selected genes are pivotal drivers of evolutionary innovation, enabling organisms to exploit new ecological niches and respond dynamically to changing environments (Moseley et al. 2018). Unlike negative (purifying) selection, which maintains genomic stability by removing deleterious mutations across long evolutionary timescales (Cvijovic et al. 2018), positive selection favors advantageous alleles that confer improved fitness—whether through enhanced metabolic efficiency, novel biochemical pathways, or refined sensory capabilities (Messer and Petrov 2013; Li et al. 2025).
Most protein‐coding genes in Aletris are under purifying selection (ω < 1; Figure 8; Table S7), consistent with the functional constraints of essential photosynthetic and ribosomal genes. However, positive selection (ω > 1) was detected in ccsA ( A. scopulorum ), cemA ( A. medogensis and A. stenoloba), and rps12 ( A. spicata and A. fauriei). These genes, involved in cytochrome synthesis, chloroplast envelope stability, and ribosomal function (Xie and Merchant 1996; Ogawa et al. 1994; Ramundo et al. 2013), have also been found to undergo positive selection in other taxa (e.g., Ficus, Populus) (Zhang et al. 2022; Shi et al. 2025). Their accelerated evolution might reflect species‐specific adaptations to environmental stressors like high‐altitude habitats or pathogen resistance (Zhang et al. 2020). The faster evolutionary rates of matK and ndhF in Aletris than in other Nartheciaceae genera further highlight lineage‐specific selective regimes, possibly linked to ecological diversification (Barthet and Hilu 2007; Zhao et al. 2017).
Conservation Implications
4.5
Despite its restricted distribution in Medog County, A. medogensis is currently classified as data deficient (DD) due to insufficient population data. The species' reliance on moss‐covered riparian habitats makes it vulnerable to climate change, which can manifest as altered stream flow regimes and the desiccation of its required microclimate, and to direct anthropogenic disturbances such as tourism development and infrastructure construction along its fragile streamside habitat (Xu et al. 2009; Zhang and Yan 2023). In‐depth surveys are needed across similar ecosystems in the Eastern Himalayas to assess the true distribution and conservation status of this endemic lineage, allowing for essential morphometric comparisons across populations to validate a suite of stable, diagnostic traits. Additionally, the genomic resources generated here, including hypervariable markers and cp genome annotations, will facilitate monitoring efforts and inform conservation strategies for this endemic lineage.
Conclusions
5
The integration of morphology, chloroplast genomics, and phylogenomics effectively resolves taxonomic conflicts in Aletris and reveals evolutionary mechanisms underlying species diversification. This work enhances our understanding of Himalayan plant diversity and provides a framework for future studies on cryptic species in evolving ecosystems.
Taxonomic Treatment
6
** Aletris medogensis ** W.B.Ju, Y.L.Qiu & Bo Xu, sp. nov.
Type
6.1
CHINA. Xizang: Motuo County, Beibeng Xiang, from Hanmi to Xiaoyandong on the opposite side, ca. 2931 m, 29°23′25.24′′, 95°06′00.75′′, occurring on moss‐covered rocks along riparian zones. June 9, 2024, Ju Wen‐Bin, Li Jiang‐Tao, Zeng Li‐Ting, and Deng Hui‐Wen YLZB12018 (holotype: CDBI0298322; Figure S1).
Diagnosis
6.2
Aletris medogensis is morphologically most similar to Aletris alpestris, but it can be distinguished from the latter in having narrower leaves (0.45–0.9 mm wide vs. 1–2.5 mm wide) and an obtuse apex (vs. an acuminate apex); the flower pedicels are extremely short, the rachis has densely glandular hairs (vs. sparsely puberulent), the two bracts are unequal in length and shorter than the flowers (vs. one of them 1–4 × flower length). Lobes are obtuse to rounded at the apex (vs. the apex being obtuse to acute).
Description
6.3
Perennial herb, tillering with developing creeping stolons. Roots are usually fibrous. Leaves in basal rosette, occasionally, leaf primordia split from the basal region, giving rise to new leaves, linear, 2.5–9 cm × 0.45–0.9 mm, apex obtuse, glabrous. Scape 3.5–16 cm × 0.7–1 mm, glandular are sparse near the inflorescence and progressively become glabrous toward the lower part, bract‐like leaves 0.2–0.6 cm long in the middle and lower part, lanceolate or ovate‐lanceolate, apex acute. Raceme 1.5–2.5 cm, densely 10–14‐flowered, pedicels extremely short, rachis densely glandular hairs; bract and bracteole lanceolate, 2–4 mm, borne at the apex of the pedicel, shorter than the flower. Perianth white, glabrous, split to the middle; tube campanulate; lobes recurved or erect, ovate, apex obtuse to rounded, 1.2–2 × 0.8–1.5 mm. Filaments of stamens adnate to perianth, 0.5 mm, anthers elliptical, 0.5 mm, the style abruptly constricted into a short style, stigma not or only slightly thickened, capitate. Ovary ovate, ridged. Fruits capsular, 3‐locular (Figures 9 and 10).
Line drawing of Aletris medogensis. Lateral (a) and front (b) views of the flower. (c) Stamens. (d) Ovary and stigma. (e) Inflorescence. (f, g) Plants.
Aletris medogensis. Lateral (a), front (b), and back (c) views of the flower. (d) Stamens. (e) Ovary and stigma. (f) Inflorescence. (g) Scape. (h) Bracteate leaf. (i) Leaf. (k) Roots. (l) Plant.
Phenology
6.4
Flowering from June to July, fruiting from August to September.
Etymology
6.5
Located in southeastern Xizang Autonomous Region, Medog (Pinyin spelling “motuo”) County is one of the biodiversity hotspots in China, which has rich plant diversity in the Eastern Himalaya (Qiu et al. 2022). The new species, Aletris medogensis, is found in this region and is named after the geographic location. Its Chinese name, mo tuo fen tiao er cai (墨脱粉条儿菜).
Distribution and Ecology
6.6
This new species is currently known only from the opposite bank of Hanmi to the Xiaoyandong direction in Motuo County, Nyingchi City, Xizang (Tibet), China, at an elevation of 2200–2400 m. Aletris medogensis grows on moss‐covered rocks along streams in moist environments.
Conservation Status
6.7
Currently known from a small population at the type locality, approximately 150 mature individuals have been observed. The data available for this new species are still insufficient to assess its conservation status. According to the IUCN 2023 Red List criteria (International Union for Conservation of Nature 2023), the species is classified as Data Deficient (DD). Further collection and monitoring efforts are required to enable more conclusive assessments of the species' rarity and vulnerability.
Author Contributions
Xiong Li: data curation (lead), software (equal), visualization (equal), writing – original draft (lead), writing – review and editing (lead). Yong‐Ling Qiu: data curation (equal), software (equal), writing – original draft (equal), writing – review and editing (supporting). Jiang‐Tao Li: investigation (equal), writing – review and editing (supporting). Bo Xu: writing – original draft (supporting), writing – review and editing (supporting). Qi Yu: writing – review and editing (supporting). Wen‐Bin Ju: investigation (lead), project administration (lead), resources (lead), writing – review and editing (equal).
Funding
This work was supported by the Science and Technology Major Project of Xizang (XZ202501ZY05151), the Special Investigation and Monitoring of the Yarlung Zangbo Grand Canyon National Nature Reserve in Xizang (GZFCG202314256), and the Second Tibetan Plateau Scientific Expedition and Research (STEP) program (2024QZKK0200).
Ethics Statement
This study's material collections and experimental research followed the relevant institutional, national, and international guidelines and legislation.
Consent
The authors have nothing to report.
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Appendix S1–S2: ece372654‐sup‐0001‐AppendixS1‐S2.docx.
Figures S1–S2: ece372654‐sup‐0002‐FigureS1‐S2.docx.
Tables S1–S7: ece372654‐sup‐0003‐TableS1‐S7.xlsx.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Adolf, E. 1905. Botanische Jahrbücher für Systematik, Pflanzengeschichte und Pflanzengeographie. Vol. 36. Forgotten Books.
- 2Akahori, A. , F. Yasuda , and T. Okanishi . 1971. “Steroidal Sapogenins of Aletris spicata (Thunb.) Franchet.” Chemical & Pharmaceutical Bulletin 19: 2409–2411.
- 3Ambrose, J. D. 1980. “A Re‐Evaluation of the Melanthioideae (Liliaceae) Using Numerical Analyses.” In Pelaloid Monocotyledons. Linnean Society Symposium Series, edited by C. D. Brickell and M. Gregory , vol. 8, 65–82. Academic Press.
- 4Amiryousefi, A. , J. Hyvonen , and P. Poczai . 2018. “Irscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes.” Bioinformatics 34, no. 17: 3030–3031.29659705 10.1093/bioinformatics/bty 220 · doi ↗ · pubmed ↗
- 5Barthet, M. M. , and K. W. Hilu . 2007. “Expression of Matk: Functional and Evolutionary Implications.” American Journal of Botany 94, no. 8: 1402–1412.21636508 10.3732/ajb.94.8.1402 · doi ↗ · pubmed ↗
- 6Beier, S. , T. Thiel , T. Munch , U. Scholz , and M. Mascher . 2017. “Misa‐Web: A Web Server for Microsatellite Prediction.” Bioinformatics 33, no. 16: 2583–2585.28398459 10.1093/bioinformatics/btx 198PMC 5870701 · doi ↗ · pubmed ↗
- 7Brotherus, V. F. , and H. R. E. Handel‐Mazzetti . 1936. Symbolae Sinicae: Botanische Ergebnisse Der Expedition Der Akademie Der Wissenschaften in Wein Nach Südwest‐China, 1914–1918. Vol. 7. Akademie der Wissenschaften.
- 8Caddick, L. R. , P. J. Rudall , P. Wilkin , T. A. J. Hedderson , and M. W. Chase . 2002. “Phylogenetics of Dioscoreales Based on Combined Analyses of Morphological and Molecular Data.” Botanical Journal of the Linnean Society 138, no. 2: 123–144.